Co-reporter:Weijing Yang, Yifeng Xia, Yan Zou, Fenghua Meng, Jian Zhang, and Zhiyuan Zhong
Chemistry of Materials October 24, 2017 Volume 29(Issue 20) pp:8757-8757
Publication Date(Web):September 29, 2017
DOI:10.1021/acs.chemmater.7b02953
Rapidly evolving protein technology has generated hundreds of therapeutic proteins that are promising for treating various human diseases. The clinical use of protein drugs remains, however, limited due to the absence of viable vehicles. Here, we report that anisamide-functionalized bioresponsive chimaeric nanopolymersomes (Anis-BCPs) can efficiently load granzyme B (GrB), a potent apoptotic protein, and enable targeted and efficacious protein therapy for H460 human lung cancer in vivo. Anis-BCPs are readily obtained from poly(ethylene glycol)-b-poly(N-2-hydroxypropyl methacrylamide-g-lipoic acid)-b-poly(acrylic acid) triblock copolymer. Notably, GrB-loaded Anis-BCPs a display superior antitumor effect toward sigma receptor-overexpressing H460 lung cancer cells (IC50 = 7.8 nM). The in vivo studies reveal that Anis-BCPs have a long circulation time and remarkable tumor accumulation. Interestingly, GrB-loaded Anis-BCPs at 6.24 nmol GrB equiv/kg dose, given either in four injections or one single injection, effectively inhibit H460 tumor growth and significantly improve the survival rate for mice. These robust, bioresponsive, and nontoxic chimaeric nanopolymersomes provide a potential platform for cancer protein therapy as well as basic research on intracellular functional proteins.
Co-reporter:Yaqin Zhu, Jian Zhang, Fenghua Meng, Chao Deng, Ru Cheng, Jan Feijen, and Zhiyuan Zhong
ACS Applied Materials & Interfaces October 18, 2017 Volume 9(Issue 41) pp:35651-35651
Publication Date(Web):September 27, 2017
DOI:10.1021/acsami.7b12439
The application of cell-penetrating peptides like TAT for in vivo targeted delivery is limited because the penetration behavior is not cell-specific. Herein, we designed cRGD and TAT comodified cross-linkable micelles (cRGD/TAT CMs), in which the TAT peptide was shielded by relatively long poly(ethylene glycol) (PEG) chains. Docetaxel (DTX)-loaded cRGD/TAT CMs were very stable with minimal drug leakage under physiological conditions, whereas rapid DTX release took place in a reductive environment. Flow cytometry showed that the cRGD/TAT CMs with molar ratios of 20% cRGD and 10% TAT (cRGD20/TAT10 CMs) were selectively and efficiently taken up by ανβ3-overexpressing U87MG glioma cells, with 8.3-fold and 18.3-fold higher uptake than cRGD20 CMs and PEG CMs, respectively. DTX-loaded cRGD20/TAT10 CMs exhibited a high cytotoxicity in U87MG cells, leading to rapid apoptosis of the tumor cells. Uptake mechanism studies revealed that cRGD20/TAT10 CMs mainly employed the caveolae-mediated endocytotic pathway and efficiently escaped from the lysosomes. Notably, cRGD20/TAT10 CMs had a long circulating time of 6.25 h in vivo, due to cross-linking of the micelles and shielding of the TAT peptide. Moreover, DTX-loaded cRGD20/TAT10 CMs exhibited a significantly higher accumulation and deeper penetration in subcutaneous U87MG glioma tissue compared to cRGD20 CMs and PEG CMs, leading to superior antitumor efficacy in vivo. Therefore, this dual-ligand strategy provides an effective way to realize tumor-specific penetration and inhibition.Keywords: anticancer drug; Biodegradable micelles; reduction-sensitive; targeted delivery; tumor penetration;
Co-reporter:Zhiyuan Zhong
Biomacromolecules November 13, 2017 Volume 18(Issue 11) pp:3652-3652
Publication Date(Web):November 13, 2017
DOI:10.1021/acs.biomac.7b01459
Co-reporter:Jing Chen, Jia Ouyang, Qijun Chen, Chao Deng, Fenghua Meng, Jian Zhang, Ru Cheng, Qing Lan, and Zhiyuan Zhong
ACS Applied Materials & Interfaces July 19, 2017 Volume 9(Issue 28) pp:24140-24140
Publication Date(Web):July 4, 2017
DOI:10.1021/acsami.7b06879
Protein drugs with intracellular targets like Granzyme B (GrB) have demonstrated great proliferative inhibition activity in cancer cells. Their clinical translation, however, relies on the development of safe, efficient, and selective protein-delivery vehicles. Here, we report that epidermal growth factor receptor (EGFR) and CD44 dual-targeted multifunctional hyaluronic acid nanogels (EGFR/CD44-NGs) boost protein delivery to ovarian and breast cancers in vitro and in vivo. EGFR/CD44-NGs obtained via nanoprecipitation and photoclick chemistry from hyaluronic acid derivatives with tetrazole, GE11 peptide/tetrazole, and cystamine methacrylate groups had nearly quantitative loading of therapeutic proteins like cytochrome C (CC) and GrB, a small size of ca. 165 nm, excellent stability in serum, and fast protein release under a reductive condition. Flow cytometry assays showed that EGFR/CD44-NGs exhibited over 6-fold better uptake in CD44 and EGFR-positive SKOV-3 ovarian cancer cells than CD44-NGs. In accordance, GrB-loaded EGFR/CD44-NGs (GrB-EGFR/CD44-NGs) displayed enhanced caspase activity and growth inhibition in SKOV-3 cells as compared to GrB-loaded CD44-NGs (GrB-CD44-NGs) control. Intriguingly, the therapeutic studies in SKOV-3 human ovarian carcinoma and MDA-MB-231 human breast tumor xenografted in nude mice revealed that GrB-EGFR/CD44-NGs at a low dose of 3.85 nmol GrB equiv/kg induced nearly complete growth suppression of both tumors, which was obviously more effective than GrB-CD44-NGs, without causing any adverse effects. EGFR and CD44 dual-targeted multifunctional hyaluronic acid nanogels have appeared as a safe and efficacious platform for cancer protein therapy.Keywords: cancer therapy; dual-targeting; nanogels; protein delivery; redox-sensitive;
Co-reporter:Min Qiu, Jia Ouyang, Huanli Sun, Fenghua Meng, Ru Cheng, Jian Zhang, Liang Cheng, Qing Lan, Chao Deng, and Zhiyuan Zhong
ACS Applied Materials & Interfaces August 23, 2017 Volume 9(Issue 33) pp:27587-27587
Publication Date(Web):August 7, 2017
DOI:10.1021/acsami.7b10533
Poly(ethylene glycol)-b-polypeptide block copolymer micelles, with excellent safety, are one of the most clinically studied nanocarriers for anticancer drug delivery. Notably, self-assembled nanosystems based on hydrophobic polypeptides showing typically a low drug loading and burst drug release are limited to preclinical studies. Here, we report that poly(ethylene glycol)-b-poly(α-aminopalmitic acid) (PEG-b-PAPA) block copolymer could be easily prepared with tailored Mn through ring-opening polymerization of α-aminopalmitic acid N-carboxyanhydride (APA-NCA). Interestingly, PEG-b-PAPA copolymers exhibited superb solubility in common organic solvents (including CHCl3, CH2Cl2, and THF), while stable nanomicelles were formed in phosphate buffer, with a small size of 59 nm and a low critical micelle concentration of 2.38 mg/L. These polylipopeptide micelles (Lipep-Ms) allowed facile loading of a potent anticancer drug, docetaxel (DTX), likely due to the existence of a strong interaction between the lipophilic drug and polylipopeptide in the core. Notably, cRGD-peptide-functionalized Lipep-Ms (cRGD-Lipep-Ms) were also obtained with similar biophysical characteristics. The in vitro studies showed efficient cellular uptake of DTX-loaded cRGD-Lipep-Ms by B16F10 cells and fast intracellular drug release due to the enzymatic degradation of PAPA blocks in endo/lysosome, leading to a pronounced anticancer effect (IC50 = 0.15 μg DTX equiv/mL). The in vivo therapy studies showed that DTX–cRGD-Lipep-Ms exhibited superior tumor growth inhibition of B16F10 melanoma, improved survival rate, and little side effects as compared to free DTX. These polylipopeptide micelles appear as a promising and robust nanoplatform for anticancer drug delivery.Keywords: cancer therapy; docetaxel; drug delivery; lipopeptide; micelles; polypeptide;
Co-reporter:Jintian Wu, Jian Zhang, Chao Deng, Fenghua MengRu Cheng, Zhiyuan Zhong
ACS Applied Materials & Interfaces 2017 Volume 9(Issue 4) pp:
Publication Date(Web):January 12, 2017
DOI:10.1021/acsami.6b15105
PLGA-based nanomedicines have enormous potential for targeted cancer therapy. To boost their stability, targetability, and intracellular drug release, here we developed novel multifunctional PLGA anticancer nanomedicines by combining a reductively cleavable surfactant (RCS), vitamin E–SS–oligo(methyl diglycol l-glutamate), with covalent hyaluronic acid (HA) coating. Reduction-sensitive HA-coated PLGA nanoparticles (rHPNPs) were obtained with small sizes of 55–61 nm and ζ potentials of −26.7 to −28.8 mV at 18.4–40.3 wt % RSC. rHPNPs were stable against dilution and 10% FBS while destabilized under reductive condition. The release studies revealed significantly accelerated docetaxel (DTX) release in the presence of 10 mM glutathione. DTX–rHPNPs exhibited potent and specific antitumor effect to CD44 + A549 lung cancer cells (IC50 = 0.52 μg DTX equiv/mL). The in vivo studies demonstrated that DTX–rHPNPs had an extended circulation time and greatly enhanced tolerance in mice. Strikingly, DTX–rHPNPs completely inhibited growth of orthotopic human A549-Luc lung tumor in mice, leading to a significantly improved survival rate and reduced adverse effect as compared to free DTX. This study highlights that advanced nanomedicines can be rationally designed by combining functional surfactants and surface coating.Keywords: docetaxel; lung cancer; PLGA nanoparticles; reduction-responsive; surface coating; surfactant;
Co-reporter:Yan Zou;Meng Zheng;Weijing Yang;Fenghua Meng;Kanjiro Miyata;Hyun Jin Kim;Kazunori Kataoka
Advanced Materials 2017 Volume 29(Issue 42) pp:
Publication Date(Web):2017/11/01
DOI:10.1002/adma.201703285
AbstractSmall interfering RNA (siRNA) offers a highly selective and effective pharmaceutical for various life-threatening diseases, including cancers. The clinical translation of siRNA is, however, challenged by its short plasma life, poor cell uptake, and cumbersome intracellular trafficking. Here, cNGQGEQc peptide-functionalized reversibly crosslinked chimaeric polymersomes (cNGQ/RCCPs) is shown to mediate high-efficiency targeted delivery of Polo-like kinase1 specific siRNA (siPLK1) to orthotopic human lung cancer in nude mice. Strikingly, siRNA is completely and tightly loaded into the aqueous lumen of the polymersomes at an unprecedentedly low N/P ratio of 0.45. cNGQ/RCCPs loaded with firefly luciferase specific siRNA (siGL3) or siPLK1 are efficiently taken up by α3β1-integrin-overexpressing A549 lung cancer cells and quickly release the payloads to the cytoplasm, inducing highly potent and sequence-specific gene silencing in vitro. The in vivo studies using nude mice bearing orthotopic A549 human lung tumors reveal that siPLK1-loaded cNGQ/RCCPs boost long circulation, superb tumor accumulation and selectivity, effective suppression of tumor growth, and significantly improved survival time. These virus-mimicking chimaeric polymersomes provide a robust and potent platform for targeted cancer siRNA therapy.
Co-reporter:Kaiqi Wu;Ru Cheng;Jian Zhang;Fenghua Meng;Chao Deng
Journal of Materials Chemistry B 2017 vol. 5(Issue 28) pp:5658-5667
Publication Date(Web):2017/07/19
DOI:10.1039/C7TB01297G
Bortezomib (BTZ) is the first proteasome inhibitor approved for the treatment of malignant tumors. The current clinical formulation, however, shows fast clearance, low tumor accumulation, and several side effects. Here, we report that micellar nanoformulation of lipophilized bortezomib achieves significantly enhanced drug loading, prolonged circulation time, improved tolerability and targeted treatment of triple negative breast cancer in vivo. Lipophilized bortezomib, bortezomib-pinanediol (BP), was readily prepared in high yield. Interestingly, cRGD-targeted micelles based on poly(ethylene glycol)-b-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate) achieved a high drug loading content of 8.05 wt% BTZ equiv. for BP, which was more than 8-fold higher than BTZ. BP-loaded cRGD-decorated micelles (BP-cRGD-Ms) exhibited a small size (ca. 49 nm), reduction-triggered drug release, and active targeting ability to αvβ3-overexpressing MDA-MB-231 triple-negative breast cancer cells, resulting in a low IC50 of 0.986 μM. The in vivo studies displayed that BP-cRGD-Ms had a nearly 20-fold improvement in the elimination half-life and a 20-fold higher maximum-tolerated dose as compared to free BTZ. The biodistribution and therapeutic studies in MDA-MB-231 tumor-bearing nude mice demonstrated that BP-cRGD-Ms induced significantly better tumor accumulation and inhibition with fewer adverse effects than free BTZ, leading to greatly improved mice survival rates. This micellar nanoformulation of lipophilized bortezomib appears to be a novel and effective strategy to achieve targeted tumor chemotherapy with bortezomib.
Co-reporter:Ya Fang, Yu Jiang, Yan Zou, Fenghua Meng, ... Zhiyuan Zhong
Acta Biomaterialia 2017 Volume 50(Volume 50) pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.actbio.2017.01.007
Cyclic RGD peptide-functionalized reversibly core-crosslinked biodegradable poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG-PCL) micelles (cRGD-RCCMs) were designed and developed for highly potent and targeted glioma chemotherapy. To achieve crosslinkable core, dithiolane-functionalized trimethylene carbonate (DTC) was incorporated into PCL block. Interestingly, cRGD-RCCMs displayed a high doxorubicin (DOX) loading content of ∼18 wt%, small hydrodynamic size of ∼50 nm, and excellent colloidal stability with minimum drug leakage under physiological conditions while fast DOX release under cytoplasmic-mimicking reductive environments. MTT, confocal microscopy and flow cytometry measurement results pointed out that cRGD-RCCMs with 30% cRGD surface density (cRGD30-RCCMs) showed an evident selectivity, efficient cytoplasmic drug release, and superior antitumor activity to clinically used pegylated liposomal doxorubicin (DOX-LPs) in αvβ3 integrin overexpressing U87MG glioblastoma cells. Strikingly, DOX-loaded cRGD30-RCCMs demonstrated a prolonged circulation time showing an elimination half-life of ∼4.7 h, three times exceeding that of the non-crosslinked counterparts, and a remarkably enhanced tumor accumulation of 7.7%ID/g. Furthermore, in vivo therapeutic studies revealed that DOX-loaded cRGD30-RCCMs effectively suppressed tumor growth, significantly prolonged survival time, and lessened side effects in subcutaneous U87MG glioblastoma-bearing nude mice. These reversibly core-crosslinked multifunctional biodegradable micelles might be developed into advanced and clinically viable targeted anticancer nanomedicines.Statement of SignificanceNanomedicines based on biodegradable micelles and nanoparticles offer a most promising treatment for malignant tumors. The therapeutic outcomes of current nanomedicines are, however, trimmed by their instability, low tumor retention, inefficient tumor cell uptake, and inferior drug release control. We report herein that cRGD-functionalized, rapidly glutathione-responsive, and reversibly core-crosslinked biodegradable micellar doxorubicin based on PEG-PCL block copolymer mediates potent and targeted glioma chemotherapy, affording significantly better treatment efficacy and lower systemic toxicity than the non-crosslinked micellar doxorubicin and clinically used pegylated liposomal doxorubicin controls. These reversibly core-crosslinked multifunctional biodegradable micelles have emerged as a robust, simple, versatile, and safe nanoplatform that might elegantly bridge the gap between the scientific and translational anticancer nanomedicine research.Download high-res image (131KB)Download full-size image
Co-reporter:Yuan Fang, Weijing Yang, Liang Cheng, Fenghua Meng, ... Zhiyuan Zhong
Acta Biomaterialia 2017 Volume 64(Volume 64) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.actbio.2017.10.013
Liver cancer is a globally leading malignancy that has a poor five-year survival rate of less than 20%. The systemic chemotherapeutics are generally ineffective for liver cancers partly due to fast clearance and low tumor uptake. Here, we report that GE11 peptide functionalized polymersomal doxorubicin (GE11-PS-DOX) effectively targets and inhibits epidermal growth factor receptor (EGFR)-positive SMMC7721 orthotopic human liver tumor xenografts in mice. GE11-PS-DOX with a GE11 surface density of 10% displayed a high drug loading of 15.4 wt%, a small size of 78 nm, and glutathione-triggered release of DOX. MTT assays, flow cytometry and confocal microscopy studies revealed that GE11-PS-DOX mediated obviously more efficient DOX delivery into SMMC7721 cells than the non-targeting PS-DOX and clinically used liposomal doxorubicin (Lipo-DOX) controls. The in vivo studies showed that GE11-PS-DOX had a long circulation time and an extraordinary accumulation in the tumors (13.3 %ID/g). Interestingly, GE11-PS-DOX caused much better treatment of SMMC7721 orthotopic liver tumor-bearing mice as compared to PS-DOX and Lipo-DOX. The mice treated with GE11-PS-DOX (12 mg DOX equiv./kg) exhibited a significantly improved survival rate (median survival time: 130 days versus 70 and 38 days for PS-DOX at 12 mg DOX equiv./kg and Lipo-DOX at 6 mg DOX equiv./kg, respectively) and achieved 50% complete regression. Notably, GE11-PS-DOX induced obviously lower systemic toxicity than Lipo-DOX. EGFR-targeted multifunctional polymersomal doxorubicin with improved efficacy and safety has a high potential for treating human liver cancers.Statement of SignificanceLiver cancer is one of the top five leading causes of cancer death worldwide. The systemic chemotherapeutics and biotherapeutics generally have a low treatment efficacy for hepatocellular carcinoma partly due to fast clearance and/or low tumor uptake. Nanomedicines based on biodegradable micelle and polymersomes offer a most promising treatment for malignant liver cancers. Their clinical effectiveness remains, however, suboptimal owing to issues like inadequate systemic stability, low tumor accumulation and selectivity, and poor control over drug release. Here we report that GE11 peptide-functionalized, disulfide-crosslinked multifunctional polymersomal doxorubicin (GE11-PS-DOX) can effectively suppress the growth of orthotopic SMMC7721 human liver tumors in nude mice. They showed significantly decreased systemic toxicity and improved mouse survival rate with 3.4-fold longer median survival time as compared to clinically used pegylated liposomal doxorubicin (Lipo-DOX) and achieving 50% complete regression. GE11-PS-DOX, based on PEG-PTMC is biodegradable, nontoxic, and easy to prepare, appears as a safe, robust, versatile and all-function-in-one nanoplatform that has a high potential in targeted chemotherapy of EGFR expressed hepatocellular carcinoma.Download high-res image (84KB)Download full-size image
Co-reporter:Chunfeng Hang, Yan Zou, Yinan Zhong, Zhiyuan Zhong, Fenghua Meng
Colloids and Surfaces B: Biointerfaces 2017 Volume 158(Volume 158) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.colsurfb.2017.07.041
•DOX loaded HA-CM nanogels can be facilely prepared.•Both UV and NIR can trigger DOX release from HA-CM nanogels.•HA-CM nanogels enter CD44+ cancer cells via receptor mediated endocytosis.•NIR triggers nanogels to release DOX intracellularly causing tumor cell death.Hyaluronic acid (HA) is an endogenous polysaccharide that shows intrinsic targetability to CD44+ cancer cells. Here, we developed NIR and UV-responsive degradable nanogels from hyaluronic acid-g-7-N,N-diethylamino-4-hydroxymethylcoumarin (HA-CM) for CD44 targeted and remotely controlled intracellular doxorubicin (DOX) delivery. Nanometer-sized HA-CM nanogels could readily load DOX, and both NIR and UV irradiation could significantly enhance DOX release from the nanogels, resulting from light-triggered cleavage of urethane bonds that connect CM to HA. MTT assays showed that DOX-loaded HA-CM nanogels combined with NIR irradiation induced much higher antitumor activity to MCF-7 cells (CD44+) than to U-87MG cells (CD44-) and free HA pretreated MCF-7 cells. CLSM observations confirmed that DOX-loaded HA-CM nanogels were internalized by CD44+ cells via receptor mediated endocytosis mechanism, and intracellular DOX release was triggered by NIR. These HA-CM nanogels with easy preparation, CD44 targetability and photo-controlled intracellular drug release are interesting for cancer chemotherapy.Download high-res image (213KB)Download full-size image
Co-reporter:Zhiyuan Zhong, Jan Feijen
Journal of Controlled Release 2017 Volume 259(Volume 259) pp:
Publication Date(Web):10 August 2017
DOI:10.1016/j.jconrel.2017.06.014
Co-reporter:Ping Zhong, Hao Meng, Jie Qiu, Jian Zhang, Huanli Sun, Ru Cheng, Zhiyuan Zhong
Journal of Controlled Release 2017 Volume 259(Volume 259) pp:
Publication Date(Web):10 August 2017
DOI:10.1016/j.jconrel.2016.12.011
Antibody-maytansinoid conjugates (AMCs) have emerged as one of the most promising active targeting cancer therapeutics. Their clinical use is, however, challenged by their low drug content, poor stability, high cost and potential immune response. Here, we designed and developed robust, cRGD-functionalized, reduction-sensitive polymeric micellar mertansine (DM1) prodrug (cRGD-MMP) that showed targeted treatment of B16F10 melanoma-bearing C57BL/6 mice. cRGD-MMP was obtained with a superb drug content of ~ 40 wt.% and a small size of ~ 45 nm from poly(ethylene glycol)-b-(poly(trimethylene carbonate)-graft-SSDM1) (PEG-P(TMC-g-SSDM1)) and cRGD-functionalized PEG-P(TMC-g-SSDM1) copolymers. cRGD-MMP exhibited excellent stability in 10% fetal bovine serum and cell culture medium while fast swelling and markedly accelerated drug release under a reductive environment. Confocal microscopy, flow cytometry and MTT assays indicated receptor-mediated uptake and high antitumor effect of cRGD-MMP in αvβ3 integrin over-expressing B16F10 melanoma cells. Notably, cRGD-MMP displayed a long elimination half-life of 5.25 h and 4-fold better maximum-tolerated dose than free DM1. The in vivo studies demonstrated that cRGD-MMP effectively inhibited B16F10 melanoma growth and greatly improved mice survival rate as compared to free DM1 and non-targeted MMP control. cRGD-MMP with superior stability, drug loading, and αvβ3 targetability offers an attractive alternative to AMCs for malignant tumor therapy.cRGD-functionalized and reduction-sensitive polymeric micellar mertansine prodrug (cRGD-MMP) mediates targeted treatment of malignant B16F10 melanoma-bearing C57BL/6 mice, resulting in effective suppression of tumor growth and markedly improved survival time.Download high-res image (234KB)Download full-size image
Co-reporter:Jintian Wu, Chao Deng, Fenghua Meng, Jian Zhang, Huanli Sun, Zhiyuan Zhong
Journal of Controlled Release 2017 Volume 259(Volume 259) pp:
Publication Date(Web):10 August 2017
DOI:10.1016/j.jconrel.2016.12.024
PLGA nanotherapeutics though representing a most promising platform for targeted cancer therapy are confronted with low stability and insufficient tumor cell uptake. Here, we report that hyaluronic acid (HA) coated PLGA nanoparticulate docetaxel (DTX-HPLGA) is particularly robust and can effectively target and suppress orthotopic human lung cancer. DTX-HPLGA was easily prepared with a small size of 154 nm and negative surface charge of − 22.7 mV by nanoprecipitation and covalent coating with HA. DTX-HPLGA displayed a low IC50 of 0.91 μg/mL in CD44 + A549 cells and a prolonged elimination half-life of 4.13 h in nude mice. Interestingly, DTX-HPLGA demonstrated 4.4-fold higher accumulation in the cancerous lung than free DTX, reaching a remarkable level of 13.7 %ID/g at 8 h post-injection, in orthotopic human A549 lung cancer-bearing mice. Accordingly, DTX-HPLGA exhibited significantly better inhibition of tumor growth than free DTX, leading to healthy mice growth and markedly improved survival time. DTX-HPLGA with easy fabrication, excellent stability and tumor accumulation, effective tumor suppression, and low side effects is of particular interest for targeted chemotherapy of lung cancers.Hyaluronic acid coating endows PLGA nanoparticulate docetaxel enhanced stability and superior tumor cell selectivity, leading to long circulation time, high drug accumulation in the cancerous lung, effective tumor inhibition and reduced systemic toxicity.Download high-res image (108KB)Download full-size image
Co-reporter:Yaqin Zhu, Jian Zhang, Fenghua Meng, Chao Deng, Ru Cheng, Jan Feijen, Zhiyuan Zhong
Journal of Controlled Release 2016 Volume 233() pp:29-38
Publication Date(Web):10 July 2016
DOI:10.1016/j.jconrel.2016.05.014
Biodegradable micelles are one of the most studied systems for the delivery of hydrophobic anticancer drugs. Their therapeutic efficacy in vivo is, however, suboptimal, partly due to poor tumor cell uptake as well as slow intracellular drug release. Here, we show that cRGD-functionalized intracellularly shell-sheddable biodegradable PEG-SS-PCL micelles mediate enhanced doxorubicin (DOX) delivery to U87MG glioma xenografts in vivo, resulting in significantly improved tumor growth inhibition as compared to reduction-insensitive cRGD/PEG-PCL controls. cRGD/PEG-SS-PCL micelles revealed a small size of ca. 61 nm, a decent DOX loading of 14.9 wt%, and triggered drug release in a reductive environment (10 mM glutathione). Flow cytometry, confocal microscopy, and MTT assays demonstrated that cRGD/PEG-SS-PCL micelles with a cRGD ligand density of 20% efficiently delivered and released DOX into αvβ3 integrin overexpressing U87MG cells. The in vivo pharmacokinetics studies displayed that DOX-loaded cRGD20/PEG-SS-PCL micelles had a prolonged elimination half-life time of 3.51 h, which was comparable to that of cRGD20/PEG-PCL counterparts, indicating that disulfide bonds in the PEG-SS-PCL micelles are stable in the circulation. Notably, in vivo imaging and biodistribution studies in U87MG glioma xenografts showed that cRGD20/PEG-SS-PCL micelles led to efficient accumulation as well as fast drug release in the tumor. The therapeutic outcomes demonstrated that DOX-loaded cRGD20/PEG-SS-PCL micelles exhibited little side effects and superior tumor growth inhibition as compared to non-targeting PEG-SS-PCL and reduction-insensitive cRGD20/PEG-PCL counterparts. The reduction-sensitive shell-sheddable biodegradable micelles have appeared as a fascinating platform for targeted tumor chemotherapy.
Co-reporter:Yinan Zhong, Mathias Dimde, Daniel Stöbener, Fenghua Meng, Chao Deng, Zhiyuan Zhong, and Rainer Haag
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 41) pp:27530
Publication Date(Web):September 27, 2016
DOI:10.1021/acsami.6b09204
Cancer nanomedicines are typically stealthed by a poly(ethylene glycol) layer that is important to obtain extended blood circulation and elevated tumor accumulation. PEG stealth, however, also leads to poor tumor cell selectivity and uptake thereby reducing treatment efficacy. Here, we report that biodegradable micelles with sheddable dendritic polyglycerol sulfate (dPGS) shells show an unusual tumor targetability and chemotherapy in vivo. The self-assembly of dPGS-SS-poly(ε-caprolactone) amphiphilic block copolymer with an Mn of 4.8–3.7 kg mol–1 affords negatively charged and small sized micelles (dPGS-SS-PCL Ms). dPGS-SS-PCL Ms reveal a low cytotoxicity, decent doxorubicin (DOX) loading, and accelerated drug release under a reductive condition. Notably, DOX-loaded dPGS-SS-PCL Ms exhibit a high tolerable dosage of more than 40 mg kg–1, a long plasma half-life of ca. 2.8 h, and an extraordinary tumor accumulation. Intriguingly, therapeutic results demonstrate that DOX-loaded dPGS-SS-PCL Ms induce complete tumor suppression, significantly improved survival rate, and diminishing adverse effects as compared to free drug (DOX·HCl) in MCF-7 human mammary carcinoma models. Dendritic polyglycerol sulfate with a superior tumor homing ability appears to be an attractive alternative to PEG in formulating targeted cancer nanomedicines.Keywords: biodegradable micelles; cancer chemotherapy; polyglycerol; reduction-sensitive; tumor targeting
Co-reporter:Shuai Li, Jian Zhang, Chao Deng, Fenghua Meng, Lin Yu, and Zhiyuan Zhong
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 33) pp:21155
Publication Date(Web):August 10, 2016
DOI:10.1021/acsami.6b05775
In spite of their high specificity and potency, few protein therapeutics are applied in clinical cancer therapy owing to a lack of safe and efficacious delivery systems. Here, we report that redox-sensitive and intrinsically fluorescent photoclick hyaluronic acid nanogels (HA-NGs) show highly efficient loading and breast tumor-targeted delivery of cytochrome c (CC). HA-NGs were obtained from hyaluronic acid-graft-oligo(ethylene glycol)-tetrazole (HA-OEG-Tet) via inverse nanoprecipitation and catalyst-free photoclick cross-linking with l-cystine dimethacrylamide (MA-Cys-MA). HA-NGs exhibited a superb CC loading content of up to 40.6 wt %, intrinsic fluorescence (λem = 510 nm), and a small size of ca. 170 nm. Notably, CC-loaded nanogels (CC-NGs) showed a fast glutathione-responsive protein release behavior. Importantly, released CC maintained its bioactivity. MTT assays revealed that CC-NGs were highly potent with a low IC50 of 3.07 μM to CD44+ MCF-7 human breast tumor cells. Confocal microscopy observed efficient and selective internalization of fluorescent HA-NGs into MCF-7 cells. Interestingly, HA-NGs exhibited also effective breast tumor penetration. The therapeutic results demonstrated that CC-NGs effectively inhibited the growth of MCF-7 breast tumor xenografts at a particularly low dose of 80 or 160 nmol CC equiv./kg. Moreover, CC-NGs did not cause any change in mice body weight, corroborating their low systemic side effects. Redox-sensitive and intrinsically fluorescent photoclick hyaluronic acid nanogels have appeared as a “smart” protein delivery nanoplatform enabling safe, efficacious, traceable, and targeted cancer protein therapy in vivo.Keywords: cancer therapy; click reaction; nanogels; protein delivery; reduction-sensitive; tumor targeting
Co-reporter:Yan Zou, Ya Fang, Hao Meng, Fenghua Meng, Chao Deng, Jian Zhang, Zhiyuan Zhong
Journal of Controlled Release 2016 Volume 244(Part B) pp:326-335
Publication Date(Web):28 December 2016
DOI:10.1016/j.jconrel.2016.05.060
Nanomedicines based on biodegradable micelles offer a most promising treatment for malignant tumors. Their clinical effectiveness, however, remains to be improved. Here, we report that self-crosslinkable and intracellularly decrosslinkable micellar nanoparticles (SCID-Ms) self-assembled from novel amphiphilic biodegradable poly(ethylene glycol)-b-poly(dithiolane trimethylene carbonate) block copolymer achieve high-efficiency targeted cancer chemotherapy in vivo. Interestingly, doxorubicin (DOX)-loaded SCID-Ms showed favorable features of superb stability, minimal drug leakage, long circulation time, triggered drug release inside the tumor cells, and an unprecedented maximum-tolerated dose (MTD) of over 100 mg DOX equiv./kg in mice, which was at least 10 times higher than free drug. The in vivo studies in malignant B16 melanoma-bearing C57BL/6 mice revealed that DOX-SCID-Ms at a dosage of 30 mg DOX equiv./kg could effectively suppress tumor growth and prolong mice survival time without causing obvious systemic toxicity. Moreover, DOX-SCID-Ms could be readily decorated with a targeting ligand like cRGD peptide. The biodistribution studies showed that cRGD20/DOX-SCID-Ms had a high tumor accumulation of 6.13% ID/g at 6 h post injection, which was ca. 3-fold higher than that for clinically used pegylated liposomal doxorubicin (DOX-LPs). Accordingly, cRGD20/DOX-SCID-Ms exhibited significantly better therapeutic efficacy and lower side effects than DOX-LPs in B16 melanoma-bearing mice. These self-regulating biodegradable micellar nanoparticles offer a robust, multifunctional and viable nanoplatform for targeted cancer chemotherapy.Cyclic RGD peptide-decorated disulfide-crosslinked micellar doxorubicin exhibits excellent stability, high maximum-tolerated dose, and superior targetability and therapeutic efficacy to pegylated liposomal doxorubicin in αvβ3 overexpressing B16 melanoma-bearing mice.
Co-reporter:Yaqin Zhu, Xiuxiu Wang, Jing Chen, Jian Zhang, Fenghua Meng, Chao Deng, Ru Cheng, Jan Feijen, Zhiyuan Zhong
Journal of Controlled Release 2016 Volume 244(Part B) pp:229-239
Publication Date(Web):28 December 2016
DOI:10.1016/j.jconrel.2016.08.027
Nanotheranostics is a rapidly growing field combining disease diagnosis and therapy, which ultimately may add in the development of ‘personalized medicine’. Here, we designed and developed bioresponsive and fluorescent hyaluronic acid-iodixanol nanogels (HAI-NGs) for targeted X-ray computed tomography (CT) imaging and chemotherapy of MCF-7 human breast tumors. HAI-NGs were obtained with a small size of ca. 90 nm, bright green fluoresence and high serum stability from hyaluronic acid-cystamine-tetrazole and reductively degradable polyiodixanol-methacrylate via nanoprecipitation and a photo-click crosslinking reaction. Notably, paclitaxel (PTX)-loaded HAI-NGs showed a fast glutathione-responsive drug release. Confocal microscopy displayed efficient uptake of HAI-NGs by CD44 overexpressing MCF-7 cells via a receptor-mediated mechanism. MTT assays revealed that HAI-NGs were nontoxic to MCF-7 cells even at a high concentration of 1 mg/mL whereas PTX-loaded HAI-NGs exhibited strong inhibition of MCF-7 cells. The in vivo pharmcokinetics, near-infrared imaging and biodistribution studies revealed that HAI-NGs significantly prolonged the blood circulation time and enhanced tumor accumulation of PTX. Interestingly, significantly enhanced CT imaging was observed for MCF-7 breast tumors in nude mice via either intratumoral or intravenous injection of HAI-NGs as compared to iodixanol. HAI-NGs fluoresence was distributed thoughout the whole tumor indicating deep tumor penetration. PTX-loaded HAI-NGs showed effective suppression of tumor growth with little systemic toxicity. HAI-NGs appear as a “smart” theranostic nanoplatform for the treatment of CD44 positive tumors.Hyaluronic acid-iodixanol nanogels (HAI-NGs) have integrated multiple functions including excellent biocompatibility, bright green fluoresence, high stability, CD44-targetability and fast glutathione-responsive drug release. Notably, HAI-NGs do not only show significantly enhanced in vivo CT imaging of the tumor but also highly efficient and targeted delivery of paclitaxel to MCF-7 human breast tumor in nude mice, presenting a CD44-specific multifunctional theranostic nanoplatform.
Co-reporter:Yan Zou, Fenghua Meng, Chao Deng, Zhiyuan Zhong
Journal of Controlled Release 2016 Volume 239() pp:149-158
Publication Date(Web):10 October 2016
DOI:10.1016/j.jconrel.2016.08.022
Pegylated liposomal doxorubicin (Lipo-Dox) is one of the few clinically used cancer nanomedicines. Here we show that tumor-homing, redox-responsive and reversibly crosslinked multifunctional biodegradable polymersomes are a better alternative to liposomes for Dox delivery. Cyclic peptide cNGQGEQc-decorated polymersomes (cNGQ-PS) are easily prepared with a small size and high Dox loading. Dox-loaded cNGQ-PS (cNGQ-PS-Dox) shows superb stability with minimal drug leakage under physiological conditions while spontaneous disassembly and quick drug release in response to 10 mM glutathione. MTT assays, flow cytometry and confocal microscopy clearly display efficient receptor-mediated internalization of cNGQ-PS-Dox, fast intracellular drug release, and high antitumor activity in α3β1 integrin-overexpressing A549 lung cancer cells. Intriguingly, cNGQ-PS-Dox presents a remarkably high maximum-tolerated dose of over 100 mg/kg, over 6-fold higher than Lipo-Dox. The in vivo pharmacokinetics and biodistribution studies reveal that cNGQ-PS-Dox has a long circulation time and significantly enhanced tumor accumulation (8.60%ID/g) as compared to Lipo-Dox and non-targeting PS-Dox controls. Notably, cNGQ-PS-Dox shows superior treatment of both subcutaneous and orthotopic A549 human lung cancer bearing nude mice to Lipo-Dox, resulting in effective tumor suppression, significantly improved survival time, and markedly reduced adverse effects. cNGQ-PS appears to be a clinically viable system for targeted lung cancer chemotherapy.
Co-reporter:Bingfeng Sun, Chao Deng, Fenghua Meng, Jian Zhang, Zhiyuan Zhong
Acta Biomaterialia 2016 Volume 45() pp:223-233
Publication Date(Web):November 2016
DOI:10.1016/j.actbio.2016.08.048
Abstract
The clinical success of cancer nanomedicines critically depends on availability of simple, safe and highly efficient nanocarriers. Here, we report that robust and multifunctional nanoparticles self-assembled from hyaluronic acid-g-poly(γ-benzyl-l-glutamate)-lipoic acid conjugates achieve a remarkably high loading (up to 25.8 wt.%) and active targeted delivery of doxorubicin (DOX) to human breast tumor xenograft in vivo. DOX-loaded nanoparticles following auto-crosslinking (DOX-CLNPs) are highly stable with little drug leakage under physiological conditions while quickly release ca. 92% DOX in 30 h under a cytoplasmic-mimicking reductive environment. The in vitro assays reveal that DOX-CLNPs possess a superior selectivity and antitumor activity to clinically used pegylated liposomal doxorubicin hydrochloride (DOX-LPs) in CD44 receptor overexpressing MCF-7 human breast cancer cells. Strikingly, DOX-CLNPs exhibit a superb tolerated dose of over 100 mg DOX equiv./kg, which is more than 5 times higher than DOX-LPs, and an extraordinary breast tumor accumulation of 8.6%ID/g in mice. The in vivo therapeutic studies in MCF-7 human breast tumor-bearing nude mice show that DOX-CLNPs effectively inhibit tumor growth, improve survival rate, and significantly decrease adverse effects as compared to DOX-LPs. DOX-CLNPs based on natural endogenous materials with high drug loading, great stability and CD44-targetability are highly promising for precision cancer chemotherapy.
Statement of Significance
We demonstrate that with rational design, simple and multifunctional anticancer nanotherapeutics can be developed to achieve highly efficient and targeted cancer chemotherapy. Doxorubicin-loaded multifunctional nanoparticles based on hyaluronic acid-g-poly(γ-benzyl-l-glutamate)-lipoic acid conjugates exhibit a high drug loading, superior stability, fast bioresponsivity, high tolerability, and obvious selectivity toward CD44-overexpressing tumors in vivo. These nanotherapeutics achieve effective tumor suppression, drastically improved survival rate and reduced side effects as compared to clinically used pegylated liposomal doxorubicin in MCF-7 human breast tumor-bearing nude mice. Unlike previously reported multifunctional nanomedicines, the present nanotherapeutics primarily based on natural endogenous materials are simple and straightforward to fabricate, which makes them potentially interesting for clinical translation.
Co-reporter:Ping Zhong, Jian Zhang, Chao Deng, Ru Cheng, Fenghua Meng, and Zhiyuan Zhong
Biomacromolecules 2016 Volume 17(Issue 11) pp:3602
Publication Date(Web):October 10, 2016
DOI:10.1021/acs.biomac.6b01094
Low tolerability and tumor selectivity restricts many potent anticancer drugs including mertansine from wide clinical use. Here, glutathione-activatable hyaluronic acid-SS-mertansine prodrug (HA-SS-DM1) was designed and developed to achieve enhanced tolerability and targeted therapy of CD44+ human breast tumor xenografts. DM1 was readily conjugated to HA using 2-(2-pyridyldithio)-ethylamine as a linker. Notably, HA-SS-DM1 with a high DM1 content of 20 wt % had a mean size of ∼170 nm at concentrations above 0.2 mg/mL while transformed into unimers upon dilution to 0.04 mg/mL. HA-SS-DM1 exhibited a superior targetability to MCF-7 cancer cells with an exceptionally low IC50 of 0.13 μg DM1/mL. The pharmacokinetic studies displayed that Cy5-labeled HA-SS-DM1 had an elimination half-life of 2.12 h. Notably, HA-SS-DM1 displayed better tolerability with a maximum-tolerated dose 4-fold higher than free DM1. Cy5-labeled HA-SS-DM1 quickly accumulated in the MCF-7 tumor, the fluorescence intensity of which was maximized at 24 h post injection and kept strong in 48 h. The tumor Cy5 level reached 8.17%ID/g at 24 h. The therapeutic results demonstrated that HA-SS-DM1 effectively inhibited tumor growth at 800 μg DM1 equiv/kg while causing reduced side effects as compared to free DM1. Glutathione-cleavable HA-SS-DM1 prodrug with superior drug content, excellent targetability, enhanced tolerability, and easy large-scale synthesis appears to be a highly promising alternative to clinically used Trastuzumab emtansine (T-DM1) for targeted breast tumor therapy.
Co-reporter:Yinan Zhong, Katharina Goltsche, Liang Cheng, Fang Xie, Fenghua Meng, Chao Deng, Zhiyuan Zhong, Rainer Haag
Biomaterials 2016 84() pp: 250-261
Publication Date(Web):April 2016
DOI:10.1016/j.biomaterials.2016.01.049
The therapeutic efficacy of nanoscale anticancer drug delivery systems is severely truncated by their low tumor-targetability and inefficient drug release at the target site. Here, we report the design and development of novel endosomal pH-activatable paclitaxel prodrug micelles based on hyaluronic acid-b-dendritic oligoglycerol (HA-dOG-PTX-PM) for active targeting and effective treatment of CD44-overexpressing human breast cancer xenografts in nude mice. HA-dOG-PTX-PM had a high drug content of 20.6 wt.% and an average diameter of 155 nm. The release of PTX was slow at pH 7.4 but greatly accelerated at endosomal pH. MTT assays, flow cytometry and confocal experiments showed that HA-dOG-PTX-PM possessed a high targetability and antitumor activity toward CD44 receptor overexpressing MCF-7 human breast cancer cells. The in vivo pharmacokinetics and biodistribution studies showed that HA-dOG-PTX-PM had a prolonged circulation time in the nude mice and a remarkably high accumulation in the MCF-7 tumor (6.19%ID/g at 12 h post injection). Interestingly, HA-dOG-PTX-PM could effectively treat mice bearing MCF-7 human breast tumor xenografts with little side effects, resulting in complete inhibition of tumor growth and a 100% survival rate over an experimental period of 55 days. These results indicate that hyaluronic acid-shelled acid-activatable PTX prodrug micelles have a great potential for targeted chemotherapy of CD44-positive cancers.
Co-reporter:Xiuxiu Wang, Jian Zhang, Ru Cheng, Fenghua Meng, Chao Deng, and Zhiyuan Zhong
Biomacromolecules 2016 Volume 17(Issue 3) pp:
Publication Date(Web):January 26, 2016
DOI:10.1021/acs.biomac.5b01578
Reductively degradable biopolymers have emerged as a unique class of smart biomedical materials. Here, a functional coupling agent, cystamine diisocyanate (CDI), was designed to offer a facile access to reductively degradable biopolymers via polycondensation with various diols. CDI was readily obtained with a decent yield of 46% by reacting cystamine dihydrochloride with triphosgene. The polycondensation of oligo(ethylene glycol) diol (Mn = 0.4 or 1.5 kg/mol) or oligo(ε-caprolactone) diol (Mn = 0.53 kg/mol) with CDI in N,N-dimethylformamide at 60 °C using dibutyltin dilaurate as a catalyst afforded reductively degradable poly(ethylene glycol) (SSPEG, Mn = 6.2–76.8 kg/mol) or poly(ε-caprolactone) (SSPCL, Mn = 6.8–16.3 kg/mol), in which molecular weights were well controlled by diol/CDI molar ratios. Moreover, PEG-SSPCL-PEG triblock copolymers could be readily prepared by reacting dihydroxyl-terminated SSPCL with PEG-isocyanate derivative. PEG-SSPCL-PEG with an Mn of 5.0–16.3–5.0 kg/mol formed small-sized micelles with an average diameter of about 85 nm in PB buffer. The in vitro release studies using doxorubicin (DOX) as a model drug showed that, in sharp contrast to reduction-insensitive PEG-PCL(HDI)-PEG controls, drug release from PEG-SSPCL-PEG micelles was fast and nearly complete in 24 h under a reductive condition containing 10 mM glutathione. The confocal microscopy experiments in drug-resistant MCF-7 cells (MCF-7/ADR) displayed efficient cytoplasmic DOX release from PEG-SSPCL-PEG micelles. MTT assays revealed that DOX-loaded PEG-SSPCL-PEG micelles were much more potent against MCF-7/ADR cells than reduction-insensitive PEG-PCL(HDI)-PEG controls (IC50: 6.3 vs 55.4 μg/mL). It should further be noted that blank PEG-SSPCL-PEG micelles were noncytotoxic up to a tested concentration of 1 mg/mL. Hence, cystamine diisocyanate appears to be an innovative coupling agent that facilitates versatile synthesis of biocompatible and reductively degradable biopolymers.
Co-reporter:Jintian Wu, Jian Zhang, Chao Deng, Fenghua Meng, and Zhiyuan Zhong
Biomacromolecules 2016 Volume 17(Issue 7) pp:
Publication Date(Web):June 15, 2016
DOI:10.1021/acs.biomac.6b00380
Poly(d,l-lactide-co-glycolide) (PLGA) nanoparticles have attracted an enormous interest for controlled drug delivery. Their clinical applications are, however, partly hindered by lack of biocompatible, biodegradable and functional surfactants. Here, we designed and developed a novel biocompatible surfactant based on amphiphilic vitamin E-oligo(methyl diglycol l-glutamate) (VEOEG) for facile fabrication of robust and tumor-targeting PLGA-based nanomedicines. VEOEG was prepared with controlled Mn of 1.7–2.6 kg/mol and low molecular weight distribution (Đ = 1.04–1.16) via polymerization of methyl diglycol l-glutamate N-carboxyanhydride using vitamin E-ethylenediamine derivative (VE-NH2) as an initiator. VEOEG had a hydrophile–lipophile balance data of 13.8–16.1 and critical micellar concentration of 189.3–203.8 mg/L depending on lengths of oligopeptide. Using VEOEG as a surfactant, PLGA nanoparticles could be obtained via nanoprecipitation method with a small and uniform hydrodynamic size of 135 nm and positive surface charge of +26.6 mV, in accordance with presence of amino groups at the surface. The resulting PLGA nanoparticles could be readily coated with hyaluronic acid (HA) to form highly stable, small-sized (143 nm), monodisperse, and negatively charged nanoparticles (HA-PLGA NPs). Notably, paclitaxel-loaded HA-PLGA NPs (PTX-HA-PLGA NPs) exhibited better antitumor effects in CD44-positive MCF-7 breast tumor cells than Taxol (a clinical paclitaxel formulation). The in vivo pharmacokinetics assay in nude mice displayed that PTX-HA-PLGA NPs possessed a long plasma half-life of 3.14 h. The in vivo biodistribution studies revealed that PTX-HA-PLGA NPs had a high tumor PTX level of 8.4% ID/g, about 6 times better than that of Taxol. Interestingly, therapeutic studies showed that PTX-HA-PLGA NPs caused significantly more effective tumor growth inhibition, better survival rate and lower adverse effect than Taxol. VEOEG has emerged as a versatile and functional surfactant for the fabrication of advanced anticancer nanomedicines.
Co-reporter:Xin Ma, Zhiwei He, Fengxuan Han, Zhiyuan Zhong, Liang Chen, Bin Li
Colloids and Surfaces B: Biointerfaces 2016 Volume 143() pp:81-87
Publication Date(Web):1 July 2016
DOI:10.1016/j.colsurfb.2016.03.025
•Hybrid hydrogels composed of collagen, hydroxyapatite and alendronate.•Formed under physiological condition using genipin as crosslinker.•Gelation time ranged from 5 to 37 min.•Had improved mechanical property and tunable enzymatical degradation.•Supported osteoblastic cell adhesion and growth.Development of biomimetic scaffolds represents a promising direction in bone tissue engineering. In this study, we designed a two-step process to prepare a type of biomimetic hybrid hydrogels that were composed of collagen, hydroxyapatite (HAP) and alendronate (ALN), an anti-osteoporosis drug. First, water-soluble ALN-conjugated HAP (HAP-ALN) containing 4.0 wt.% of ALN was synthesized by treating HAP particles with ALN. Hydrogels were then formed from HAP-ALN conjugate and collagen under physiological conditions using genipin (GNP) as the crosslinker. Depending on the ALN/collagen molar ratio and GNP concentration, the gelation time of hydrogels ranged from 5 to 37 min. Notably, these hybrid hydrogels exhibited markedly improved mechanical property (storage modulus G′= 38–187 kPa), higher gel contents, and lower swelling ratios compared to the hydrogels prepared from collagen alone under similar conditions. Moreover, they showed tunable degradation behaviors against collagenase. The collagen/HAP-ALN hybrid hydrogels supported the adhesion and growth of murine MC3T3-E1 osteoblastic cells well. Such tough yet enzymatically degradable hybrid hydrogels hold potential as scaffolds for bone tissue engineering.Biomimetic hybrid hydrogels composed of collagen and alendronate (ALN)-functionalized hydroxyapatite (HAP) were synthesized and used as scaffolds for bone tissue engineering.
Co-reporter:Jing Chen, Yan Zou, Chao Deng, Fenghua Meng, Jian Zhang, and Zhiyuan Zhong
Chemistry of Materials 2016 Volume 28(Issue 23) pp:
Publication Date(Web):November 16, 2016
DOI:10.1021/acs.chemmater.6b04404
Protein therapeutics offer a most effective treatment for many human diseases, including diabetes, cardiovascular diseases, and malignant tumors. Unlike most chemotherapeutics that often cause notorious side effects, many protein drugs possess a high specificity and reduced systemic toxicity. Notably, clinically used protein drugs are mostly limited to those that have extracellular effects. Protein drugs that have intracellular targets do represent a large family of protein biologics that have not been introduced into the clinic, because of the absence of translatable intracellular protein delivery vehicles. Here we report efficient and targeted cancer protein therapy in vivo by bioresponsive fluorescent photoclick hyaluronic acid (HA) nanogels. Two intracellular protein drugs, cytochrome c (CC) and granzyme B (GrB), are loaded into the nanogels with preserved bioactivity. CC- and GrB-loaded HA nanogels can effectively target and release proteins to CD44 positive MCF-7 and A549 cancer cells, yielding striking antitumor effects with a half-maximal inhibitory concentration thousands of times lower than those of clinical chemotherapeutics. Remarkably, GrB-loaded HA nanogels at a low dose of 3.8–5.7 nmol of GrB equivalents/kg exhibit complete suppression of tumor growth and minimal adverse effects in nude mice bearing subcutaneous MCF-7 human breast tumor and orthotopic A549 human lung tumor xenografts.
Co-reporter:Wei Chen, Fenghua Meng, Ru Cheng, Chao Deng, Jan Feijen and Zhiyuan Zhong
Journal of Materials Chemistry A 2015 vol. 3(Issue 11) pp:2308-2317
Publication Date(Web):27 Jan 2015
DOI:10.1039/C4TB01962H
Glycopolymer-b-poly(ε-caprolactone) (GP-PCL) block copolymer micelles (‘glycomicelles’) with tailored lactose functionalities were developed and investigated for hepatoma-targeted doxorubicin (DOX) delivery. Amphiphilic GP-PCL copolymers were readily prepared with controlled lactobionic acid (LBA) functionalities of 20%, 40%, 80%, and 100% (denoted as GP20-PCL, GP40-PCL, GP80-PCL, and GP100-PCL, respectively) through post-polymerization modification of the poly(acryloyl cyclic carbonate)-b-poly(ε-caprolactone) (PAC-b-PCL, 11.6–6.4 kg mol−1) block copolymer with thiolated LBA (LBA-SH) and 2-(2-methoxyethoxy)ethanethiol ((EO)2-SH) via the Michael-type addition reaction. These self-assembled glycomicelles had mean hydrodynamic diameters ranging from 31.9 to 76.8 nm depending on LBA densities, and exhibited high DOX loading efficiencies of 83.0–89.2%. In vitro release studies showed that the DOX release rate depended on the pH and LBA content. Flow cytometric analyses revealed that asialoglycoprotein receptor (ASGP-R) over-expressed HepG2 liver cancer cells following 4 h treatment with DOX-loaded glycomicelles had a 6.6–17.1-fold higher DOX level, depending on LBA densities, as compared to those treated with the corresponding DOX-loaded non-glycomicelles (100% substitution with (EO)2-SH) under otherwise the same conditions. MTT assays demonstrated that DOX-loaded GP20-PCL, GP40-PCL, GP80-PCL and GP100-PCL micelles had much lower half maximal inhibitory concentration (IC50) values of 2.05, 0.75, 0.45 and 0.43 μg DOX equiv. mL−1, respectively, in HepG2 cells than DOX-loaded non-glycomicelles (IC50: 6.55 μg mL−1 DOX equiv. mL−1). Competitive inhibition experiments showed that after the incubation with DOX-loaded glycomicelles for 4 h, more efficient killing activity against free HepG2 cells (−LBA) was observed, as compared to that against LBA-blocked HepG2 cells (+LBA) after a subsequent 72 h incubation. Glycomicelles with tailored LBA functionalities, high drug loading capacity, and high uptake by ASGP-R positive cells are promising candidates for liver cancer chemotherapy.
Co-reporter:Yinan Zhong, Jian Zhang, Ru Cheng, Chao Deng, Fenghua Meng, Fang Xie, Zhiyuan Zhong
Journal of Controlled Release 2015 Volume 205() pp:144-154
Publication Date(Web):10 May 2015
DOI:10.1016/j.jconrel.2015.01.012
The existence of drug resistance poses a major obstacle for the treatment of various malignant human cancers. Here, we report on reduction-sensitive reversibly crosslinked hyaluronic acid (HA) nanoparticles based on HA-Lys-LA conjugates (Lys: l-lysine methyl ester, LA: lipoic acid) for active targeting delivery of doxorubicin (DOX) to CD44 + breast cancers in vitro and in vivo, effectively overcoming drug resistance (ADR). HA-Lys-LA with degrees of substitution of 5, 10 and 28% formed robust nano-sized nanoparticles (152–219 nm) following auto-crosslinking. DOX-loaded crosslinked nanoparticles revealed inhibited DOX release under physiological conditions while fast drug release in the presence of 10 mM glutathione (GSH). Notably, MTT assays showed that DOX-loaded crosslinked HA-Lys-LA10 nanoparticles possessed an apparent targetability and a superior antitumor activity toward CD44 receptor overexpressing DOX-resistant MCF-7 human breast cancer cells (MCF-7/ADR). The in vivo pharmacokinetics and biodistribution studies in MCF-7/ADR tumor xenografts in nude mice showed that DOX-loaded crosslinked HA-Lys-LA10 nanoparticles had a prolonged circulation time and a remarkably high accumulation in the tumor (12.71% ID/g). Notably, DOX-loaded crosslinked HA-Lys-LA10 nanoparticles exhibited effective inhibition of tumor growth while continuous tumor growth was observed for mice treated with free drug. The Kaplan–Meier survival curves showed that in contrast to control groups, all mice treated with DOX-loaded crosslinked HA-Lys-LA10 nanoparticles survived over an experimental period of 44 days. Importantly, DOX-loaded crosslinked HA nanoparticles caused low side effects. The reversibly crosslinked hyaluronic acid nanoparticles with excellent biocompatibility, CD44-targetability, and effective reversal of drug resistance have a great potential in cancer therapy.Robust, biocompatible and multifunctional hyaluronic acid nanoparticles mediate specific and efficient delivery and release of doxorubicin to CD44-positive drug-resistant human breast cancers in vitro and in vivo, inducing effective reversal of drug resistance, high therapeutic efficacy and low side effects.
Co-reporter:Zhiyuan Zhong, Jan Feijen
Journal of Controlled Release 2015 Volume 205() pp:1-2
Publication Date(Web):10 May 2015
DOI:10.1016/j.jconrel.2015.03.032
Co-reporter:Zhiyuan Zhong
Journal of Controlled Release 2015 Volume 205() pp:3-6
Publication Date(Web):10 May 2015
DOI:10.1016/j.jconrel.2015.03.013
Co-reporter:Wentao Lu, Xiuxiu Wang, Ru Cheng, Chao Deng, Fenghua Meng and Zhiyuan Zhong
Polymer Chemistry 2015 vol. 6(Issue 33) pp:6001-6010
Publication Date(Web):30 Jun 2015
DOI:10.1039/C5PY00828J
α-Amino acid-based functional biopolymers are highly appealing for various biomedical applications including controlled drug delivery. In this paper, we report the design and development of novel reductively biodegradable α-amino acid-based poly(disulfide urethane)s, denoted as AAPU(SS)s, as well as PEG-AAPU(SS)-PEG triblock copolymer micelles for triggered intracellular doxorubicin (DOX) release. AAPU(SS)s were synthesized with controlled Mn ranging from 4.6 to 35.7 kg mol−1via polycondensation reaction between two α-amino acid derivatives, disulfide-linked bis(ethyl L-serinate) (SS-BSER) and L-lysine ethyl ester diisocyanate (LDI). AAPU(SS)s are amorphous with a glass transition temperature (Tg) of 31.7–49.2 °C and were degraded into low molecular weight fragments under a reductive condition. PEG-AAPU(SS)-PEG copolymer could be readily obtained by treating AAPU(SS) with mPEG-NCO. PEG-AAPU(SS)-PEG formed micelles with a mean diameter of 155 nm. The in vitro release studies showed that drug release from DOX-loaded PEG-AAPU(SS)-PEG micelles was significantly accelerated in the presence of 10 mM glutathione (GSH). MTT assays revealed that DOX-loaded PEG-AAPU(SS)-PEG micelles caused effective growth inhibition of both RAW 264.7 and drug resistant MCF-7 cells (MCF-7/ADR) while the corresponding blank micelles were non-cytotoxic even at a high concentration of 1.0 mg mL−1. Confocal microscopy showed that PEG-AAPU(SS)-PEG micelles efficiently transported and released DOX into the perinuclear and nuclear regions of MCF-7/ADR cells. These biocompatible and bioreducible α-amino acid-based poly(disulfide urethane) micelles have appeared to be a particularly interesting platform for triggered intracellular anticancer drug delivery.
Co-reporter:Guohui Xu, Xiaolin Wang, Chao Deng, Xiaomei Teng, Erik J. Suuronen, Zhenya Shen, Zhiyuan Zhong
Acta Biomaterialia 2015 Volume 15() pp:55-64
Publication Date(Web):15 March 2015
DOI:10.1016/j.actbio.2014.12.016
Abstract
Injectable biodegradable hybrid hydrogels were designed and developed based on thiolated collagen (Col-SH) and multiple acrylate containing oligo(acryloyl carbonate)-b-poly(ethylene glycol)-b-oligo(acryloyl carbonate) (OAC-PEG-OAC) copolymers for functional cardiac regeneration. Hydrogels were readily formed under physiological conditions (37 °C and pH 7.4) from Col-SH and OAC-PEG-OAC via a Michael-type addition reaction, with gelation times ranging from 0.4 to 8.1 min and storage moduli from 11.4 to 55.6 kPa, depending on the polymer concentrations, solution pH and degrees of substitution of Col-SH. The collagen component in the hybrid hydrogels retained its enzymatic degradability against collagenase, and the degradation time of the hydrogels increased with increasing polymer concentration. In vitro studies showed that bone marrow mesenchymal stem cells (BMSCs) exhibited rapid cell spreading and extensive cellular network formation on these hybrid hydrogels. In a rat infarction model, the infarcted left ventricle was injected with PBS, hybrid hydrogels, BMSCs or BMSC-encapsulating hybrid hydrogels. Echocardiography demonstrated that the hybrid hydrogels and BMSC-encapsulating hydrogels could increase the ejection fraction at 28 days compared to the PBS control group, resulting in improved cardiac function. Histology revealed that the injected hybrid hydrogels significantly reduced the infarct size and increased the wall thickness, and these were further improved with the BMSC-encapsulating hybrid hydrogel treatment, probably related to the enhanced engraftment and persistence of the BMSCs when delivered within the hybrid hydrogel. Thus, these injectable hybrid hydrogels combining intrinsic bioactivity of collagen, controlled mechanical properties and enhanced stability provide a versatile platform for functional cardiac regeneration.
Co-reporter:Jiaolong Lv, Huanli Sun, Yan Zou, Fenghua Meng, Aylvin A. Dias, Marc Hendriks, Jan Feijen and Zhiyuan Zhong
Biomaterials Science 2015 vol. 3(Issue 7) pp:1134-1146
Publication Date(Web):08 Apr 2015
DOI:10.1039/C4BM00436A
Novel reductively degradable α-amino acid-based poly(ester amide)-graft-galactose (SSPEA-Gal) copolymers were designed and developed to form smart nano-vehicles for active hepatoma-targeting doxorubicin (DOX) delivery. SSPEA-Gal copolymers were readily synthesized via solution polycondensation reaction of di-p-toluenesulfonic acid salts of bis-L-phenylalanine 2,2-thiodiethanol diester and bis-vinyl sulfone functionalized cysteine hexanediol diester with dinitrophenyl ester of adipic acid, followed by conjugating with thiol-functionalized galactose (Gal-SH) via the Michael addition reaction. SSPEA-Gal formed unimodal nanoparticles (PDI = 0.10 − 0.12) in water, in which average particle sizes decreased from 138 to 91 nm with increasing Gal contents from 31.6 wt% to 42.5 wt%. Notably, in vitro drug release studies showed that over 80% DOX was released from SSPEA-Gal nanoparticles within 12 h under an intracellular mimicking reductive conditions, while low DOX release (<20%) was observed for reduction-insensitive PEA-Gal nanoparticles under otherwise the same conditions and SSPEA-Gal nanoparticles under non-reductive conditions. Notably, SSPEA-Gal nanoparticles exhibited high specificity to asialoglycoprotein receptor (ASGP-R)-overexpressing HepG2 cells. MTT assays using HepG2 cells showed that DOX-loaded SSPEA-Gal had a low half maximal inhibitory concentration (IC50) of 1.37 μg mL−1, approaching that of free DOX. Flow cytometry and confocal laser scanning microscopy studies confirmed the efficient uptake of DOX-loaded SSPEA-Gal nanoparticles by HepG2 cells as well as fast intracellular DOX release. Importantly, SSPEA-Gal and PEA-Gal nanoparticles were non-cytotoxic to HepG2 and MCF-7 cells up to a tested concentration of 1.0 mg mL−1. These tumor-targeting and reduction-responsive degradable nanoparticles have appeared as an interesting multi-functional platform for advanced drug delivery.
Co-reporter:Ling Lu, Yan Zou, Weijing Yang, Fenghua Meng, Chao Deng, Ru Cheng, and Zhiyuan Zhong
Biomacromolecules 2015 Volume 16(Issue 6) pp:
Publication Date(Web):May 4, 2015
DOI:10.1021/acs.biomac.5b00193
In spite of their high potency and specificity, few protein drugs have advanced to the clinical settings due to lack of safe and efficient delivery vehicles. Here, novel anisamide-decorated pH-sensitive degradable chimaeric polymersomes (Anis-CPs) were designed, prepared, and investigated for efficient and targeted delivery of apoptotic protein, granzyme B (GrB), to lung cancer cells. Anis-CPs were readily prepared with varying Anis surface densities from anisamide end-capped poly(ethylene glycol)-b-poly(2,4,6- trimethoxybenzylidene-1,1,1-tris(hydroxymethyl)ethane methacrylate)-b-poly(acrylic acid) (Anis-PEG-PTTMA-PAA) and PEG-PTTMA-PAA copolymers. Using cytochrome C (CC) as a model protein, Anis-CPs displayed high protein loading efficiencies (40.5–100%) and loading contents (up to 16.8 wt %). CC-loaded Anis-CPs had narrow distribution (PDI 0.04–0.13) and small sizes ranging from 152 to 171 nm, which increased with increasing CC contents. Notably, the release of proteins from Anis-CPs was accelerated under mildly acidic conditions, due to the hydrolysis of acetal bonds in PTTMA. MTT assays showed that GrB-loaded Anis-CPs (GrB-Anis-CPs) displayed high targetability to sigma receptor overexpressing cancer cells such as H460 and PC-3 cells. For example, GrB-Anis-CPs exhibited increasing antitumor efficacy to H460 cells with increasing Anis contents from 0 to 80%. The antitumor activity of GrB-Anis-CPs was significantly reduced upon pretreating H460 cells with haloperidol (a competitive antagonist). Notably, the half-maximal inhibitory concentrations (IC50) of GrB-Anis70-CPs were determined to be 6.25 and 5.94 nM for H460 and PC-3 cells, respectively, which were 2–3 orders of magnitude lower than that of chemotherapeutic drugs, such as paclitaxel. Flow cytometry studies demonstrated that GrB-Anis70-CPs induced widespread apoptosis of H460 cells. The confocal laser scanning microscopy (CLSM) experiments using FITC-labeled CC-loaded Anis-CPs confirmed fast internalization and intracellular protein release into H460 cells. GrB-Anis-CPs with high potency and specificity are particularly interesting for targeted therapy of lung cancers.
Co-reporter:Peipei Chen, Min Qiu, Chao Deng, Fenghua Meng, Jian Zhang, Ru Cheng, and Zhiyuan Zhong
Biomacromolecules 2015 Volume 16(Issue 4) pp:
Publication Date(Web):March 11, 2015
DOI:10.1021/acs.biomac.5b00113
pH-Responsive chimaeric polypeptide-based polymersomes (refer to as pepsomes) were designed and developed from asymmetric poly(ethylene glycol)-b-poly(l-leucine)-b-poly(l-glutamic acid) (PEG-PLeu-PGA, PEG is longer than PGA) triblock copolymers for efficient encapsulation and triggered intracellular delivery of doxorubicin hydrochloride (DOX·HCl). PEG-PLeu-PGA was conveniently prepared by sequential ring-opening polymerization of l-leucine N-carboxyanhydride and γ-benzyl-l-glutamate N-carboxyanhydride using PEG-NH2 as an initiator followed by deprotection. Pepsomes formed from PEG-PLeu-PGA had unimodal distribution and small sizes of 64–71 nm depending on PLeu block lengths. Interestingly, these chimaeric pepsomes while stable at pH 7.4 were quickly disrupted at pH 5.0, likely due to alternation of ionization state of the carboxylic groups in PGA that shifts PGA blocks from hydrophilic and random coil structure into hydrophobic and α-helical structure. DOX·HCl could be actively loaded into the watery core of pepsomes with a high loading efficiency. Remarkably, the in vitro release studies revealed that release of DOX·HCl was highly dependent on pH, in which about 24.0% and 75.7% of drug was released at pH 7.4 and 5.0, respectively, at 37 °C in 24 h. MTT assays demonstrated that DOX·HCl-loaded pepsomes exhibited high antitumor activity, similar to free DOX·HCl in RAW 264.7 cells. Moreover, they were also potent toward drug-resistant MCF-7 cancer cells (MCF-7/ADR). Confocal microscopy studies showed that DOX·HCl-loaded pepsomes delivered and released drug into the cell nuclei of MCF-7/ADR cells in 4 h, while little DOX·HCl fluorescence was observed in MCF-7/ADR cells treated with free drug under otherwise the same conditions. These chimaeric pepsomes with facile synthesis, efficient drug loading, and pH-triggered drug release behavior are an attractive alternative to liposomes for targeted cancer chemotherapy.
Co-reporter:Hua He, Yugang Bai, Jinhui Wang, Qiurong Deng, Lipeng Zhu, Fenghua Meng, Zhiyuan Zhong, and Lichen Yin
Biomacromolecules 2015 Volume 16(Issue 4) pp:
Publication Date(Web):March 10, 2015
DOI:10.1021/acs.biomac.5b00180
Polycations often suffer from the irreconcilable inconsistency between transfection efficiency and toxicity. Polymers with high molecular weight (MW) and cationic charge feature potent gene delivery capabilities, while in the meantime suffer from strong chemotoxicity, restricted intracellular DNA release, and low stability in vivo. To address these critical challenges, we herein developed pH-responsive, reversibly cross-linked, polyetheleneimine (PEI)-based polyplexes coated with hyaluronic acid (HA) for the effective and targeted gene delivery to cancer cells. Low-MW PEI was cross-linked with the ketal-containing linker, and the obtained high-MW analogue afforded potent gene delivery capabilities during transfection, while rapidly degraded into low-MW segments upon acid treatment in the endosomes, which promoted intracellular DNA release and reduced material toxicity. HA coating of the polyplexes shielded the surface positive charges to enhance their stability under physiological condition and simultaneously reduced the toxicity. Additionally, HA coating allowed active targeting to cancer cells to potentiate the transfection efficiencies in cancer cells in vitro and in vivo. This study therefore provides an effective approach to overcome the efficiency-toxicity inconsistence of nonviral vectors, which contributes insights into the design strategy of effective and safe vectors for cancer gene therapy.
Co-reporter:Huanli Sun, Ru Cheng, Chao Deng, Fenghua Meng, Aylvin A. Dias, Marc Hendriks, Jan Feijen, and Zhiyuan Zhong
Biomacromolecules 2015 Volume 16(Issue 2) pp:
Publication Date(Web):January 2, 2015
DOI:10.1021/bm501652d
A novel and versatile family of enzymatically and reductively degradable α-amino acid-based poly(ester amide)s (SS-PEAs) were developed from solution polycondensation of disulfide-containing di-p-toluenesulfonic acid salts of bis-l-phenylalanine diesters (SS-Phe-2TsOH) with di-p-nitrophenyl adipate (NA) in N,N-dimethylformamide (DMF). SS-PEAs with Mn ranging from 16.6 to 23.6 kg/mol were obtained, depending on NA/SS-Phe-2TsOH molar ratios. The chemical structures of SS-PEAs were confirmed by 1H NMR and FTIR spectra. Thermal analyses showed that the obtained SS-PEAs were amorphous with a glass transition temperature (Tg) in the range of 35.2–39.5 °C. The in vitro degradation studies of SS-PEA films revealed that SS-PEAs underwent surface erosion in the presence of 0.1 mg/mL α-chymotrypsin and bulk degradation under a reductive environment containing 10 mM dithiothreitol (DTT). The preliminary cell culture studies displayed that SS-PEA films could well support adhesion and proliferation of L929 fibroblast cells, indicating that SS-PEAs have excellent cell compatibility. The nanoparticles prepared from SS-PEA with PVA as a surfactant had an average size of 167 nm in phosphate buffer (PB, 10 mM, pH 7.4). SS-PEA nanoparticles while stable under physiological environment undergo rapid disintegration under an enzymatic or reductive condition. The in vitro drug release studies showed that DOX release was accelerated in the presence of 0.1 mg/mL α-chymotrypsin or 10 mM DTT. Confocal microscopy observation displayed that SS-PEA nanoparticles effectively transported DOX into both drug-sensitive and -resistant MCF-7 cells. MTT assays revealed that DOX-loaded SS-PEA nanoparticles had a high antitumor activity approaching that of free DOX in drug-sensitive MCF-7 cells, while more than 10 times higher than free DOX in drug-resistant MCF-7/ADR cells. These enzymatically and reductively degradable α-amino acid-based poly(ester amide)s have provided an appealing platform for biomedical technology in particular controlled drug delivery applications.
Co-reporter:Fushi Huang, Ru Cheng, Fenghua Meng, Chao Deng, and Zhiyuan Zhong
Biomacromolecules 2015 Volume 16(Issue 7) pp:
Publication Date(Web):June 25, 2015
DOI:10.1021/acs.biomac.5b00625
Polyurethanes are a unique class of biomaterials that are widely used in medical devices. In spite of their easy synthesis and excellent biocompatibility, polyurethanes are less explored for controlled drug delivery due to their slow or lack of degradation. In this paper, we report the design and development of novel acid degradable poly(acetal urethane) (PAU) and corresponding triblock copolymer micelles for pH-triggered intracellular delivery of a model lipophilic anticancer drug, doxorubicin (DOX). PAU with Mn ranging from 4.3 to 12.3 kg/mol was conveniently prepared from polycondensation reaction of lysine diisocyanate (LDI) and a novel diacetal-containing diol, terephthalilidene-bis(trimethylolethane) (TPABTME) using dibutyltin dilaurate (DBTDL) as a catalyst in N,N-dimethylformamide (DMF). The thiol-ene click reaction of Allyl-PAU-Allyl with thiolated PEG (Mn = 5.0 kg/mol) afforded PEG-PAU-PEG triblock copolymers that readily formed micelles with average sizes of about 90−120 nm in water. The dynamic light scattering (DLS) measurements revealed fast swelling and disruption of micelles under acidic pH. UV/vis spectroscopy corroborated that acetal degradation was accelerated at pH 4.0 and 5.0. The in vitro release studies showed that doxorubicin (DOX) was released in a controlled and pH-dependent manner, in which ca. 96%, 73%, and 30% of drug was released within 48 h at pH 4.0, 5.0, and 7.4, respectively. Notably, MTT assays displayed that DOX-loaded PEG-PAU-PEG micelles had a high in vitro antitumor activity in both RAW 264.7 and drug-resistant MCF-7/ADR cells. The confocal microscopy and flow cytometry experiments demonstrated that PEG-PAU-PEG micelles mediated efficient cytoplasmic delivery of DOX. Importantly, blank PEG-PAU-PEG micelles were shown to be nontoxic to RAW 264.7 and MCF-7/ADR cells even at a high concentration of 1.5 mg/mL. Hence, micelles based on poly(acetal urethane) have appeared as a new class of biocompatible and acid-degradable nanocarriers for efficient intracellular drug delivery.
Co-reporter:Ru Cheng, Fenghua Meng, Chao Deng, Zhiyuan Zhong
Nano Today 2015 Volume 10(Issue 5) pp:656-670
Publication Date(Web):October 2015
DOI:10.1016/j.nantod.2015.09.005
•Gives an overview on biosignals existing in the tumor and tumor cells.•Illustrates the potential impacts and uses of biosignals on anticancer drug delivery.•Highlights the design rationale and recent exciting development of bioresponsive polymeric nanotherapeutics for targeted cancer chemotherapy.•Presents personal perspectives on the challenges and future development of translational nanomedicine based on bioresponsive polymeric nanotherapeutics.In recent years, bioresponsive polymeric nanotherapeutics that facilitate tumor cell uptake and trigger drug release at the target site have emerged as a fascinating platform for safe and efficient cancer therapy. The naturally occurring environments such as tumor acidity, tumor extracellular enzymes like matrix metalloproteases (MMP), endo/lysosomal pH, elevated glutathione levels in the cytoplasm and cell nucleus, lysosomal enzymes, as well as reactive oxygen species (ROS) in the mitochondria have been exploited as potential internal stimuli to achieve active drug and protein release in the tumor tissue or cancer cells. These bioresponsive nanosystems present several unique features such as no need of an external device, precision control over site of response (from tumor tissue down to cellular organelle level) following accumulation in the tumor via either passive or active targeting, and spontaneous activation in the tumor site or inside the tumor cells. In this review, we highlight the design rationale and recent exciting development of bioresponsive polymeric nanotherapeutics for enhanced cancer treatments with low side effects.
Co-reporter:Chao Deng, Jintian Wu, Ru Cheng, Fenghua Meng, Harm-Anton Klok, Zhiyuan Zhong
Progress in Polymer Science 2014 Volume 39(Issue 2) pp:330-364
Publication Date(Web):February 2014
DOI:10.1016/j.progpolymsci.2013.10.008
Polypeptides derived from naturally occurring α-amino acids have emerged as a unique and versatile family of bio-inspired biomaterials that can be tailor-made for varying biomedical applications such as controlled drug release, gene delivery, tissue engineering and regenerative medicine. In contrast to traditional biodegradable polymers such as aliphatic polyesters and polycarbonates, polypeptides are inherently functional, allow precise control over polarity and charges, show excellent stability against hydrolysis, and are prone to rapid biodegradation in vivo by specific enzymes. Ring-opening polymerization (ROP) of α-amino acid N-carboxyanhydrides (NCAs) is the most straightforward and practical approach for large-scale production of high molecular weight polypeptides. In the past decade, a remarkable progress has been made in controlled NCA polymerization, which offers an unprecedented access to precision polypeptide and hybrid materials by combining with living radical polymerization, click chemistry, and/or post-polymerization modification. Notably, several micellar anti-cancer drugs based on poly(ethylene glycol)-polypeptides have been already advanced to the clinical evaluation. In this review paper, we give an overview on de novo design, controlled synthesis and emerging biomedical applications of functional polypeptide and hybrid materials.
Co-reporter:Yan Zou, Yuan Song, Weijing Yang, Fenghua Meng, Haiyan Liu, Zhiyuan Zhong
Journal of Controlled Release 2014 Volume 193() pp:154-161
Publication Date(Web):10 November 2014
DOI:10.1016/j.jconrel.2014.05.016
In this study, we designed and developed galactose-installed photo-crosslinked pH-sensitive degradable micelles (Gal-CLMs) for active targeting chemotherapy of hepatocellular carcinoma in mice. Gal-CLMs were readily obtained from co-self-assembly of poly(ethylene glycol)-b-poly(mono-2,4,6-trimethoxy benzylidene-pentaerythritol carbonate-co-acryloyl carbonate) (PEG-b-P(TMBPEC-co-AC)) and Gal-PEG-b-poly(ε-caprolactone) (Gal-PEG-b-PCL) copolymers followed by photo-crosslinking. Notably, paclitaxel (PTX)-loaded Gal-CLMs (Gal-PTX-CLMs) showed a narrow distribution (PDI = 0.08–0.12) with average sizes ranging from 92.1 to 136.3 nm depending on the Gal contents. The release of PTX from Gal-CLMs while inhibited at physiological pH was enhanced under endosomal pH conditions. MTT assays in asialoglycoprotein receptor (ASGP-R) over-expressing HepG2 cells demonstrated that half-maximal inhibitory concentration (IC50) values of Gal-PTX-CLMs decreased from 11.7 to 2.9 to 1.1 μg/mL with increasing Gal contents from 10% to 20% to 30%, supporting receptor-mediated endocytosis mechanism. The in vivo biodistribution studies in human hepatoma SMMC-7721 tumor-bearing nude mice displayed that Gal20-PTX-CLMs resulted in significantly enhanced drug accumulation in the tumors over non-targeting PTX-CLM counterpart. In accordance, Gal20-PTX-CLMs caused much greater tumor growth inhibition than non-targeting PTX-CLMs as well as non-crosslinking Gal20-PTX-NCLM controls (average tumor volume: ca. 35 mm3versus 144 mm3 and 130 mm3, respectively). Histological analysis showed that Gal20-PTX-CLMs induced more extensive apoptosis of tumor cells while less damage to normal liver and kidney compared to Taxol. Ligand-installed photo-crosslinked pH-responsive degradable micelles have a great potential for targeted cancer chemotherapy.Ligand-directed photo-crosslinked pH-sensitive degradable micelles simultaneously resolve dilemmas of stability versus intracellular drug release and stealth versus efficient internalization by target tumor cells, resulting in potent inhibition of tumor growth in vivo as well as alleviation of systemic side effects.
Co-reporter:Yinan Zhong, Chao Wang, Ru Cheng, Liang Cheng, Fenghua Meng, Zhuang Liu, Zhiyuan Zhong
Journal of Controlled Release 2014 Volume 195() pp:63-71
Publication Date(Web):10 December 2014
DOI:10.1016/j.jconrel.2014.07.054
cRGD-directed, NIR-responsive and robust AuNR/PEG–PCL hybrid nanoparticles (cRGD-HNs) were designed and developed for targeted chemotherapy of human glioma xenografts in mice. As expected, cRGD-HNs had excellent colloidal stability. The in vitro release studies showed that drug release from DOX-loaded cRGD-HNs (cRGD-HN-DOX) was minimal under physiological conditions but markedly accelerated upon NIR irradiation at a low power density of 0.2 W/cm2, due to photothermally induced phase transition of PCL regime. MTT assays showed that the antitumor activity of cRGD-HN-DOX in αvβ3 integrin over-expressed human glioblastoma U87MG cells was greatly boosted by mild NIR irradiation, which was significantly more potent than non-targeting HN-DOX counterpart under otherwise the same conditions and was comparable or superior to free DOX, supporting receptor-mediated endocytosis mechanism. The in vivo pharmacokinetics studies showed that cRGD-HN-DOX had much longer circulation time than free DOX. The in vivo imaging and biodistribution studies revealed that cRGD-HN-DOX could actively target human U87MG glioma xenograft in nude mice. The therapeutic studies in human U87MG glioma xenografts exhibited that cRGD-HN-DOX in combination with NIR irradiation completely inhibited tumor growth and possessed much lower side effects than free DOX. The Kaplan–Meier survival curves showed that all mice treated with cRGD-HN-DOX plus NIR irradiation survived over an experimental period of 48 days while control groups treated with PBS, cRGD-HN-DOX, cRGD-HNs with NIR irradiation, free DOX, or HN-DOX with NIR irradiation (non-targeting control) had short life spans of 15–40 days. Ligand-directed AuNR/PEG–PCL hybrid nanoparticles with evident tumor-targetability as well as superior spatiotemporal and rate control over drug release have emerged as an appealing platform for cancer chemotherapy in vivo.cRGD-functionalized and NIR-responsive AuNR/PEG–PCL hybrid nanoparticles mediate targeted delivery as well as remotely controlled release of doxorubicin into human glioblastoma xenografts in mice, leading to complete inhibition of tumor growth with little adverse effects and 100% mice survival over an experimental period of 48 days.
Co-reporter:Huanli Sun, Fenghua Meng, Ru Cheng, Chao Deng, Zhiyuan Zhong
Acta Biomaterialia 2014 Volume 10(Issue 5) pp:2159-2168
Publication Date(Web):May 2014
DOI:10.1016/j.actbio.2014.01.010
Abstract
The clinical applications of protein drugs are restricted because of the absence of viable protein delivery vehicles. Here, we report on reduction- and pH--sensitive crosslinked polymersomes based on the poly(ethylene glycol)–poly(acrylic acid)–poly(2-(diethyl amino)ethyl methacrylate) (PEG–PAA–PDEA) triblock copolymer for efficient intracellular delivery of proteins and the potent induction of cancer cell apoptosis. PEG–PAA–PDEA (1.9–0.8–8.2 kg mol−1) was synthesized by controlled reversible addition-fragmentation chain transfer polymerization and further modified with cysteamine to yield the thiol-containing PEG–PAA(SH)–PDEA copolymer. PEG–PAA(SH)–PDEA was water-soluble at acidic and physiological pH but formed robust and monodisperse polymersomes with an average size of ∼35 nm upon increasing the pH to 7.8 or above followed by oxidative crosslinking. These disulfide-crosslinked polymersomes, while exhibiting excellent colloidal stability, were rapidly dissociated in response to 10 mM glutathione at neutral or mildly acidic conditions. Notably, these polymersomes could efficiently load proteins like bovine serum albumin and cytochrome C (CC). The in vitro release studies revealed that protein release was fast and nearly quantitative under the intracellular-mimicking reducing environment. Confocal microscopy observations showed that these dual-sensitive polymersomes efficiently released fluorescein isothiocyanate-CC into MCF-7 cells in 6 h. Most remarkably, MTT assays showed that CC-loaded dual-sensitive polymersomes induced potent cancer cell apoptosis, in which markedly decreased cell viabilities of 11.3%, 8.1% and 52.7% were observed for MCF-7, HeLa and 293T cells, respectively, at a CC dosage of 160 μg ml−1. In contrast, free CC caused no cell death under otherwise the same conditions. These dual-bioresponsive polymersomes have appeared as a multifunctional platform for active intracellular protein release.
Co-reporter:Weiwei Guo;Meng Zheng;Yinan Zhong;Fenghua Meng;Chao Deng
Chinese Journal of Chemistry 2014 Volume 32( Issue 1) pp:57-65
Publication Date(Web):
DOI:10.1002/cjoc.201300611
Abstract
Water soluble poly(ethylene oxide)-graft-methotrexate (PEO-g-MTX) conjugates with a robust amide linkage via the amine or carboxylic acid groups of MTX were designed, prepared and investigated for in vitro anti-tumor effects. MTX was conjugated to multi-functional PEO containing multiple pendant carboxylic acid (PEO-g-COOH) or amine groups (PEO-g-NH2) via the carbodiimide chemistry, which afforded PEO-g-MTX conjugates with an amide bond to the aminopteridine ring or carboxylic acid groups of MTX (denoted as PEO-g-MTX(COOH) and PEO-g-MTX(NH2), respectively). Dynamic light scattering (DLS) revealed that all PEO-g-MTX conjugates, with MTX contents varying from 4.8 to 19.6 wt%, existed as unimers in phosphate buffer (PB, pH 7.4, 20 mmol·L−1). Interestingly, MTT assays showed that PEO-g-MTX(COOH) exhibited potent anti-tumor activity in HeLa, A549, KB and NIH3T3 cells with cytotoxicity profiles comparable to that of free MTX. In contrast, PEO-g-MTX(NH2) revealed diminishing cytostatic effect with IC50 (half maximal inhibitory concentration) ten to hundred times higher than that of PEO-g-MTX(COOH). Moreover, PEO-g-MTX(COOH) conjugates allowed facile conjugation with targeting ligands. Notably, folate-decorated PEO-g-MTX(COOH) macromolecular drugs showed apparent targetability to folate receptor-overexpressing KB cells with an IC50 over 12-fold lower than non-targeting PEO-g-MTX(COOH) control and about 2-fold lower than free MTX under otherwise the same conditions.
Co-reporter:Yanjiao Jiang, Jing Chen, Chao Deng, Erik J. Suuronen, Zhiyuan Zhong
Biomaterials 2014 35(18) pp: 4969-4985
Publication Date(Web):
DOI:10.1016/j.biomaterials.2014.03.001
Co-reporter:Yinan Zhong, Fenghua Meng, Chao Deng, and Zhiyuan Zhong
Biomacromolecules 2014 Volume 15(Issue 6) pp:
Publication Date(Web):May 5, 2014
DOI:10.1021/bm5003009
In recent years, polymeric nanoparticles have appeared as a most viable and versatile delivery system for targeted cancer therapy. Various in vivo studies have demonstrated that virus-sized stealth particles are able to circulate for a prolonged time and preferentially accumulate in the tumor site via the enhanced permeability and retention (EPR) effect (so-called “passive tumor-targeting”). The surface decoration of stealth nanoparticles by a specific tumor-homing ligand, such as antibody, antibody fragment, peptide, aptamer, polysaccharide, saccharide, folic acid, and so on, might further lead to increased retention and accumulation of nanoparticles in the tumor vasculature as well as selective and efficient internalization by target tumor cells (termed as “active tumor-targeting”). Notably, these active targeting nanoparticulate drug formulations have shown improved, though to varying degrees, therapeutic performances in different tumor models as compared to their passive targeting counterparts. In addition to type of ligands, several other factors such as in vivo stability of nanoparticles, particle shape and size, and ligand density also play an important role in targeted cancer chemotherapy. In this review, concept and recent development of polymeric nanoparticles conjugated with specific targeting ligands, ranging from proteins (e.g., antibodies, antibody fragments, growth factors, and transferrin), peptides (e.g., cyclic RGD, octreotide, AP peptide, and tLyp-1 peptide), aptamers (e.g., A10 and AS1411), polysaccharides (e.g., hyaluronic acid), to small biomolecules (e.g., folic acid, galactose, bisphosphonates, and biotin), for active tumor-targeting drug delivery in vitro and in vivo are highlighted and discussed. With promise to maximize therapeutic efficacy while minimizing systemic side effects, ligand-mediated active tumor-targeting treatment modality has become an emerging and indispensable platform for safe and efficient cancer therapy.
Co-reporter:Wei Chen, Yan Zou, Fenghua Meng, Ru Cheng, Chao Deng, Jan Feijen, and Zhiyuan Zhong
Biomacromolecules 2014 Volume 15(Issue 3) pp:
Publication Date(Web):January 27, 2014
DOI:10.1021/bm401749t
Reduction-sensitive shell-sheddable glyco-nanoparticles were designed and developed based on poly(ε-caprolactone)-graft-SS-lactobionic acid (PCL-g-SS-LBA) copolymer for efficient hepatoma-targeting delivery of doxorubicin (DOX). PCL-g-SS-LBA was prepared by ring-opening copolymerization of ε-caprolactone and pyridyl disulfide carbonate followed by postpolymerization modification with thiolated lactobionic acid (LBA-SH) via thiol-disulfide exchange reaction. The dynamic light scattering (DLS) and transmission electron microscopy (TEM) showed that PCL-g-SS-LBA was self-assembled into monodisperse nanoparticles (SS-GNs) with a mean diameter of about 80 nm. SS-GNs while remaining stable under physiological conditions (37 °C, pH 7.4) were prone to rapid shell-shedding and aggregation in the presence of 10 mM dithiothreitol (DTT). DOX was loaded into SS-GNs with a decent loading content of 12.0 wt %. Notably, in vitro release studies revealed that about 80.3% DOX was released from DOX-loaded SS-GNs in 24 h under a reductive condition while low drug release (<21%) was observed for DOX-loaded PCL-g-LBA nanoparticles (reduction-insensitive control) under otherwise the same condition and for DOX-loaded SS-GNs under a nonreductive condition. The flow cytometry and confocal microscopy observations indicated that SS-GNs were efficiently taken up by asialoglycoprotein receptor (ASGP-R)-overexpressing HepG2 cells likely via a receptor-mediated endocytosis mechanism and DOX was released into the nuclei of cells following 4 h incubation. MTT assays showed that DOX-loaded SS-GNs exhibited a high antitumor activity toward HepG2 cells, which was comparable to free DOX and about 18-fold higher than their reduction-insensitive counterparts, while blank SS-GNs were nontoxic up to a tested concentration of 1.0 mg/mL. These shell-sheddable glyco-nanoparticles are promising for hepatoma-targeting chemotherapy.
Co-reporter:Wei Chen, Ping Zhong, Fenghua Meng, Ru Cheng, Chao Deng, Jan Feijen, Zhiyuan Zhong
Journal of Controlled Release 2013 Volume 169(Issue 3) pp:171-179
Publication Date(Web):10 August 2013
DOI:10.1016/j.jconrel.2013.01.001
Redox and pH dual-responsive biodegradable micelles were developed based on poly(ethylene glycol)-SS-poly(2,4,6-trimethoxybenzylidene-pentaerythritol carbonate) (PEG-SS-PTMBPEC) copolymer and investigated for intracellular doxorubicin (DOX) release. PEG-SS-PTMBPEC copolymer with an Mn of 5.0–4.1 kg/mol formed micellar particles with an average diameter of 140 nm and a low polydispersity of 0.12. DOX was loaded into PEG-SS-PTMBPEC micelles with a decent drug loading content of 11.3 wt.%. The in vitro release studies showed that under physiological conditions only ca. 24.5% DOX was released from DOX-loaded micelles in 21 h. The release of DOX was significantly accelerated at pH 5.0 or in the presence of 10 mM glutathione (GSH) at pH 7.4, in which 62.8% and 74.3% of DOX was released, respectively, in 21 h. The drug release was further boosted under 10 mM GSH and pH 5.0 conditions, with 94.2% of DOX released in 10 h. Notably, DOX release was also facilitated by 2 or 4 h incubation at pH 5.0 and then at pH 7.4 with 10 mM GSH, which mimics the intracellular pathways of endocytosed micellar drugs. Confocal microscopy observation indicated that DOX was delivered and released into the nuclei of HeLa cells following 8 h incubation with DOX-loaded PEG-SS-PTMBPEC micelles, while DOX was mainly located in the cytoplasm for reduction-insensitive PEG-PTMBPEC controls. MTT assays revealed that DOX-loaded PEG-SS-PTMBPEC micelles had higher anti-tumor activity than reduction-insensitive controls, with low IC50 of 0.75 and 0.60 μg/mL for HeLa and RAW 264.7 cells, respectively, following 48 h incubation. PEG-SS-PTMBPEC micelles displayed low cytotoxicity up to a concentration of 1.0 mg/mL. These redox and pH dual-bioresponsive degradable micelles have appeared as a promising platform for targeted intracellular anticancer drug release.pH and redox dual-responsive biodegradable micelles trigger drug release not only in the acidic endosomal compartments but also in the reducing cytoplasms, resulting in superior antitumor effect.Figure optionsDownload full-size imageDownload high-quality image (294 K)Download as PowerPoint slide
Co-reporter:Zhiyuan Zhong, Jan Feijen
Journal of Controlled Release 2013 Volume 169(Issue 3) pp:163-164
Publication Date(Web):10 August 2013
DOI:10.1016/j.jconrel.2013.04.011
Co-reporter:Yinan Zhong, Weijing Yang, Huanli Sun, Ru Cheng, Fenghua Meng, Chao Deng, and Zhiyuan Zhong
Biomacromolecules 2013 Volume 14(Issue 10) pp:
Publication Date(Web):September 2, 2013
DOI:10.1021/bm401098w
The therapeutic performance of biodegradable micellar drugs is far from optimal due to existing challenges like poor tumor cell uptake and intracellular drug release. Here, we report on ligand-directed reduction-sensitive shell-sheddable biodegradable micelles based on poly(ethylene glycol)-poly(ε-caprolactone) (PEG-PCL) copolymer actively delivering doxorubicin (DOX) into the nuclei of target cancer cells, inducing superb in vitro antitumor effects. The micelles were constructed from PEG-SS-PCL and galactose-PEG-PCL (Gal-PEG-PCL) block copolymers, in which Gal-PEG-PCL was designed with a longer PEG than that in PEG-SS-PCL (6.0 vs 5.0 kDa) to fully expose Gal ligands onto the surface of micelles for effective targeting to hepatocellular carcinoma cells. PEG-SS-PCL combining with 10 or 20 wt % of Gal-PEG-PCL formed uniform micelles with average sizes of 56.1 and 58.2 nm (denoted as PEG-SS-PCL/Gal10 and PEG-SS-PCL/Gal20, respectively). The in vitro release studies showed that about 81.1 and 75.0% DOX was released in 12 h from PEG-SS-PCL/Gal10 and PEG-SS-PCL/Gal20 micelles under a reducing condition containing 10 mM dithiothreitol (DTT). In contrast, minimal DOX release (<12%) was observed for PEG-SS-PCL/Gal10 and PEG-SS-PCL/Gal20 micelles under nonreducing conditions as well as for reduction-insensitive Gal-PEG-PCL and PEG-PCL/Gal20 micelles in the presence of 10 mM DTT. MTT assays in HeLa and HepG2 cells showed that DOX-loaded PEG-SS-PCL/Gal20 micelles exhibited apparent targetability and significantly enhanced antitumor efficacy toward asialoglycoprotein receptor (ASGP-R)-overexpressing HepG2 cells with a particularly low half maximal inhibitory concentration (IC50) of 1.58 μg DOX equiv/mL, which was comparable to free DOX and approximately six times lower than that for nontargeting PEG-SS-PCL counterparts under otherwise the same conditions. Interestingly, confocal microscopy observations using FITC-labeled PEG-SS-PCL/Gal20 micelles showed that DOX was efficiently delivered and released into the nuclei of HepG2 cells in 8 h. Flow cytometry revealed that cellular DOX level in HepG2 cells treated with DOX-loaded PEG-SS-PCL/Gal20 micelles was much greater than that with reduction-insensitive PEG-PCL/Gal20 and nontargeting PEG-SS-PCL controls, signifying the importance of combining shell-shedding and active targeting. Ligand-directed, reduction-sensitive, shell-sheddable, and biodegradable micelles have emerged as a versatile and potent platform for targeted cancer chemotherapy.
Co-reporter:Yinan Zhong, Chao Wang, Liang Cheng, Fenghua Meng, Zhiyuan Zhong, and Zhuang Liu
Biomacromolecules 2013 Volume 14(Issue 7) pp:
Publication Date(Web):May 23, 2013
DOI:10.1021/bm400530d
Gold nanorod-cored biodegradable micelles were prepared by coating gold nanorods (AuNRs) with lipoylated poly(ethylene glycol)-b-poly(ε-caprolactone) (PEG-PCL-LA) block copolymer and investigated for remotely triggered release of doxorubicin (DOX) and effective inhibition of drug-sensitive and multidrug-resistant (MDR) cancer cells. The micelles had uniform sizes and excellent colloidal stability. The in vitro release studies showed that drug release from DOX-loaded AuNR-cored micelles (AuNR-M-DOX) was minimal under physiological conditions but markedly enhanced upon NIR irradiation at a low power density of 0.2 W/cm2, most likely due to photothermally induced phase transition of PCL regime. As revealed by confocal microscopy and flow cytometry, NIR could also trigger effective DOX release in drug-sensitive as well as drug-resistant MCF-7 cells. MTT assays showed that antitumor activity of AuNR-M-DOX to drug-sensitive MCF-7 cells was significantly boosted by mild NIR irradiation, reaching a comparable level to free DOX. Most remarkably, AuNR-M-DOX combined with NIR irradiation could also effectively kill drug-resistant MCF-7 cells, in which a cell viability of 38% was observed at a DOX dosage of 10 μg equiv/mL, whereas 100% cell viability was maintained for cells treated with free DOX under otherwise the same conditions. These AuNR-cored biodegradable micelles with high stability, photo-triggered drug release, and effective reversal of multidrug resistance in cancer cells have appeared as a novel platform for targeted cancer therapy.
Co-reporter:Wei Chen, Meng Zheng, Fenghua Meng, Ru Cheng, Chao Deng, Jan Feijen, and Zhiyuan Zhong
Biomacromolecules 2013 Volume 14(Issue 4) pp:
Publication Date(Web):March 12, 2013
DOI:10.1021/bm400206m
In situ forming reduction-sensitive degradable nanogels were designed and developed based on poly(ethylene glycol)-b-poly(2-(hydroxyethyl) methacrylate-co-acryloyl carbonate) (PEG-P(HEMA-co-AC)) block copolymers for efficient loading as well as triggered intracellular release of proteins. PEG-P(HEMA-co-AC) copolymers were prepared with controlled Mn of 9.1, 9.5, and 9.9 kg/mol and varying numbers of AC units per molecule of 7, 9 and 11, respectively (denoted as copolymer 1, 2, and 3) by reversible addition–fragmentation chain transfer copolymerization. These copolymers were freely soluble in phosphate buffer but formed disulfide-cross-linked nanogels with defined sizes ranging from 72.5 to 124.1 nm in the presence of cystamine via ring-opening reaction with cyclic carbonate groups. The sizes of nanogels decreased with increasing AC units as a result of increased cross-linking density. Dynamic light scattering studies showed that these nanogels though stable at physiological conditions were rapidly dissociated in response to 10 mM dithiothreitol (DTT). Interestingly, FITC-labeled cytochrome C (FITC–CC) could be readily loaded into nanogels with remarkable loading efficiencies (up to 98.2%) and loading contents (up to 48.2 wt.%). The in vitro release studies showed that release of FITC–CC was minimal under physiological conditions but significantly enhanced under reductive conditions in the presence of 10 mM DTT with about 96.8% of FITC–CC released in 22 h from nanogel 1. In contrast, protein release from 1,4-butanediamine cross-linked nanogels (reduction-insensitive control) remained low under otherwise the same conditions. MTT assays showed that these nanogels were nontoxic to HeLa cells up to a tested concentration of 2 mg/mL. Confocal microscopy results showed that nanogel 1 delivered and released FITC–CC into the perinuclei region of HeLa cells following 8 h incubation. CC-loaded reductively degradable nanogels demonstrated apparently better apoptotic activity than free CC as well as reduction-insensitive controls. These in situ forming, surfactant and oil-free, and reduction-sensitive degradable nanogels are highly promising for targeted protein therapy.
Co-reporter:Wei Chen, Yan Zou, Junna Jia, Fenghua Meng, Ru Cheng, Chao Deng, Jan Feijen, and Zhiyuan Zhong
Macromolecules 2013 Volume 46(Issue 3) pp:699-707
Publication Date(Web):January 30, 2013
DOI:10.1021/ma302499a
Pyridyl disulfide-functionalized cyclic carbonate (PDSC) monomer was obtained in four straightforward steps from 3-methyl-3-oxetanemethanol and exploited for facile preparation of functional poly(ε-caprolactone) (PCL) containing pendant pyridyl disulfide (PDS) groups via ring-opening copolymerization with ε-caprolactone. The results showed that PDS-functionalized PCL polymers were prepared with controlled molecular weights and functionalities. The exchange reaction between PDS-functionalized PCL and thiolated poly(ethylene glycol) (PEG-SH) at a PEG-SH/PDS molar ratio of 2/1 afforded PCL-g-SS-PEG graft copolymers in high yields. The dynamic light scattering (DLS) analyses showed that PCL-g-SS-PEG copolymer self-assembled into micelles with a diameter of 110–120 nm and a low polydispersity (PDI) in phosphate buffer (pH 7.4, 10 mM). PCL-g-SS-PEG micelles while sufficiently stable under physiological conditions were prone to rapid shell shedding and aggregation under a reductive condition. Doxorubicin (DOX) was loaded into PCL-g-SS-PEG micelles with a decent drug loading content of 10.1 wt %. Notably, in vitro release studies revealed that ca. 82.1% DOX was released in 12 h under a reductive environment analogous to that of the intracellular compartments such as cytosol and the cell nucleus whereas only ca. 17.5% DOX was released in 24 h under nonreductive conditions. Confocal microscopy observation indicated that DOX was delivered into the nuclei of HeLa cells following 8 h incubation with DOX-loaded PCL-g-SS-PEG micelles. MTT assays in HeLa cells demonstrated that DOX-loaded PCL-g-SS-PEG micelles retained high antitumor activity with low IC50 (half-maximal inhibitory concentration) of 0.82–0.95 μg DOX equiv/mL while blank PCL-g-SS-PEG micelles were nontoxic up to a tested concentration of 1.0 mg/mL. This study presents a versatile and controlled synthesis of PDS-functionalized biodegradable polymers and reduction-sensitive biodegradable graft copolymer micelles that are of particular interest for active intracellular drug release.
Co-reporter:Liangliang Wu, Yan Zou, Chao Deng, Ru Cheng, Fenghua Meng, Zhiyuan Zhong
Biomaterials 2013 34(21) pp: 5262-5272
Publication Date(Web):
DOI:10.1016/j.biomaterials.2013.03.035
Co-reporter:Ru Cheng, Fenghua Meng, Chao Deng, Harm-Anton Klok, Zhiyuan Zhong
Biomaterials 2013 34(14) pp: 3647-3657
Publication Date(Web):
DOI:10.1016/j.biomaterials.2013.01.084
Co-reporter:Yudan Gu, Yinan Zhong, Fenghua Meng, Ru Cheng, Chao Deng, and Zhiyuan Zhong
Biomacromolecules 2013 Volume 14(Issue 8) pp:
Publication Date(Web):June 18, 2013
DOI:10.1021/bm400615n
Endosomal pH-activatable paclitaxel (PTX) prodrug micellar nanoparticles were designed and prepared by conjugating PTX onto water-soluble poly(ethylene glycol)-b-poly(acrylic acid) (PEG-PAA) block copolymers via an acid-labile acetal bond to the PAA block and investigated for potent growth inhibition of human cancer cells in vitro. PTX was readily conjugated to PEG-PAA with high drug contents of 21.6, 27.0, and 42.8 wt % (denoted as PTX prodrugs 1, 2, and 3, respectively) using ethyl glycol vinyl ether (EGVE) as a linker. The resulting PTX conjugates had defined molecular weights and self-assembled in phosphate buffer (PB, pH 7.4, 10 mM) into monodisperse micellar nanoparticles with average sizes of 158.3–180.3 nm depending on PTX contents. The in vitro release studies showed that drug release from PTX prodrug nanoparticles was highly pH-dependent, in which ca. 86.9%, 66.4% and 29.0% of PTX was released from PTX prodrug 3 at 37 °C in 48 h at pH 5.0, 6.0, and pH 7.4, respectively. MTT assays showed that these pH-sensitive PTX prodrug nanoparticles exhibited high antitumor effect to KB and HeLa cells (IC50 = 0.18 and 0.9 μg PTX equiv/mL, respectively) as well as PTX-resistant A549 cells. Notably, folate-decorated PTX prodrug micellar nanoparticles based on PTX prodrug 3 and 20 wt % folate-poly(ethylene glycol)-b-poly(d,l-lactide) (FA-PEG-PLA) displayed apparent targetability to folate receptor-overexpressing KB cells with IC50 over 12 times lower than nontargeting PTX prodrug 3 under otherwise the same conditions. Furthermore, PTX prodrug nanoparticles could also load doxorubicin (DOX) to simultaneously release PTX and DOX under mildly acidic pH. These acetal-linked PTX prodrug micellar nanoparticles have appeared as a highly versatile and potent platform for cancer therapy.
Co-reporter:Yaping Fan, Chao Deng, Ru Cheng, Fenghua Meng, and Zhiyuan Zhong
Biomacromolecules 2013 Volume 14(Issue 8) pp:
Publication Date(Web):July 2, 2013
DOI:10.1021/bm400637s
In situ forming hydrogels were developed from 4-arm poly(ethylene glycol)–methacrylate (PEG-4-MA) and −tetrazole (PEG-4-Tet) derivatives through catalyst-free and bioorthogonal “tetrazole–alkene” photo-click chemistry. PEG-4-MA and PEG-4-Tet (Mn = 10 kg/mol) were soluble at 37 °C in phosphate buffer (PB, pH 7.4, 10 mM) at total polymer concentrations ranging from 20 to 60 wt % but formed fluorescent hydrogels upon 365 nm UV irradiation at an intensity of 20.6, 30.7, or 60 mW/cm2. The gelation times ranged from ca. 50 s to 5 min, and storage moduli varied from 0.65 to 25.2 kPa depending on polymer concentrations and degrees of Tet substitution in PEG-4-Tet conjugates. The cell experiments via an indirect contact assay demonstrated that these “tetrazole–alkene” photo-click PEG hydrogels were noncytotoxic. The high specificity of photo-click reaction renders thus obtained PEG hydrogels particularly interesting for controlled protein release. Notably, in vitro release studies showed that cytochrome c (CC), γ-globulins (Ig), and recombinant human interleukin-2 (rhIL-2) all were released from PEG hydrogels in a sustained and quantitative manner over a period of 14–20 days. Importantly, released CC and rhIL-2 exhibited comparable biological activities to native CC and rhIL-2, respectively. These results confirm that “tetrazole–alkene” photo-click reaction is highly compatible with these loaded proteins. This photo-controlled, specific, efficient, and catalyst-free click chemistry provides a new and versatile strategy to in situ forming hydrogels that hold tremendous potentials for protein delivery and tissue engineering.
Co-reporter:Jianren Zhou, Peipei Chen, Chao Deng, Fenghua Meng, Ru Cheng, and Zhiyuan Zhong
Macromolecules 2013 Volume 46(Issue 17) pp:
Publication Date(Web):August 21, 2013
DOI:10.1021/ma4014669
Vinyl sulfone-substituted l-cysteine N-carboxyanhydride (VSCys-NCA) monomer was designed and developed to afford a novel and versatile family of vinyl sulfone (VS)-functionalized polypeptides, which further offer a facile access to functional polypeptide-based materials including glycopolypeptides, functional polypeptide coatings, and in situ forming polypeptide hydrogels through Michael-type addition chemistry under mild conditions. VSCys-NCA was obtained in two straightforward steps with a high overall yield of 76%. The copolymerization of γ-benzyl l-glutamate NCA (BLG-NCA), N-benzyloxycarbonyl-l-lysine NCA (ZLL-NCA), or l-leucine NCA (Leu-NCA) with VSCys-NCA using 1,1,1-trimethyl-N-2-propenylsilanamine (TMPS) as an initiator proceeded smoothly in DMF at 40 °C, yielding P(BLG-co-VSCys), P(ZLL-co-VSCys), or P(Leu-co-VSCys) with defined functionalities, controlled molecular weights, and moderate polydispersities (PDI = 1.15–1.50). The acidic deprotection of P(BLG-co-VSCys) and P(ZLL-co-VSCys) furnished water-soluble VS-functionalized poly(l-glutamic acid) (P(Glu-co-VSCys)) and VS-functionalized poly(l-lysine) (P(LL-co-VSCys)), respectively. These VS-functionalized polypeptides were amenable to direct, efficient, and selective postpolymerization modification with varying thiol-containing molecules such as 2-mercaptoethanol, 2-mercaptoethylamine hydrochloride, l-cysteine, and thiolated galactose providing functional polypeptides containing pendant hydroxyl, amine, amino acid, and saccharide, respectively. The contact angle and fluorescence measurements indicated that polymer coatings based on P(Leu-co-VSCys) allowed direct functionalization with thiol-containing molecules under aqueous conditions. Moreover, hydrogels formed in situ upon mixing aqueous solutions of P(Glu-co-VSCys) and thiolated glycol chitosan at 37 °C. These vinyl sulfone-functionalized polypeptides have opened a new avenue to a broad range of advanced polypeptide-based materials.
Co-reporter:Xiaoyan Wang, Huanli Sun, Fenghua Meng, Ru Cheng, Chao Deng, and Zhiyuan Zhong
Biomacromolecules 2013 Volume 14(Issue 8) pp:
Publication Date(Web):July 1, 2013
DOI:10.1021/bm4007248
Hepatoma-targeting reduction-sensitive chimaeric biodegradable polymersomes were designed and developed based on galactose–poly(ethylene glycol)–poly(ε-caprolactone) (Gal-PEG-PCL), PEG–PCL–poly(2-(diethylamino)ethyl methacrylate) (PEG-PCL-PDEA, asymmetric), and PEG-SS-PCL for facile loading and triggered intracellular delivery of proteins. The chimaeric polymersomes formed from PEG-PCL-PDEA and PEG-SS-PCL had a monodisperse distribution with average sizes ranging from 95.5 to 199.2 nm depending on PEG-SS-PCL contents. Notably, these polymersomes displayed decent loading of bovine serum albumin (BSA), ovalbumin (OVA), and cytochrome C (CC) proteins likely due to presence of electrostatic and hydrogen bonding interactions between proteins and PDEA block located in the interior of polymersomes. The in vitro release studies showed that protein release was largely accelerated under a reductive condition containing 10 mM dithiothreitol (DTT). For example, ca. 77.2 and 22.1% of FITC-BSA were released from CP(SS50) (chimaeric polymersomes containing 50 wt % PEG-SS-PCL) at 37 °C in 12 h in the presence and absence of 10 mM DTT, respectively. Confocal microscopy showed that FITC-CC-loaded Gal-decorated CP(SS40) could efficiently deliver and release FITC-CC into HepG2 cells following 24 h treatment, in contrast to little or negligible fluorescence detected in HepG2 cells treated with FITC-CC-loaded nontargeting polymersomes or free CC. MTT assays revealed that CC-loaded Gal-decorated CP(SS40) exhibited apparent targetability and pronounced antitumor activity to HepG2 cells, in which cell viabilities decreased from 81.9, 60.6, 49.5, 42.2 to 31.5% with increasing Gal-PEG-PCL contents from 0, 10, 20, 30 to 40 wt %. Most remarkably, granzyme B-loaded Gal-decorated chimaeric polymersomes effectively caused apoptosis of HepG2 cells with a markedly low half-maximal inhibitory concentration (IC50) of 2.7 nM. These reduction-responsive chimaeric biodegradable polymersomes offer a multifunctional platform for efficient intracellular protein delivery.
Co-reporter:Fenghua Meng, Ru Cheng, Chao Deng, Zhiyuan Zhong
Materials Today 2012 Volume 15(Issue 10) pp:436-442
Publication Date(Web):October 2012
DOI:10.1016/S1369-7021(12)70195-5
In order to elicit therapeutic effects, many drugs including small molecule anticancer drugs, proteins, siRNA, and DNA have to be delivered and released into the specific cellular compartments typically the cytoplasm or nucleus of target cells. Intracellular environment-responsive nanosystems that exhibit good extracellular stability while rapidly releasing drugs inside cancer cells have been actively pursued for effective cancer therapy. Here, we highlight novel designs of smart nanosystems that release drugs in response to an intracellular biological signal of cancer cells such as acidic pH in endo/lysosomal compartments, enzymes in lysosomes, and redox potential in cytoplasm and the cell nucleus.
Co-reporter:Ru Cheng, Xiaoyan Wang, Wei Chen, Fenghua Meng, Chao Deng, Haiyan Liu and Zhiyuan Zhong
Journal of Materials Chemistry A 2012 vol. 22(Issue 23) pp:11730-11738
Publication Date(Web):03 Apr 2012
DOI:10.1039/C2JM30700F
Biodegradable micelles were prepared from poly(ε-caprolactone)-g-poly(2-hydroxyethyl methacrylate) (PCL-g-PHEMA) graft copolymers and investigated for controlled release of doxorubicin (DOX). PCL-g-PHEMA copolymers were readily obtained by controlled ring-opening copolymerization of acryloyl cyclic carbonate and ε-caprolactone, Michael-type conjugate addition reaction with cysteamine, coupling reaction with 4-cyanopentanoic acid dithionaphthalenoate (CPADN) via carbodiimide chemistry, and reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-hydroxyethyl methacrylate (HEMA). 1H NMR analyses showed that Mn of PHEMA ranged from 8.7, 16.3 to 33.8 kg mol−1, in proximity to the design as well as those determined by gel permeation chromatography (GPC). Differential scanning calorimetry (DSC) revealed that all three PCL-g-PHEMA graft copolymers had depressed melting temperatures (Tm = 31.3–32.5 °C) and low crystallinities (Xc = 3.05–5.66%). Dynamic light scattering (DLS) showed that PCL-g-PHEMA formed monodisperse micelles with low polydispersity indexes of 0.04–0.16 and average sizes ranging from 80.5 to 179.7 nm depending on PHEMA chain lengths. These graft copolymers displayed low critical micelle concentrations (CMCs) of 0.051–0.151 μM. The micellar sizes decreased following loading with DOX while PDI remained low. Interestingly, in vitro drug release studies showed that DOX-loaded PCL-g-PHEMA micelles exhibited superior pH-responsive release behaviors, in which up to 94.5% of DOX was released in 3 d at pH 5.0 while DOX release was significantly slower at pH 7.4 (maximum 54.1% release in 3 d). MTT assays with HeLa cells demonstrated that DOX-loaded PCL-g-PHEMA micelles retained high anti-tumor activity with low IC50 (half inhibitory concentration) of 1.47–1.74 μg DOX equiv. mL−1 while PCL-g-PHEMA micelles were practically non-toxic up to a tested concentration of 80 mg mL−1. These novel biodegradable PCL-g-PHEMA graft copolymer micelles with low CMC, small and tunable sizes, high drug loading, and pH-responsive drug release have emerged as superior nanocarriers for “smart” tumor-targeting drug delivery.
Co-reporter:Yali Wu, Wei Chen, Fenghua Meng, Zhongjuan Wang, Ru Cheng, Chao Deng, Haiyan Liu, Zhiyuan Zhong
Journal of Controlled Release 2012 Volume 164(Issue 3) pp:338-345
Publication Date(Web):28 December 2012
DOI:10.1016/j.jconrel.2012.07.011
The extracellular stability versus intracellular drug release dilemma has been a long challenge for micellar drug delivery systems. Here, core-crosslinked pH-sensitive degradable micelles were developed based on poly(ethylene glycol)-b-poly(mono-2,4,6-trimethoxy benzylidene-pentaerythritol carbonate-co-acryloyl carbonate) (PEG-b-P(TMBPEC-co-AC)) diblock copolymer that contains acid-labile acetal and photo-crossslinkable acryloyl groups in the hydrophobic polycarbonate block for intracellular paclitaxel (PTX) release. The micelles following photo-crosslinking while displaying high stability at pH 7.4 were prone to rapid hydrolysis at mildly acidic pHs of 4.0 and 5.0, with half lives of ca. 12.5 and 38.5 h, respectively. Notably, these micelles showed high drug loading efficiencies of 76.0–93.2% at theoretical PTX loading contents of 5–15 wt.%. Depending on drug loading contents, PTX-loaded micelles had average sizes varying from 132.2 to 171.6 nm, which were decreased by 17–22 nm upon photo-crosslinking. The in vitro release studies showed that PTX release at pH 7.4 was greatly inhibited by crosslinking of micelles. Notably, rapid drug release was obtained under mildly acidic conditions, in which 90.0% and 78.1% PTX was released in 23 h at pH 4.0 and 5.0, respectively. MTT assays showed that PTX-loaded crosslinked micelles retained high anti-tumor activity with a cell viability of 9.2% observed for RAW 264.7 cells following 72 h incubation, which was comparable to PTX-loaded non-crosslinked counterparts (cell viability 7.5%) under otherwise the same conditions, supporting efficient drug release from PTX-loaded crosslinked micelles inside the tumor cells. These core-crosslinked pH-responsive biodegradable micelles with superior extracellular stability and rapid intracellular drug release provide a novel platform for tumor-targeting drug delivery.Photo-crosslinked pH-sensitive degradable micelles while exhibit superior extracellular stability “actively” release paclitaxel under a mildly acidic condition mimicking that of the endo/lysosomal compartments, elegantly resolving extracellular stability and inrtracellular drug release dilemma of micellar drug delivery systems.
Co-reporter:Wei Wang, Huanli Sun, Fenghua Meng, Shoubao Ma, Haiyan Liu and Zhiyuan Zhong
Soft Matter 2012 vol. 8(Issue 14) pp:3949-3956
Publication Date(Web):28 Feb 2012
DOI:10.1039/C2SM07461C
The reduction-sensitive shedding of hydrophilic shells has recently emerged as a simple, effective and general approach to achieve markedly improved intracellular drug release from micelles. Here, the effects of disulfide content on reduction-sensitivity, triggered drug release and the anti-tumor activity of shell-sheddable micelles self-assembled from a mixture of reducible poly(ethylene glycol)–SS–poly(ε-caprolactone) (PEG–SS–PCL) and non-reducible poly(ethylene glycol)–poly(ε-caprolactone) (PEG–PCL) block copolymers were systematically investigated. Interestingly, in contrast to the rapid aggregation of PEG–SS–PCL micelles, mixed micelles containing 10–90 wt% PEG–PCL displayed little size change in response to 10 mM dithiothreitol (DTT). The in vitro release studies showed that under intracellular-mimicking reductive environments, the doxorubicin (DOX) release rate increased with increasing PEG–SS–PCL content in the micelles, in which about 29.4, 42.7, 77.9 and 86.9% DOX was released within 12 h from micelles containing 30, 50, 70 and 90 wt% PEG–SS–PCL, respectively. In contrast, DOX release was limited (<20%) under non-reductive physiological conditions. Notably, flow cytometry displayed clear correlation between cellular DOX levels and PEG–SS–PCL content in DOX-loaded micelles. Moreover, confocal laser scanning microscopy (CLSM) observations indicated progressively stronger DOX fluorescence in RAW 264.7 cells following 12 h treatment with DOX-loaded micelles containing increasing PEG–SS–PCL contents. In addition, MTT assays in RAW 264.7 cells showed that the cytotoxicity of DOX-loaded micelles was augmented proportionally to PEG–SS–PCL content, signifying the role of reduction-triggered “active” drug release in cells. These results have shown that the intracellular drug release and therefore anti-tumor activity of micellar drugs can be precisely controlled by the extent of reduction-triggered shedding of hydrophilic shells.
Co-reporter:Caihong Zhu, Meng Zheng, Fenghua Meng, Frauke Martina Mickler, Nadia Ruthardt, Xiulin Zhu, and Zhiyuan Zhong
Biomacromolecules 2012 Volume 13(Issue 3) pp:
Publication Date(Web):January 26, 2012
DOI:10.1021/bm201693j
Reversibly shielded DNA polyplexes based on bioreducible poly(dimethylaminoethyl methacrylate)-SS-poly(ethylene glycol)-SS-poly(dimethylaminoethyl methacrylate) (PDMAEMA-SS-PEG-SS-PDMAEMA) triblock copolymers were designed, prepared and investigated for in vitro gene transfection. Two PDMAEMA-SS-PEG-SS-PDMAEMA copolymers with controlled compositions, 6.6–6–6.6 and 13–6–13 kDa, were obtained by reversible addition–fragmentation chain transfer (RAFT) polymerization of dimethylaminoethyl methacrylate (DMAEMA) using CPADN-SS-PEG-SS-CPADN (CPADN: 4-cyanopentanoic acid dithionaphthalenoate; PEG: 6 kDa) as a macro-RAFT agent. Like their nonreducible PDMAEMA-PEG-PDMAEMA analogues, PDMAEMA-SS-PEG-SS-PDMAEMA triblock copolymers could effectively condense DNA into small particles with average diameters less than 120 nm and close to neutral zeta potentials (0 ∼ +6 mV) at and above an N/P ratio of 3/1. The resulting polyplexes showed excellent colloidal stability against 150 mM NaCl, which contrasts with polyplexes of 20 kDa PDMAEMA homopolymer. In the presence of 10 mM dithiothreitol (DTT), however, polyplexes of PDMAEMA-SS-PEG-SS-PDMAEMA were rapidly deshielded and unpacked, as revealed by significant increase of positive surface charges as well as increase of particle sizes to over 1000 nm. Release of DNA in response to 10 mM DTT was further confirmed by gel retardation assays. These polyplexes, either stably or reversibly shielded, revealed a low cytotoxicity (over 80% cell viability) at and below an N/P ratio of 12/1. Notably, in vitro transfection studies showed that reversibly shielded polyplexes afforded up to 28 times higher transfection efficacy as compared to stably shielded control under otherwise the same conditions. Confocal laser scanning microscope (CLSM) studies revealed that reversibly shielded polyplexes efficiently delivered and released pDNA into the perinuclei region as well as nuclei of COS-7 cells. Hence, reduction-sensitive reversibly shielded DNA polyplexes based on PDMAEMA-SS-PEG-SS-PDMAEMA are highly promising for nonviral gene transfection.
Co-reporter:Meng Zheng, Zhihong Zhong, Lei Zhou, Fenghua Meng, Rui Peng, and Zhiyuan Zhong
Biomacromolecules 2012 Volume 13(Issue 3) pp:
Publication Date(Web):February 17, 2012
DOI:10.1021/bm2017965
Poly(ethylene oxide) grafted with 1.8 kDa branched polyethylenimine (PEO-g-PEI) copolymers with varying compositions, that is, PEO(13k)-g-10PEI, PEO(24k)-g-10PEI, and PEO(13k)-g-22PEI, were prepared and investigated for in vitro nonviral gene transfer. Gel electrophoresis assays showed that PEO(13k)-g-10PEI, PEO(24k)-g-10PEI, and PEO(13k)-g-22PEI could completely inhibit DNA migration at an N/P ratio of 4/1, 4/1, and 3/1, respectively. Dynamic light scattering (DLS) and zeta potential measurements revealed that all three graft copolymers were able to effectively condense DNA into small-sized (80–245 nm) particles with moderate positive surface charges (+7.2 ∼ +24.1 mV) at N/P ratios ranging from 5/1 to 40/1. The polyplex sizes and zeta-potentials intimately depended on PEO molecular weights and PEI graft densities. Notably, unlike 25 kDa PEI control, PEO-g-PEI polyplexes were stable against aggregation under physiological salt as well as 20% serum conditions due to the shielding effect of PEO. MTT assays in 293T cells demonstrated that PEO-g-PEI polyplexes had decreased cytotoxicity with increasing PEO molecular weights and decreasing PEI graft densities, wherein low cytotoxicities (cell viability >80%) were observed for polyplexes of PEO(13k)-g-22PEI, PEO(13k)-g-10PEI, and PEO(24k)-g-10PEI up to an N/P ratio of 20/1, 30/1, and 40/1, respectively. Interestingly, in vitro transfection results showed that PEO(13k)-g-10PEI polyplexes have the best transfection activity. For example, PEO(13k)-g-10PEI polyplexes formed at an N/P ratio of 20/1, which were essentially nontoxic (100% cell viability), displayed over 3- and 4-fold higher transfection efficiencies in 293T cells than 25 kDa PEI standard under serum-free and 10% serum conditions, respectively. Confocal laser scanning microscopy (CLSM) studies using Cy5-labeled DNA confirmed that these PEO-g-PEI copolymers could efficiently deliver DNA into the perinuclei region as well as into nuclei of 293T cells at an N/P ratio of 20/1 following 4 h transfection under 10% serum conditions. PEO-g-PEI polyplexes with superior colloidal stability, low cytotoxicity, and efficient transfection under serum conditions are highly promising for safe and efficient in vitro as well as in vivo gene transfection applications.
Co-reporter:Rongran Wei, Liang Cheng, Meng Zheng, Ru Cheng, Fenghua Meng, Chao Deng, and Zhiyuan Zhong
Biomacromolecules 2012 Volume 13(Issue 8) pp:
Publication Date(Web):June 29, 2012
DOI:10.1021/bm3006819
Reduction-sensitive reversibly core-cross-linked micelles were developed based on poly(ethylene glycol)-b-poly(N-2-hydroxypropyl methacrylamide)–lipoic acid (PEG-b-PHPMA-LA) conjugates and investigated for triggered doxorubicin (DOX) release. Water-soluble PEG-b-PHPMA block copolymers were obtained with Mn,PEG of 5.0 kg/mol and Mn,HPMA varying from 1.7 and 4.1 to 7.0 kg/mol by reversible addition–fragmentation chain transfer (RAFT) polymerization. The esterification of the hydroxyl groups in the PEG-b-PHPMA copolymers with lipoic acid (LA) gave amphiphilic PEG-b-PHPMA-LA conjugates with degrees of substitution (DS) of 71–86%, which formed monodispersed micelles with average sizes ranging from 85.3 to 142.5 nm, depending on PHPMA molecular weights, in phosphate buffer (PB, 10 mM, pH 7.4). These micelles were readily cross-linked with a catalytic amount of dithiothreitol (DTT). Notably, PEG-b-PHPMA(7.0k)-LA micelles displayed superior DOX loading content (21.3 wt %) and loading efficiency (90%). The in vitro release studies showed that only about 23.0% of DOX was released in 12 h from cross-linked micelles at 37 °C at a low micelle concentration of 40 μg/mL, whereas about 87.0% of DOX was released in the presence of 10 mM DTT under otherwise the same conditions. MTT assays showed that DOX-loaded core-cross-linked PEG-b-PHPMA-LA micelles exhibited high antitumor activity in HeLa and HepG2 cells with low IC50 (half inhibitory concentration) of 6.7 and 12.8 μg DOX equiv/mL, respectively, following 48 h incubation, while blank micelles were practically nontoxic up to a tested concentration of 1.0 mg/mL. Confocal laser scanning microscope (CLSM) studies showed that DOX-loaded core-cross-linked micelles released DOX into the cell nuclei of HeLa cells in 12 h. These reduction-sensitive disassemblable core-cross-linked micelles with excellent biocompatibility, superior drug loading, high extracellular stability, and triggered intracellular drug release are promising for tumor-targeted anticancer drug delivery.
Co-reporter:Yinfeng Du, Wei Chen, Meng Zheng, Fenghua Meng, Zhiyuan Zhong
Biomaterials 2012 33(29) pp: 7291-7299
Publication Date(Web):
DOI:10.1016/j.biomaterials.2012.06.034
Co-reporter:Jinchao Zhang, Liangliang Wu, Fenghua Meng, Zhongjuan Wang, Chao Deng, Haiyan Liu, and Zhiyuan Zhong
Langmuir 2012 Volume 28(Issue 4) pp:2056-2065
Publication Date(Web):December 21, 2011
DOI:10.1021/la203843m
pH and reduction dual-bioresponsive nanosized polymersomes based on poly(ethylene glycol)-SS-poly(2-(diethyl amino)ethyl methacrylate) (PEG-SS-PDEA) diblock copolymers were developed for efficient encapsulation and triggered intracellular release of proteins. PEG-SS-PDEA copolymers with PDEA-block molecular weights ranging from 4.7, 6.8, to 9.2 kg/mol were synthesized in a controlled manner via reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-(diethyl amino)ethyl methacrylate (DEAEMA) using PEG-SS-CPADN (CPADN = 4-cyanopentanoic acid dithionaphthalenoate; Mn PEG = 1.9 kg/mol) as a macro-RAFT agent. These copolymers existed as unimers in water at mildly acidic pH (<7.2) conditions, but readily formed monodisperse nanosized polymersomes (54.5–66.8 nm) when adjusting solution pH to 7.4. These polymersomes were highly sensitive to intracellular pH and reductive environments, which resulted in fast dissociation and aggregation of polymersomes, respectively. Notably, both fluorescein isothiocyanate (FITC)-labeled bovine serum albumin (FITC-BSA) and cytochrome C (FITC-CC) proteins could facilely be encapsulated into polymersomes with excellent protein-loading efficiencies, likely as a result of electrostatic interactions between proteins and PDEA. The in vitro release studies showed that protein release was minimal (<20% in 8 h) at pH 7.4 and 37 °C. The release of proteins was significantly enhanced at pH 6.0 due to collapse of polymersomes. Notably, the fastest protein release was observed under intracellular-mimicking reductive environments (10 mM dithiothreitol, pH 7.4). MTT assays in RAW 264.7 and MCF-7 cells indicated that PEG-SS-PDEA (9.2 k) polymersomes had low cytotoxicity up to a polymer concentration of 300 μg/mL. Confocal laser scanning microscope (CLSM) observations revealed that FITC-CC-loaded PEG-SS-PDEA (9.2 k) polymersomes efficiently delivered and released proteins into MCF-7 cells following 6 h of incubation. Importantly, flow cytometry assays showed that CC-loaded PEG-SS-PDEA (9.2 k) polymersomes induced markedly enhanced apoptosis of MCF-7 cells as compared to free CC and CC-loaded PEG–PDEA (8.9 k) polymersomes (reduction-insensitive control). These dual-bioresponsive polymersomes have appeared to be highly promising for intracellular delivery of protein drugs.
Co-reporter:Meng Zheng;Chunmei Yang;Fenghua Meng;Rui Peng
Macromolecular Research 2012 Volume 20( Issue 3) pp:327-334
Publication Date(Web):2012 March
DOI:10.1007/s13233-012-0063-9
Co-reporter:Juan Xiong, Fenghua Meng, Chao Wang, Ru Cheng, Zhuang Liu and Zhiyuan Zhong
Journal of Materials Chemistry A 2011 vol. 21(Issue 15) pp:5786-5794
Publication Date(Web):07 Mar 2011
DOI:10.1039/C0JM04410E
The poor stability of micellar drug delivery systems in vivo due to large volume dilution and interactions with blood pool often leads to premature drug release with low targetability and therapeutic efficacy. Here, we designed folate-conjugated interfacially crosslinked biodegradable micelles consisting of poly(ethylene glycol)-b-poly(acryloyl carbonate)-b-poly(D,L-lactide) (PEG-PAC-PLA) and folate-PEG-PLA (FA-PEG-PLA) block copolymers for receptor-mediated delivery of paclitaxel (PTX) into KB cells. Micelles with varying amounts of folate ligands were prepared at 0–20 wt.% of FA-PEG-PLA. The resulting micelles, either with or without PTX loading, were readily crosslinked by UV irradiation. The crosslinked micelles had much smaller sizes and better stability as compared to the non-crosslinked controls. Notably, these micelles achieved high drug loading efficiencies of 70–88% at an initial PTX loading content of 10 wt.%. The in vitro release studies revealed that crosslinked micelles exhibited significantly inhibited PTX release at low micelle concentrations. MTT assays in KB cells showed that the crosslinked micelles were non-toxic while the toxicity of PTX-loaded micelles, either crosslinked or non-crosslinked, increased with increasing folate contents. Remarkably, at 12 h incubation time folate-decorated PTX-loaded crosslinked micelles composed of 20 wt.% of FA-PEG-PLA displayed markedly higher toxicity to KB cells than free PTX (33% versus 50% cell viability), which is most likely due to their much more efficient cellular uptake through FA receptor-mediated endocytosis. Flow cytometry studies showed that folate-decorated FITC-labeled crosslinked micelles were much more efficiently taken up by KB cells than controls without folate ligands. These results indicate that ligand-conjugated interfacially crosslinked PEG-PLA micelles have great potential in targeted cancer therapy.
Co-reporter:Ru Cheng, Fenghua Meng, Shoubao Ma, Haifei Xu, Haiyan Liu, Xiabin Jing and Zhiyuan Zhong
Journal of Materials Chemistry A 2011 vol. 21(Issue 47) pp:19013-19020
Publication Date(Web):31 Oct 2011
DOI:10.1039/C1JM13536H
The study of biological functions of proteins in cells as well as therapeutic exploration of many protein drugs demands efficient and nontoxic intracellular protein delivery systems. Herein, reduction and temperature dual-responsive crosslinked polymersomes were developed for the facile encapsulation of various proteins under mild conditions as well as rapid release of proteins in cancer cells. Two thermo-sensitive triblock copolymers, PEG5k-PAA1.7k-PNIPAM22k and PEG5k-PAA0.7k-PNIPAM12k (denoted as polymer 1 and 2, respectively), were prepared by controlled reversible addition–fragmentation chain-transfer (RAFT) polymerization. Interestingly, polymers 1 and 2 exhibited lower critical solution temperatures (LCST) of 39 and 38 °C in PBS (pH 7.4, 20 mM, 150 mM NaCl) and 34 and 32 °C in MES (pH 5.5, 20 mM), respectively. Increasing the temperature of polymer solutions in MES to 40 °C yielded robust polymersomes with average diameters of ca. 150∼170 nm following crosslinking the PAA segment with cystamine (Cys) viacarbodiimide chemistry. These crosslinked polymersomes kept their structures in PBS at 37 °C but rapidly dissociated into unimers in response to 10 mM dithiothreitol (DTT). Remarkably, various proteins including bovine serum albumin (BSA), lysozyme (Lys), cytochrome C (CC), and ovalbumin (Ova) could be conveniently loaded into the polymersomes with markedly high protein loading efficiencies of 60∼100% at theoretical protein loading contents of 10∼50 wt%. The in vitro release studies using Cys-crosslinked polymersome 1 showed that release of BSA, Lys, and CC was minimal (ca. 20%) in 11 h in PBS at 37 °C, while fast protein release of over 70% was observed under an intracellular mimicking reductive environment. MTT assays revealed that these polymersomes were practically non-toxic to HeLa and MCF-7 cells up to a tested concentration of 200 μg mL−1. Confocal laser scanning microscope (CLSM) observations showed that FITC-CC loaded Cys-crosslinked polymersomes efficiently delivered and released FITC-CC into the cytosol of MCF-7 cells after 12 h incubation. In contrast, little FITC-CC fluorescence was observed in MCF-7 cells treated with free FITC-CC as well as FITC-CC loaded 1,4-butadiamine crosslinked polymersomes (reduction-insensitive control). Flow cytometry studies showed that CC loaded Cys-crosslinked polymersomes induced markedly enhanced apoptosis of MCF-7 cells as compared to free CC and the reduction-insensitive controls. These novel reduction and temperature dual-responsive crosslinked polymersomes have opened a new avenue to targeted intracellular protein delivery.
Co-reporter:Meng Zheng, Yinan Zhong, Fenghua Meng, Rui Peng, and Zhiyuan Zhong
Molecular Pharmaceutics 2011 Volume 8(Issue 6) pp:2434-2443
Publication Date(Web):September 17, 2011
DOI:10.1021/mp2003797
The clinical success of gene therapy intimately relies on the development of safe and efficient gene carrier systems. We found here that 1.8 kDa polyethylenimine (PEI) following hydrophobic modification with lipoic acid (LA) mediated nontoxic and highly potent in vitro gene transfection in both HeLa and 293T cells. 1.8 kDa PEI–LA conjugates were prepared with controlled degree of substitution (DS) by coupling LA to PEI using carbodiimide chemistry. Gel electrophoresis measurements showed that the DNA binding ability of 1.8 kDa PEI was impaired by lipoylation, in which an N/P ratio of 2/1 and 4–6/1 was required for 1.8 kDa PEI and 1.8 kDa PEI–LA conjugates, respectively, to completely inhibit DNA migration. Interestingly, dynamic light scattering measurements (DLS) revealed that PEI–LA conjugates condensed DNA into much smaller sizes (183–84 nm) than unmodified 1.8 kDa PEI (444–139 nm) at N/P ratios ranging from 20/1 to 60/1. These polyplexes revealed similar surface charges of ca. +22 to +30 mV. 1.8 kDa PEI–LA2 polyplexes formed at an N/P ratio of 10/1 were stable against exchange with 12-fold excess of negatively charged dextran sodium sulfate (DSS) relative to DNA phosphate groups while 1.8 kDa PEI controls dissociated at 6-fold excess of DSS, indicating that lipoylation of 1.8 kDa PEI resulted in stronger binding with DNA. Importantly, DNA was released from 1.8 kDa PEI–LA2 polyplexes upon addition of 10 mM dithiothreitol (DTT). Reduction-triggered unpacking of 1.8 kDa PEI–LA2 polyplexes was also confirmed by DLS. MTT assays demonstrated that all PEI–LA conjugates and polyplexes were essentially nontoxic to HeLa and 293T cells up to a tested concentration of 50 μg/mL and an N/P ratio of 80/1, respectively. The in vitro gene transfection studies in HeLa and 293T cells showed that lipoylation of 1.8 kDa PEI markedly boosted its transfection activity. For example, 1.8 kDa PEI–LA2 polyplexes displayed 400-fold and 500-fold higher levels of gene expression than unmodified 1.8 kDa PEI controls, which were ca. 2-fold and 3-fold higher than 25 kDa PEI controls, in serum-free and 10% serum media, respectively. The transfection efficiency decreased with increasing DS, following an order of 1.8 kDa PEI–LA2 > 1.8 kDa PEI–LA4 > 1.8 kDa PEI–LA6 ≫ 1.8 kDa PEI. Confocal laser scanning microscopy (CLSM) studies corroborated that 1.8 kDa PEI–LA2 delivered and released DNA into the nuclei of HeLa cells more efficiently than 25 kDa PEI. These nontoxic 1.8 kDa PEI–LA conjugates form a superb basis for the development of targeting, biocompatible and highly efficient carriers of gene delivery.Keywords: gene delivery; hydrophobic modification; plasmid DNA; polyethylenimine; polyplexes; reduction-sensitive;
Co-reporter:Yuexin Yu;Chao Deng;Fenghua Meng;Qin Shi;Jan Feijen
Journal of Biomedical Materials Research Part A 2011 Volume 99A( Issue 2) pp:316-326
Publication Date(Web):
DOI:10.1002/jbm.a.33199
Abstract
Novel injectable biodegradable glycol chitosan hydrogels were developed based on thiolated glycol chitosan (GC-SH) and water soluble oligo(acryloyl carbonate)-b- poly(ethylene glycol)-b-oligo(acryloyl carbonate) (OAC-PEG-OAC) triblock copolymers via Michael-type addition reaction. The rheology measurements showed that robust hydrogels were formed rapidly upon mixing aqueous solutions of GC-SH and OAC-PEG-OAC at remarkably low total polymer concentrations of 1.5–4.5 wt % under physiological conditions. The gelation times (varying from 10 s to 17 min) and storage moduli (100 to 4300 Pa) of hydrogels could be controlled by degrees of substitution (DS) of GC-SH, solution pH, and polymer concentration. These glycol chitosan hydrogels had microporous structures, low swelling and slow hydrolytic degradation (stable for over 6 months) under physiological conditions. Notably, these hydrogels were prone to enzymatic degradation with lysozyme. The multiple acryloyl functional groups of OAC-PEG-OAC allowed facile conjugation with thiol-containing biomolecules prior to gelation endowing hydrogels with specific bioactivity. The preliminary cell culture studies revealed that these glycol chitosan hydrogels were cell non-adhesive while Gly-Arg-Gly-Asp-Cys (GRGDC) peptide modified hydrogels could well support adhesion and growth of both MG63 osteoblast and L929 fibroblast cells. These rapidly in situ forming enzymatically biodegradable hybrid hydrogels have great potentials in the development of injectable cell-specific bioactive extracellular matrices for tissue engineering. © 2011 Wiley Periodicals, Inc. J Biomed Mater Res Part A:, 2011.
Co-reporter:Jie Chen;Wei Chen;Chao Deng;Fenghua Meng
Journal of Polymer Science Part A: Polymer Chemistry 2011 Volume 49( Issue 20) pp:
Publication Date(Web):
DOI:10.1002/pola.24878
Abstract
Risedronate-anchored hydroxyapatite (HA-RIS) nanocrystals were prepared with 4.1 wt % RIS and used for controlled surface-initiated ring-opening polymerization (ROP) of L-lactide (L-LA). The strong adsorption of RIS to HA surface not only led to enhanced dispersion of HA nanocrystals in water as well as in organic solvents but also provided alkanol groups as active initiating species for ROP of L-LA. HA-RIS was characterized by thermogravimetric analysis, dynamic light scattering, 1H NMR, Fourier transform infrared spectrometer, and X-ray diffraction. The graft polymerization of L-LA onto HA-RIS took place smoothly in the presence of stannous octoate in toluene at 120 °C, resulting in HA/poly(L-LA) nanocomposites with high yields of 85–90% and high poly(L-LA) contents of up to 97.5 wt %. Notably, differential scanning calorimetry measurements revealed that the poly(L-LA) in HA/poly(L-LA) nanocomposites exhibited considerably higher melting temperatures (Tm = 173.3−178.1 °C) and higher degrees of crystallinity (Xc = 41.0−43.1%) as compared to poly(L-LA) homopolymer (Tm = 168.5 °C, Xc =25.7%). In addition, our initial results showed that these HA/poly(L-LA) nanocomposites could readily be electrospun into porous matrices. This study presented a novel and controlled synthetic strategy to HA/RIS/poly(L-LA) nanocomposites that are promising for orthopedic applications as well as for bone tissue engineering. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011
Co-reporter:Sibin Luo;Ru Cheng;Fenghua Meng;Tae Gwan Park
Journal of Polymer Science Part A: Polymer Chemistry 2011 Volume 49( Issue 15) pp:3366-3373
Publication Date(Web):
DOI:10.1002/pola.24773
Abstract
Water-soluble cationic polymers, poly(histamine acrylamide)s (PHAs), with superior buffer capacity at the endosomal pH range were designed, prepared, and investigated for non-viral gene transfection. PHAs were obtained with molecular weights ranging from 9.2 to 28.7 kDa through controlled radical polymerization of histamine acrylamide (HA). Acid–base titration results displayed that all PHA polymers had a remarkably high buffer capacity of about 70% at pH 5.1–7.2. 12.7–28.7 kDa PHAs were able to effectively condense DNA into nano-sized (<220 nm) polyplexes with moderate positive surface charges (+13–+19 mV) at N/P ratios ≥10/1. CCK assays indicated that polyplexes of 12.7 and 17.5 kDa PHAs were non-toxic to COS-7 cells up to a tested N/P ratio of 20/1. Interestingly, the in vitro transfection using pCMV-Luc and pEGFP-C1 plasmid DNA as reporter genes showed that polyplexes of 12.7 kDa PHA formed at an N/P ratio of 20/1 mediated efficient transfection in COS-7 cells under 10% serum conditions, with transfection efficiencies comparable to that of 25 kDa polyethylenimine control. Their versatile design of structures, controlled synthesis, low cytotoxicity, and high transfection activity render PHA-based cationic polymers particularly interesting for the development of safe and efficient non-viral gene delivery systems. © 2011 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem, 2011.
Co-reporter:Rong Wang, Wei Chen, Fenghua Meng, Ru Cheng, Chao Deng, Jan Feijen, and Zhiyuan Zhong
Macromolecules 2011 Volume 44(Issue 15) pp:6009-6016
Publication Date(Web):July 14, 2011
DOI:10.1021/ma200824k
The ever-growing biomedical technology such as tissue engineering, regenerative medicine, and controlled drug release intimately relies on the development of advanced functional biomaterials. Here, we report on versatile and robust synthesis of novel vinyl sulfone (VS)-functionalized biodegradable polymers that offer unprecedented access to advanced functional biodegradable polymers and coatings through selective Michael-type conjugate reaction with thiol-containing molecules. VS-functionalized biodegradable polymers including poly(ε-caprolactone) (PCL), poly(l-lactide) (PLA), and poly(trimethylene carbonate) (PTMC) were conveniently prepared with controlled molecular weights and functionalities through ring-opening copolymerization of ε-caprolactone (ε-CL), l-lactide (LA), or trimethylene carbonate (TMC) with a new cyclic carbonate monomer, vinyl sulfone carbonate (VSC), in toluene at 110 °C using isopropanol as an initiator and stannous octoate as a catalyst. Interestingly, these VS-functionalized biodegradable polymers allowed quantitative modification, without aid of a catalyst, with various thiol-containing molecules including 2-mercaptoethanol, cystamine, cysteine, GRGDC peptide, and thiolated poly(ethylene glycol) (PEG-SH) at a ligand-SH/VS molar ratio of 2/1 in DMF at room temperature, confirming that the Michael-type conjugate addition to VS is highly selective and tolerant to most other functional groups including hydroxyl, carboxyl, and amine. Remarkably, results of contact angle measurements, X-ray photoelectron spectroscopy (XPS), and fluorescence studies showed that biodegradable coatings based on these VS-functionalized polymers allowed direct, efficient, and clean (without catalyst and byproduct) surface functionalization with thiol-containing molecules in aqueous conditions, which is unprecedented and opens a new avenue to surface functionalization of medical implants as well as cell and tissue scaffolds. The preliminary cell culture studies using MG6 cells showed that unmodified VS-functionalized PCL films, similar to tissue culture plate, could well support cell attachment and growth, indicating that VS-functionalized PCL film is nontoxic and biocompatible. The surface of VS-functionalized PCL films could be elegantly engineered with thiolated nonfouling polymers (e.g., PEG and glycol chitosan) or cell adhesive motif (GRGDC peptide) to control cell attachment and growth. We are convinced that these vinyl sulfone-functionalized biodegradable polymers have a tremendous potential in biomedical engineering.
Co-reporter:Zhaozhong Liu, Meng Zheng, Fenghua Meng, Zhiyuan Zhong
Biomaterials 2011 32(34) pp: 9109-9119
Publication Date(Web):
DOI:10.1016/j.biomaterials.2011.08.017
Co-reporter:Huanli Sun, Fenghua Meng, Aylvin A. Dias, Marc Hendriks, Jan Feijen, and Zhiyuan Zhong
Biomacromolecules 2011 Volume 12(Issue 6) pp:
Publication Date(Web):April 7, 2011
DOI:10.1021/bm200043u
Currently, biomedical engineering is rapidly expanding, especially in the areas of drug delivery, gene transfer, tissue engineering, and regenerative medicine. A prerequisite for further development is the design and synthesis of novel multifunctional biomaterials that are biocompatible and biologically active, are biodegradable with a controlled degradation rate, and have tunable mechanical properties. In the past decades, different types of α-amino acid-containing degradable polymers have been actively developed with the aim to obtain biomimicking functional biomaterials. The use of α-amino acids as building units for degradable polymers may offer several advantages: (i) imparting chemical functionality, such as hydroxyl, amine, carboxyl, and thiol groups, which not only results in improved hydrophilicity and possible interactions with proteins and genes, but also facilitates further modification with bioactive molecules (e.g., drugs or biological cues); (ii) possibly improving materials biological properties, including cell–materials interactions (e.g., cell adhesion, migration) and degradability; (iii) enhancing thermal and mechanical properties; and (iv) providing metabolizable building units/blocks. In this paper, recent developments in the field of α-amino acid-containing degradable polymers are reviewed. First, synthetic approaches to prepare α-amino acid-containing degradable polymers will be discussed. Subsequently, the biomedical applications of these polymers in areas such as drug delivery, gene delivery and tissue engineering will be reviewed. Finally, the future perspectives of α-amino acid-containing degradable polymers will be evaluated.
Co-reporter:Lei Zhou, Ru Cheng, Huiquan Tao, Shoubao Ma, Weiwei Guo, Fenghua Meng, Haiyan Liu, Zhuang Liu, and Zhiyuan Zhong
Biomacromolecules 2011 Volume 12(Issue 5) pp:
Publication Date(Web):February 18, 2011
DOI:10.1021/bm101340u
Novel poly(ethylene oxide)-graft-doxorubicin (PEO-g-DOX) prodrugs with DOX covalently conjugated to PEO via a pH-sensitive hydrazone bond were developed. PEO-g-DOX conjugates could be readily prepared in the following steps: (i) anionic ring-opening copolymerization of ethylene oxide (EO) and allyl glycidyl ether (AGE) afforded functional PEO with controlled molecular weights, low polydispersities, and multiple pendant double bonds (PEO-g-allyl); (ii) conjugation of PEO-g-allyl with methyl mercaptoacetate, followed by treating with hydrazine hydrate, quantitatively transformed allyl into hydrazide groups (PEO-g-hydrazide); and (iii) DOX was covalently immobilized to PEO-g-hydrazide via acid-labile hydrazone bonds (PEO-g-DOX). Here on the basis of PEO-g-allyl4.4 (Mn GPC = 22 400, PDI = 1.19) and PEO-g-allyl7.1 (Mn GPC = 15 300, PDI = 1.16, the subscription refers to number of allyl groups per chain) two freely water-soluble PEO-g-DOX prodrugs with 2.9 and 3.6 DOX per molecule (denoted as PEO-g-DOX2.9 and PEO-g-DOX3.6, corresponding to drug loading content of 5.6 and 9.0 wt %, respectively) were obtained. The in vitro release studies confirmed much faster release of DOX at pH 5.0 and 6.0 than at pH 7.4. For example, approximately 16, 52, and 61% of drug were released in 22 h, and 23, 83, and 92% of drug were released in 120 h from PEO-g-DOX2.9 at pH 7.4, 6.0 and 5.0, respectively. Notably, confocal laser scanning microscope (CLSM) observations revealed that DOX was released and delivered into the nuclei of RAW 264.7 cells following 24 h of incubation. MTT assays demonstrated that PEO-g-DOX2.9 had pronounced cytotoxic effects to RAW 264.7, HeLa, and 4T1 breast tumor cells with IC50 values of about 26.5, 42.5, and 32.0 μg DOX equiv/mL, whereas the corresponding polymer carrier PEO-g-hydrazide4.4 was nontoxic. The In Vivo pharmacokinetics and biodistribution studies in mice showed that PEO-g-DOX2.9 prodrugs had significantly prolonged circulation time and enhanced drug accumulation in the tumor as compared with free DOX. We are convinced that endosomal pH-activatable PEO-g-DOX prodrugs have tremendous potential for targeted cancer therapy.
Co-reporter:Yuhe Wang, Meng Zheng, Fenghua Meng, Jing Zhang, Rui Peng, and Zhiyuan Zhong
Biomacromolecules 2011 Volume 12(Issue 4) pp:
Publication Date(Web):February 18, 2011
DOI:10.1021/bm101364f
Twenty-five kDa polyethylenimine (PEI) is one of the most efficient nonviral gene transfer agents currently applied as a golden standard for in vitro transfection. In this study, novel 25 kDa PEI derivatives with reductively cleavable cystamine periphery (PEI-Cys) were designed to reduce carrier-associated cytotoxicity and to enhance further the transfection activity. The Michael-type conjugate addition of 25 kDa PEI with N-tert-butoxycarbonyl-N′-acryloyl-cystamine (Ac-Cys-tBoc) and N-tert-butoxycarbonyl-N′-methacryloyl-cystamine (MAc-Cys-tBoc) followed by deprotection readily afforded PEI-Cys derivatives, denoted as PEI-(Cys)x(Ac) and PEI-(Cys)x(MAc), with degree of substitution (DS) ranging from 14 to 34 and 13 to 38, respectively. All PEI-Cys derivatives had higher buffer capacity than the parent 25 kDa PEI (21.2 to 23.1% versus 15.1%). Gel retardation and ethidium bromide exclusion assays showed that cystamine modification resulted in largely enhanced interactions with DNA. PEI-(Cys)x(Ac) could condense DNA into small-sized particles of 80−90 nm at and above an N/P ratio of 5/1, which were smaller than polyplexes of 25 kDa PEI (100−130 nm). In comparison, PEI-(Cys)x(MAc) condensed DNA into somewhat larger particles (100−180 nm at N/P ratios from 30/1 to 5/1). Gel retardation and dynamic light scattering (DLS) measurements showed that PEI-Cys polyplexes were quickly unpacked to release DNA in response to 10 mM dithiothreitol (DTT). These PEI-Cys derivatives revealed markedly decreased cytotoxicity as compared with 25 kDa PEI with IC50 values of >100 mg/L and 50−75 mg/L for HeLa and 293T cells, respectively (corresponding IC50 data of 25 kDa PEI are ca. 11 and 3 mg/L). The in vitro transfection experiments in HeLa and 293T cells using pGL3 as a reporter gene showed that gene transfection activity of PEI-Cys derivatives decreased with increasing DS and PEI-(Cys)x(MAc) exhibited higher transfection activity than PEI-(Cys)x(Ac) at similar DS. Notably, polyplexes of PEI-(Cys)14(Ac) and PEI-(Cys)13(MAc) showed significantly enhanced gene transfection efficiency (up to 4.1-fold) as compared with 25 kDa PEI formulation at an N/P ratio of 10/1 in both serum-free and 10% serum-containing conditions. The modification of PEI with reductively cleavable periphery appears to be a potential approach to develop safer and more efficient nonviral gene vectors.
Co-reporter:Fuxing Zhan, Wei Chen, Zhongjuan Wang, Wentao Lu, Ru Cheng, Chao Deng, Fenghua Meng, Haiyan Liu, and Zhiyuan Zhong
Biomacromolecules 2011 Volume 12(Issue 10) pp:
Publication Date(Web):September 9, 2011
DOI:10.1021/bm200876x
Endosomal pH-activatable doxorubicin (DOX) prodrug nanogels were designed, prepared, and investigated for triggered intracellular drug release in cancer cells. DOX prodrugs with drug grafting contents of 3.9, 5.7, and 11.7 wt % (denoted as prodrugs 1, 2, and 3, respectively) were conveniently obtained by sequential treatment of poly(ethylene glycol)-b-poly(2-hydroxyethyl methacrylate-co-ethyl glycinate methacrylamide) (PEG-b-P(HEMA-co-EGMA)) copolymers with hydrazine and doxorubicin hydrochloride. Notably, prodrugs 1, 2, and 3 formed monodispersed nanogels with average sizes of 114.4, 75.3, and 66.3 nm, respectively, in phosphate buffer (PB, 10 mM, pH 7.4). The in vitro release results showed that DOX was released rapidly and nearly quantitatively from DOX prodrug nanogels at endosomal pH and 37 °C in 48 h, whereas only a minor amount (ca. 20% or less) of drug was released at pH 7.4 under otherwise the same conditions. Confocal laser scanning microscope (CLSM) observations revealed that DOX prodrug nanogels delivered and released DOX into the cytosols as well as cell nuclei of RAW 264.7 cells following 24 h incubation. MTT assays demonstrated that prodrug 3 had pronounced cytotoxic effects to tumor cells following 72 h incubation with IC50 data determined to be 2.0 and 3.4 μg DOX equiv/mL for RAW 264.7 and MCF-7 tumor cells, respectively. The corresponding polymer carrier, PEG-b-P(HEMA-co-GMA-hydrazide), was shown to be nontoxic up to a tested concentration of 1.32 mg/mL. These endosomal pH-activatable DOX prodrug nanogels uniquely combining features of water-soluble macromolecular prodrugs and nanogels offer a promising platform for targeted cancer therapy.
Co-reporter:Rui Yang, Fenghua Meng, Shoubao Ma, Fushi Huang, Haiyan Liu, and Zhiyuan Zhong
Biomacromolecules 2011 Volume 12(Issue 8) pp:
Publication Date(Web):July 5, 2011
DOI:10.1021/bm2006856
The inferior in vivo stability of micellar drugs has been a prime challenge for their application in targeted drug delivery. Here we report on novel galactose-decorated covalently cross-linked biodegradable micelles based on photo-cross-linkable poly(ethylene glycol)-b-poly(acryloyl carbonate)-b-poly(ε-caprolactone) (PEG-PAC-PCL) and galactose-conjugated PEG-PCL (Gal-PEG-PCL) copolymers for enhanced hepatoma-targeting delivery of paclitaxel (PTX). The molecular weight of PEG in Gal-PEG-PCL was higher than that in PEG-PAC-PCL, thereby fully exposing Gal ligands at the micellar surface. These micelles, either with or without loading of PTX, were readily cross-linked by UV irradiation to afford micelles with small sizes (ca. 79–94 nm) and enhanced stability. The in vitro release studies confirmed that drug release from cross-linked micelles was significantly inhibited. Interestingly, MTT assays showed that Gal-decorated PTX-loaded cross-linked micelles retained a high antitumor activity in HepG2 cells, which was much more effective than PTX-loaded cross-linked micelles without Gal ligands and comparable to Gal-decorated PTX-loaded non-cross-linked micelles. Remarkably, the preliminary in vivo antitumor efficacy studies in SMMC-7721 tumor (human hepatoma)-bearing nude mice revealed that Gal-decorated PTX-loaded cross-linked micelles inhibited the growth of the human hepatoma more effectively than PTX-loaded cross-linked micelles as well as Gal-decorated PTX-loaded non-cross-linked micelles. These results indicate that Gal-decorated cross-linked PEG-PCL micelles have great potential in liver tumor-targeted chemotherapy.
Co-reporter:Fenghua Meng and Zhiyuan Zhong
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 13) pp:1533-1539
Publication Date(Web):June 7, 2011
DOI:10.1021/jz200007h
Polymersomes provide a highly promising platform for mimicking biological membranes as well as for controlled delivery of various pharmaceuticals and biopharmaceuticals. The recent advancement has made it possible to prepare small to giant polymersomes spanning from nano- to microscales, stimuli-sensitive polymersomes that swell or collapse in response to an external or internal stimulus, chimaeric polymersomes that contain distinct interior environments separated from the outside by an asymmetric membrane, porous polymersomes with tailored permeability, biomimetic and targeting polymersomes that selectively deliver drugs, proteins, and/or imaging probes to the sites of action. In this Perspective, we will give a brief introduction to polymersomes, highlight recent design and preparation of several sophisticated polymersomes, and discuss future challenges.
Co-reporter:Guijing Liu, Shoubao Ma, Shaoke Li, Ru Cheng, Fenghua Meng, Haiyan Liu, Zhiyuan Zhong
Biomaterials 2010 31(29) pp: 7575-7585
Publication Date(Web):
DOI:10.1016/j.biomaterials.2010.06.021
Co-reporter:Caihong Zhu, Sooyeon Jung, Sibin Luo, Fenghua Meng, Xiulin Zhu, Tae Gwan Park, Zhiyuan Zhong
Biomaterials 2010 31(8) pp: 2408-2416
Publication Date(Web):
DOI:10.1016/j.biomaterials.2009.11.077
Co-reporter:Huanli Sun, Bingnan Guo, Xiaoqing Li, Ru Cheng, Fenghua Meng, Haiyan Liu and Zhiyuan Zhong
Biomacromolecules 2010 Volume 11(Issue 4) pp:
Publication Date(Web):March 8, 2010
DOI:10.1021/bm1001069
Reduction-responsive biodegradable micelles were developed from disulfide-linked dextran-b-poly(ε-caprolactone) diblock copolymer (Dex-SS-PCL) and applied for triggered release of doxorubicin (DOX) in vitro and inside cells. Dex-SS-PCL was readily synthesized by thiol-disulfide exchange reaction between dextran orthopyridyl disulfide (Dex-SS-py, 6000 Da) and mercapto PCL (PCL-SH, 3100 Da). Dynamic light scattering (DLS) measurements showed that Dex-SS-PCL yielded micelles with an average size of about 60 nm and a low polydispersity index (PDI 0.1−0.2) in PB (50 mM, pH 7.4). Interestingly, these micelles formed large aggregates rapidly in response to 10 mM dithiothreitol (DTT), most likely due to shedding of the dextran shells through reductive cleavage of the intermediate disulfide bonds. DOX could be efficiently loaded into the micelles with a drug loading efficiency of about 70%. Notably, the in vitro release studies revealed that Dex-SS-PCL micelles released DOX quantitatively in 10 h under a reductive environment, mimicking that of the intracellular compartments such as cytosol and the cell nucleus, whereas only about 27% DOX was released from reduction insensitive Dex-PCL micelles in 11.5 h under otherwise the same conditions and about 20% DOX released from Dex-SS-PCL micelles in 20 h under the nonreductive conditions. The cell experiments using fluorescence microscopy and confocal laser scanning microscopy (CLSM) showed clearly that DOX was rapidly released to the cytoplasm as well as to the cell nucleus. MTT studies revealed a markedly enhanced drug efficacy of DOX-loaded Dex-SS-PCL micelles as compared to DOX-loaded reduction-insensitive Dex-PCL micelles. These reduction-responsive biodegradable micelles have appeared highly promising for the targeted intracellular delivery of hydrophobic chemotherapeutics in cancer therapy.
Co-reporter:Wei Chen, Huicui Yang, Rong Wang, Ru Cheng, Fenghua Meng, Wenxiang Wei and Zhiyuan Zhong
Macromolecules 2010 Volume 43(Issue 1) pp:201-207
Publication Date(Web):December 9, 2009
DOI:10.1021/ma901897y
Various functional biodegradable polymers were readily prepared based on novel cyclic carbonate monomers, acryloyl carbonate (AC) and methacryloyl carbonate (MAC), by combining ring-opening polymerization (ROP) and Michael-type conjugate addition. AC and MAC monomers were synthesized in four straightforward steps from 1,1,1-tris(hydroxymethyl)ethane with good overall yields (ca. 40%). AC and MAC were able to copolymerize with ε-caprolactone (ε-CL) and d,l-lactide (LA) in toluene at 110 °C using stannous octoate as a catalyst, yielding biodegradable copolymers with controlled (meth)acryloyl functional groups and molecular weights. The acryloyl groups were amenable to the Michael-type conjugate addition with varying thiol-containing molecules such as 2-mercaptoethanol, 3-mercaptopropanoic acid, cysteamine, cysteine, and arginine-glycine-aspartic acid-cysteine (RGDC) peptide under mild conditions, to provide biodegradable materials with vastly different functionalities (e.g., hydroxyl, carboxyl, amine, amino acid, and peptides) and properties (e.g., hydrophilicity, cell adhesion). Notably, 100% functionalization was achieved with 2-mercaptoethanol, cysteamine and cysteine. Initial cell culture studies demonstrated enhanced cell adhesion and growth on films containing functional RGDC peptides as compared to those of the parent copolymer. Therefore, combination of ROP and Michael-type conjugate addition provides a versatile access to diverse types of functional biodegradable materials.
Co-reporter:Haifei Xu, Fenghua Meng and Zhiyuan Zhong
Journal of Materials Chemistry A 2009 vol. 19(Issue 24) pp:4183-4190
Publication Date(Web):11 May 2009
DOI:10.1039/B901141B
Water-soluble temperature responsive triblock copolymers, poly(ethylene oxide)-b-poly(acrylic acid)-b-poly(N-isopropylacrylamide) (PEO-PAA-PNIPAM), were prepared in one pot by sequential reversible addition–fragmentation chain-transfer (RAFT) polymerization using a PEO–trithiocarbonate (PEO–S-1-dodecyl-S-(R,R-dimethyl-R-aceticacid) trithiocarbonate) as a macro chain transfer agent. The block copolymers with MnPEO of 5 kDa, MnPAA of 0.35–1.45 kDa, and MnPNIPAM varying from 11–39 kDa were freely soluble in water as unimers at room temperature, but quickly self-assembled into nano-sized vesicles (about 220 nm) when raising the solution temperature to 37 °C. The vesicular structure was confirmed by confocal scanning laser microscope (CSLM) and static light scattering (SLS) measurements. The size and size distribution of the polymersomes depended on the solution concentration, the molecular weight of PNIPAM, the equilibrium time and shaking. Interestingly, thus-formed vesicles could be readily cross-linked at the interface using cystamineviacarbodiimide chemistry. The crosslinked polymersomes, while showing remarkable stability against dilution, organic solvent, high salt conditions and change of temperature in water, were otherwise rapidly dissociated under reductive conditions mimicking intracellular environment. Notably, FITC–dextran, used as a model protein, was shown to be encapsulated into the polymersomes with an unprecedently high loading efficiency (>85 wt%). The release studies showed that most FITC–dextran was retained within the polymersomes after lowering the temperature to 25 °C. However, in the presence of 10 mM dithiothreitol (DTT), fast release of FITC–dextran was achieved. These reversibly crosslinked temperature responsive nano-sized polymersomes are highly promising as smart carriers for triggered intracellular delivery of biopharmaceutics such as pDNA, siRNA, pharmaceutical proteins and peptides.
Co-reporter:Yanmin Xu;Fenghua Meng;Ru Cheng
Macromolecular Bioscience 2009 Volume 9( Issue 12) pp:1254-1261
Publication Date(Web):
DOI:10.1002/mabi.200900233
Co-reporter:Fenghua Meng, Zhiyuan Zhong and Jan Feijen
Biomacromolecules 2009 Volume 10(Issue 2) pp:
Publication Date(Web):January 5, 2009
DOI:10.1021/bm801127d
In the past decade, polymersomes (also referred to as polymeric vesicles) have attracted rapidly growing interest based on their intriguing aggregation phenomena, cell and virus-mimicking dimensions and functions, as well as tremendous potential applications in medicine, pharmacy, and biotechnology. Unlike liposomes self-assembled from low molecular weight lipids, polymersomes are in general prepared from macromolecular amphiphiles of various architectures including amphiphilic diblock, triblock, graft and dendritic copolymers. Polymersomes exhibit very unique features highlighted with high stability, tunable membrane properties, versatility, and capacity of transporting hydrophilic as well as hydrophobic species such as anticancer drugs, genes, proteins, and diagnostic probes. Recently, much effort has been directed to the development of intelligent polymersomes that respond to internal or external stimuli, in particular, pH, temperature, redox potential, light, magnetic field, and ultrasound, either reversibly or nonreversibly. Stimuli-sensitive polymersomes have emerged as novel programmable delivery systems in which the release of the encapsulated contents can be readily modulated by the stimulus. The stimuli-responsive release may result in significantly enhanced therapeutic efficacy and minimized possible side effects. It is also feasible to form and disassemble polymersomes in water simply by applying an appropriate stimulus. In this article, recent advances in stimuli-sensitive polymersomes have been reviewed, and perspectives on future developments have been discussed.
Co-reporter:Wei Chen, Fenghua Meng, Feng Li, Shun-Jun Ji and Zhiyuan Zhong
Biomacromolecules 2009 Volume 10(Issue 7) pp:
Publication Date(Web):May 26, 2009
DOI:10.1021/bm900074d
pH-responsive biodegradable micelles were prepared from block copolymers comprising of a novel acid-labile polycarbonate hydrophobe and poly(ethylene glycol) (PEG). Two new cyclic aliphatic carbonate monomers, mono-2,4,6-trimethoxybenzylidene-pentaerythritol carbonate (TMBPEC, 2a) and mono-4-methoxybenzylidene-pentaerythritol carbonate (MBPEC, 2b) were designed and successfully synthesized via a two-step procedure. The ring-opening polymerization of 2a or 2b in the presence of methoxy PEG in dichloromethane at 50 °C using zinc bis[bis(trimethylsilyl)amide] as a catalyst yielded the corresponding block copolymers PEG-PTMBPEC (3a) or PEG-PMBPEC (3b) with low polydispersities (PDI 1.03−1.04). The copolymerization of d,l-lactide (DLLA) and 2a under otherwise the same conditions could also proceed smoothly to afford PEG-P(TMBPEC-co-DLLA) (3c) block copolymer. These block copolymers readily formed micelles in water with sizes of about 120 nm as determined by dynamic light scattering (DLS). The hydrolysis of the acetals of the polycarbonate was investigated using UV/vis spectroscopy. The results showed that the acetals of micelles 3a, while stable at pH 7.4 are prone to rapid hydrolysis at mildly acidic pH of 4.0 and 5.0, with a half-life of 1 and 6.5 h, respectively. The acetal hydrolysis resulted in significant swelling of micelles, as a result of change of hydrophobic polycarbonate to hydrophilic polycarbonate. In comparison, the acetals of PMBPEC of micelles 3b displayed obviously slower hydrolysis at the same pH. Both paclitaxel and doxorubicin could be efficiently encapsulated into micelles 3a achieving high drug loading content (13.0 and 11.7 wt %, respectively). The in vitro release studies showed clearly a pH dependent release behavior, that is, significantly faster drug release at mildly acidic pH of 4.0 and 5.0 compared to physiological pH. These pH-responsive biodegradable micelles are promising as smart nanovehicles for targeted delivery of anticancer drugs.
Co-reporter:Yu-Ling Li;Li Zhu;Zhaozhong Liu;Ru Cheng Dr.;Fenghua Meng Dr.;Jing-Hao Cui Dr.;Shun-Jun Ji Dr. Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 52) pp:9914-9918
Publication Date(Web):
DOI:10.1002/anie.200904260
Co-reporter:Christine Hiemstra, Zhiyuan Zhong, Mies J. van Steenbergen, Wim E. Hennink, Jan Feijen
Journal of Controlled Release 2007 Volume 122(Issue 1) pp:71-78
Publication Date(Web):11 September 2007
DOI:10.1016/j.jconrel.2007.06.011
Our previous studies showed that degradable dextran hydrogels are rapidly formed in situ upon mixing aqueous solutions of dextran vinyl sulfone (dex-VS) conjugates and tetrafunctional mercapto poly(ethylene glycol) (PEG-4-SH) by Michael addition. The hydrogel degradation time and storage modulus could be controlled by the degree of vinyl sulfone substitution (DS) and dextran molecular weight. The degradation time could further be adjusted by the spacer between the thioether and the ester bond of the dex-VS conjugates (ethyl vs. propyl, denoted as dex-Et-VS and dex-Pr-VS, respectively). In this paper, the release of three model proteins, i.e. immunoglobulin G (dh = 10.7 nm, IgG), bovine serum albumin (BSA, dh = 7.2 nm) and lysozyme (dh = 4.1 nm), as well as basic fibroblast growth factor (bFGF) from these in situ forming dextran hydrogels is studied. Proteins could be easily loaded into the hydrogels by mixing protein containing solutions of dex-VS and PEG-4-SH. The release of IgG from dex-Et-VS hydrogels followed biphasic release kinetics, with a slow, close to first order release for the first 9 days followed by an accelerated release and over 80% of IgG was released in 12 to 25 days. Interestingly, the release of IgG from dex-Pr-VS hydrogels followed close to zero order kinetics, wherein approximately 95% was released in 21 days. The release of BSA from dex-Pr-VS hydrogels followed biphasic kinetics, with almost first order release followed by close to zero order release. Approximately 75% of the entrapped BSA could be released from dex-Pr-VS hydrogels in 16 days. Dex-Pr-VS hydrogels released 40% of lysozyme in 14 days, with full preservation of the enzymatic activity of the released lysozyme, as determined by bacteria lysis experiments. The release of basic fibroblast growth factor (bFGF) from dex-Pr-VS hydrogels showed first order kinetics, with quantitative release in 28 days. These results show that the in situ forming degradable dextran hydrogels can be used for the controlled release of proteins.
Co-reporter:Chao Lin, Zhiyuan Zhong, Martin C. Lok, Xulin Jiang, Wim E. Hennink, Jan Feijen, Johan F.J. Engbersen
Journal of Controlled Release 2007 Volume 123(Issue 1) pp:67-75
Publication Date(Web):18 October 2007
DOI:10.1016/j.jconrel.2007.07.006
Poly(amido amine) (SS-PAA) random and block copolymers having bioreducible disulfide bonds in the main chain and amino groups with distinctly different basicity in the side chain were designed and synthesized by Michael addition polymerization between N, N′-cystaminebisacrylamide (CBA) and two amine monomers, i.e., histamine (HIS) and 3-(dimethylamino)-1-propylamine (DMPA). Copolymers containing variable HIS/DMPA ratios show higher ability to bind DNA than p(CBA-HIS) homopolymer and condense DNA into the polyplexes with particle sizes (< 150 nm) that are smaller than polyplexes of p(CBA-HIS) (∼ 220 nm). The buffer capacities of the copolymers increase with increasing HIS/DMPA ratio. These copolymers are able to transfect COS-7 cells in vitro with efficiencies that increase with increasing HIS/DMPA ratio. The random and block copolymers at a HIS/DMPA ratio of 70/30 combines optimal DNA condensation capability and buffer capacity, thereby inducing higher transfection efficiency in the absence and presence of serum as compared to p(CBA-HIS) homopolymer. Moreover, random and block copolymers show a similar transfection capacity, but both have higher capacity than the physical mixtures of p(CBA-HIS) and p(CBA-DMPA) homopolymers. XTT assay reveals that the polyplexes of the SS-PAA copolymers have essentially low cytotoxicity when the highest transfection activity is observed.
Co-reporter:H. L. Fairchild
Science 1918 Vol 47(1225) pp:615-617
Publication Date(Web):21 Jun 1918
DOI:10.1126/science.47.1225.615
Co-reporter:Yaqin Zhu, Xiuxiu Wang, Jian Zhang, Fenghua Meng, Chao Deng, Ru Cheng, Jan Feijen, Zhiyuan Zhong
Journal of Controlled Release (28 March 2017) Volume 250() pp:
Publication Date(Web):28 March 2017
DOI:10.1016/j.jconrel.2017.02.002
Slow drug release at the tumor tissue and poor tumor penetration are two big challenges for the successful application of nanosystems in tumor therapy. Here, we report that a high concentration of the natural reducing agent vitamin C (VC) triggers rapid extracellular PTX release from PTX-loaded shell-sheddable PEG-SS-PCL micelles (SSM) in tumors in vivo. An in vivo tolerance study showed that VC at a blood concentration of 40 mM had little toxicity to nude mice. Notably, SSM rapidly disassembled and released the payloads (Cy5 or PTX) in response to 40 mM VC. In vivo near-infrared imaging of tumor-bearing mice showed that with post-injection of VC to establish a blood concentration of 40 mM, Cy5 was quickly released from the micelles and diffused deep into the tumor tissue. Biodistribution studies revealed that 6 h after the injection of PTX-loaded micelles the highest tumor accumulation was reached, which was set as the injection time for VC. The antitumor efficacy of a combination therapy of PTX-loaded micelles and VC was evaluated in both MCF-7 and U87MG tumor models. In both tumor models, single injections of VC didn't show any antitumor effect, while sequential administration of PTX-loaded SSM and VC exhibited significantly higher tumor inhibition effects and better survival rates as compared to single treatment with PTX-loaded micelles, demonstrating that exogenous administration of VC effectively triggered the release of PTX from SSM in vivo. The combination of reduction-sensitive nanomedicines with exogenous VC appears a promising approach to achieve potent treatment of malignant tumors.Illustration of VC-triggered micelle destabilization and drug release. (A) VC triggers destabilization and PTX release from PTX-loaded SSM by cleaving the interconnecting disulfide bonds; (B) post-injection of exogenous VC after tumor accumulation of SSM in vivo leads to micelle disassembly, rapid extracellular PTX release, and deep tumor penetration.
Co-reporter:Jiaolong Lv, Huanli Sun, Yan Zou, Fenghua Meng, Aylvin A. Dias, Marc Hendriks, Jan Feijen and Zhiyuan Zhong
Biomaterials Science (2013-Present) 2015 - vol. 3(Issue 7) pp:NaN1146-1146
Publication Date(Web):2015/04/08
DOI:10.1039/C4BM00436A
Novel reductively degradable α-amino acid-based poly(ester amide)-graft-galactose (SSPEA-Gal) copolymers were designed and developed to form smart nano-vehicles for active hepatoma-targeting doxorubicin (DOX) delivery. SSPEA-Gal copolymers were readily synthesized via solution polycondensation reaction of di-p-toluenesulfonic acid salts of bis-L-phenylalanine 2,2-thiodiethanol diester and bis-vinyl sulfone functionalized cysteine hexanediol diester with dinitrophenyl ester of adipic acid, followed by conjugating with thiol-functionalized galactose (Gal-SH) via the Michael addition reaction. SSPEA-Gal formed unimodal nanoparticles (PDI = 0.10 − 0.12) in water, in which average particle sizes decreased from 138 to 91 nm with increasing Gal contents from 31.6 wt% to 42.5 wt%. Notably, in vitro drug release studies showed that over 80% DOX was released from SSPEA-Gal nanoparticles within 12 h under an intracellular mimicking reductive conditions, while low DOX release (<20%) was observed for reduction-insensitive PEA-Gal nanoparticles under otherwise the same conditions and SSPEA-Gal nanoparticles under non-reductive conditions. Notably, SSPEA-Gal nanoparticles exhibited high specificity to asialoglycoprotein receptor (ASGP-R)-overexpressing HepG2 cells. MTT assays using HepG2 cells showed that DOX-loaded SSPEA-Gal had a low half maximal inhibitory concentration (IC50) of 1.37 μg mL−1, approaching that of free DOX. Flow cytometry and confocal laser scanning microscopy studies confirmed the efficient uptake of DOX-loaded SSPEA-Gal nanoparticles by HepG2 cells as well as fast intracellular DOX release. Importantly, SSPEA-Gal and PEA-Gal nanoparticles were non-cytotoxic to HepG2 and MCF-7 cells up to a tested concentration of 1.0 mg mL−1. These tumor-targeting and reduction-responsive degradable nanoparticles have appeared as an interesting multi-functional platform for advanced drug delivery.
Co-reporter:Wei Chen, Fenghua Meng, Ru Cheng, Chao Deng, Jan Feijen and Zhiyuan Zhong
Journal of Materials Chemistry A 2015 - vol. 3(Issue 11) pp:NaN2317-2317
Publication Date(Web):2015/01/27
DOI:10.1039/C4TB01962H
Glycopolymer-b-poly(ε-caprolactone) (GP-PCL) block copolymer micelles (‘glycomicelles’) with tailored lactose functionalities were developed and investigated for hepatoma-targeted doxorubicin (DOX) delivery. Amphiphilic GP-PCL copolymers were readily prepared with controlled lactobionic acid (LBA) functionalities of 20%, 40%, 80%, and 100% (denoted as GP20-PCL, GP40-PCL, GP80-PCL, and GP100-PCL, respectively) through post-polymerization modification of the poly(acryloyl cyclic carbonate)-b-poly(ε-caprolactone) (PAC-b-PCL, 11.6–6.4 kg mol−1) block copolymer with thiolated LBA (LBA-SH) and 2-(2-methoxyethoxy)ethanethiol ((EO)2-SH) via the Michael-type addition reaction. These self-assembled glycomicelles had mean hydrodynamic diameters ranging from 31.9 to 76.8 nm depending on LBA densities, and exhibited high DOX loading efficiencies of 83.0–89.2%. In vitro release studies showed that the DOX release rate depended on the pH and LBA content. Flow cytometric analyses revealed that asialoglycoprotein receptor (ASGP-R) over-expressed HepG2 liver cancer cells following 4 h treatment with DOX-loaded glycomicelles had a 6.6–17.1-fold higher DOX level, depending on LBA densities, as compared to those treated with the corresponding DOX-loaded non-glycomicelles (100% substitution with (EO)2-SH) under otherwise the same conditions. MTT assays demonstrated that DOX-loaded GP20-PCL, GP40-PCL, GP80-PCL and GP100-PCL micelles had much lower half maximal inhibitory concentration (IC50) values of 2.05, 0.75, 0.45 and 0.43 μg DOX equiv. mL−1, respectively, in HepG2 cells than DOX-loaded non-glycomicelles (IC50: 6.55 μg mL−1 DOX equiv. mL−1). Competitive inhibition experiments showed that after the incubation with DOX-loaded glycomicelles for 4 h, more efficient killing activity against free HepG2 cells (−LBA) was observed, as compared to that against LBA-blocked HepG2 cells (+LBA) after a subsequent 72 h incubation. Glycomicelles with tailored LBA functionalities, high drug loading capacity, and high uptake by ASGP-R positive cells are promising candidates for liver cancer chemotherapy.
Co-reporter:Juan Xiong, Fenghua Meng, Chao Wang, Ru Cheng, Zhuang Liu and Zhiyuan Zhong
Journal of Materials Chemistry A 2011 - vol. 21(Issue 15) pp:NaN5794-5794
Publication Date(Web):2011/03/07
DOI:10.1039/C0JM04410E
The poor stability of micellar drug delivery systems in vivo due to large volume dilution and interactions with blood pool often leads to premature drug release with low targetability and therapeutic efficacy. Here, we designed folate-conjugated interfacially crosslinked biodegradable micelles consisting of poly(ethylene glycol)-b-poly(acryloyl carbonate)-b-poly(D,L-lactide) (PEG-PAC-PLA) and folate-PEG-PLA (FA-PEG-PLA) block copolymers for receptor-mediated delivery of paclitaxel (PTX) into KB cells. Micelles with varying amounts of folate ligands were prepared at 0–20 wt.% of FA-PEG-PLA. The resulting micelles, either with or without PTX loading, were readily crosslinked by UV irradiation. The crosslinked micelles had much smaller sizes and better stability as compared to the non-crosslinked controls. Notably, these micelles achieved high drug loading efficiencies of 70–88% at an initial PTX loading content of 10 wt.%. The in vitro release studies revealed that crosslinked micelles exhibited significantly inhibited PTX release at low micelle concentrations. MTT assays in KB cells showed that the crosslinked micelles were non-toxic while the toxicity of PTX-loaded micelles, either crosslinked or non-crosslinked, increased with increasing folate contents. Remarkably, at 12 h incubation time folate-decorated PTX-loaded crosslinked micelles composed of 20 wt.% of FA-PEG-PLA displayed markedly higher toxicity to KB cells than free PTX (33% versus 50% cell viability), which is most likely due to their much more efficient cellular uptake through FA receptor-mediated endocytosis. Flow cytometry studies showed that folate-decorated FITC-labeled crosslinked micelles were much more efficiently taken up by KB cells than controls without folate ligands. These results indicate that ligand-conjugated interfacially crosslinked PEG-PLA micelles have great potential in targeted cancer therapy.
Co-reporter:Ru Cheng, Fenghua Meng, Shoubao Ma, Haifei Xu, Haiyan Liu, Xiabin Jing and Zhiyuan Zhong
Journal of Materials Chemistry A 2011 - vol. 21(Issue 47) pp:NaN19020-19020
Publication Date(Web):2011/10/31
DOI:10.1039/C1JM13536H
The study of biological functions of proteins in cells as well as therapeutic exploration of many protein drugs demands efficient and nontoxic intracellular protein delivery systems. Herein, reduction and temperature dual-responsive crosslinked polymersomes were developed for the facile encapsulation of various proteins under mild conditions as well as rapid release of proteins in cancer cells. Two thermo-sensitive triblock copolymers, PEG5k-PAA1.7k-PNIPAM22k and PEG5k-PAA0.7k-PNIPAM12k (denoted as polymer 1 and 2, respectively), were prepared by controlled reversible addition–fragmentation chain-transfer (RAFT) polymerization. Interestingly, polymers 1 and 2 exhibited lower critical solution temperatures (LCST) of 39 and 38 °C in PBS (pH 7.4, 20 mM, 150 mM NaCl) and 34 and 32 °C in MES (pH 5.5, 20 mM), respectively. Increasing the temperature of polymer solutions in MES to 40 °C yielded robust polymersomes with average diameters of ca. 150∼170 nm following crosslinking the PAA segment with cystamine (Cys) viacarbodiimide chemistry. These crosslinked polymersomes kept their structures in PBS at 37 °C but rapidly dissociated into unimers in response to 10 mM dithiothreitol (DTT). Remarkably, various proteins including bovine serum albumin (BSA), lysozyme (Lys), cytochrome C (CC), and ovalbumin (Ova) could be conveniently loaded into the polymersomes with markedly high protein loading efficiencies of 60∼100% at theoretical protein loading contents of 10∼50 wt%. The in vitro release studies using Cys-crosslinked polymersome 1 showed that release of BSA, Lys, and CC was minimal (ca. 20%) in 11 h in PBS at 37 °C, while fast protein release of over 70% was observed under an intracellular mimicking reductive environment. MTT assays revealed that these polymersomes were practically non-toxic to HeLa and MCF-7 cells up to a tested concentration of 200 μg mL−1. Confocal laser scanning microscope (CLSM) observations showed that FITC-CC loaded Cys-crosslinked polymersomes efficiently delivered and released FITC-CC into the cytosol of MCF-7 cells after 12 h incubation. In contrast, little FITC-CC fluorescence was observed in MCF-7 cells treated with free FITC-CC as well as FITC-CC loaded 1,4-butadiamine crosslinked polymersomes (reduction-insensitive control). Flow cytometry studies showed that CC loaded Cys-crosslinked polymersomes induced markedly enhanced apoptosis of MCF-7 cells as compared to free CC and the reduction-insensitive controls. These novel reduction and temperature dual-responsive crosslinked polymersomes have opened a new avenue to targeted intracellular protein delivery.
Co-reporter:Haifei Xu, Fenghua Meng and Zhiyuan Zhong
Journal of Materials Chemistry A 2009 - vol. 19(Issue 24) pp:NaN4190-4190
Publication Date(Web):2009/05/11
DOI:10.1039/B901141B
Water-soluble temperature responsive triblock copolymers, poly(ethylene oxide)-b-poly(acrylic acid)-b-poly(N-isopropylacrylamide) (PEO-PAA-PNIPAM), were prepared in one pot by sequential reversible addition–fragmentation chain-transfer (RAFT) polymerization using a PEO–trithiocarbonate (PEO–S-1-dodecyl-S-(R,R-dimethyl-R-aceticacid) trithiocarbonate) as a macro chain transfer agent. The block copolymers with MnPEO of 5 kDa, MnPAA of 0.35–1.45 kDa, and MnPNIPAM varying from 11–39 kDa were freely soluble in water as unimers at room temperature, but quickly self-assembled into nano-sized vesicles (about 220 nm) when raising the solution temperature to 37 °C. The vesicular structure was confirmed by confocal scanning laser microscope (CSLM) and static light scattering (SLS) measurements. The size and size distribution of the polymersomes depended on the solution concentration, the molecular weight of PNIPAM, the equilibrium time and shaking. Interestingly, thus-formed vesicles could be readily cross-linked at the interface using cystamineviacarbodiimide chemistry. The crosslinked polymersomes, while showing remarkable stability against dilution, organic solvent, high salt conditions and change of temperature in water, were otherwise rapidly dissociated under reductive conditions mimicking intracellular environment. Notably, FITC–dextran, used as a model protein, was shown to be encapsulated into the polymersomes with an unprecedently high loading efficiency (>85 wt%). The release studies showed that most FITC–dextran was retained within the polymersomes after lowering the temperature to 25 °C. However, in the presence of 10 mM dithiothreitol (DTT), fast release of FITC–dextran was achieved. These reversibly crosslinked temperature responsive nano-sized polymersomes are highly promising as smart carriers for triggered intracellular delivery of biopharmaceutics such as pDNA, siRNA, pharmaceutical proteins and peptides.
Co-reporter:Kaiqi Wu, Ru Cheng, Jian Zhang, Fenghua Meng, Chao Deng and Zhiyuan Zhong
Journal of Materials Chemistry A 2017 - vol. 5(Issue 28) pp:NaN5667-5667
Publication Date(Web):2017/06/14
DOI:10.1039/C7TB01297G
Bortezomib (BTZ) is the first proteasome inhibitor approved for the treatment of malignant tumors. The current clinical formulation, however, shows fast clearance, low tumor accumulation, and several side effects. Here, we report that micellar nanoformulation of lipophilized bortezomib achieves significantly enhanced drug loading, prolonged circulation time, improved tolerability and targeted treatment of triple negative breast cancer in vivo. Lipophilized bortezomib, bortezomib-pinanediol (BP), was readily prepared in high yield. Interestingly, cRGD-targeted micelles based on poly(ethylene glycol)-b-poly(trimethylene carbonate-co-dithiolane trimethylene carbonate) achieved a high drug loading content of 8.05 wt% BTZ equiv. for BP, which was more than 8-fold higher than BTZ. BP-loaded cRGD-decorated micelles (BP-cRGD-Ms) exhibited a small size (ca. 49 nm), reduction-triggered drug release, and active targeting ability to αvβ3-overexpressing MDA-MB-231 triple-negative breast cancer cells, resulting in a low IC50 of 0.986 μM. The in vivo studies displayed that BP-cRGD-Ms had a nearly 20-fold improvement in the elimination half-life and a 20-fold higher maximum-tolerated dose as compared to free BTZ. The biodistribution and therapeutic studies in MDA-MB-231 tumor-bearing nude mice demonstrated that BP-cRGD-Ms induced significantly better tumor accumulation and inhibition with fewer adverse effects than free BTZ, leading to greatly improved mice survival rates. This micellar nanoformulation of lipophilized bortezomib appears to be a novel and effective strategy to achieve targeted tumor chemotherapy with bortezomib.
Co-reporter:Ru Cheng, Xiaoyan Wang, Wei Chen, Fenghua Meng, Chao Deng, Haiyan Liu and Zhiyuan Zhong
Journal of Materials Chemistry A 2012 - vol. 22(Issue 23) pp:NaN11738-11738
Publication Date(Web):2012/04/03
DOI:10.1039/C2JM30700F
Biodegradable micelles were prepared from poly(ε-caprolactone)-g-poly(2-hydroxyethyl methacrylate) (PCL-g-PHEMA) graft copolymers and investigated for controlled release of doxorubicin (DOX). PCL-g-PHEMA copolymers were readily obtained by controlled ring-opening copolymerization of acryloyl cyclic carbonate and ε-caprolactone, Michael-type conjugate addition reaction with cysteamine, coupling reaction with 4-cyanopentanoic acid dithionaphthalenoate (CPADN) via carbodiimide chemistry, and reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-hydroxyethyl methacrylate (HEMA). 1H NMR analyses showed that Mn of PHEMA ranged from 8.7, 16.3 to 33.8 kg mol−1, in proximity to the design as well as those determined by gel permeation chromatography (GPC). Differential scanning calorimetry (DSC) revealed that all three PCL-g-PHEMA graft copolymers had depressed melting temperatures (Tm = 31.3–32.5 °C) and low crystallinities (Xc = 3.05–5.66%). Dynamic light scattering (DLS) showed that PCL-g-PHEMA formed monodisperse micelles with low polydispersity indexes of 0.04–0.16 and average sizes ranging from 80.5 to 179.7 nm depending on PHEMA chain lengths. These graft copolymers displayed low critical micelle concentrations (CMCs) of 0.051–0.151 μM. The micellar sizes decreased following loading with DOX while PDI remained low. Interestingly, in vitro drug release studies showed that DOX-loaded PCL-g-PHEMA micelles exhibited superior pH-responsive release behaviors, in which up to 94.5% of DOX was released in 3 d at pH 5.0 while DOX release was significantly slower at pH 7.4 (maximum 54.1% release in 3 d). MTT assays with HeLa cells demonstrated that DOX-loaded PCL-g-PHEMA micelles retained high anti-tumor activity with low IC50 (half inhibitory concentration) of 1.47–1.74 μg DOX equiv. mL−1 while PCL-g-PHEMA micelles were practically non-toxic up to a tested concentration of 80 mg mL−1. These novel biodegradable PCL-g-PHEMA graft copolymer micelles with low CMC, small and tunable sizes, high drug loading, and pH-responsive drug release have emerged as superior nanocarriers for “smart” tumor-targeting drug delivery.








![2,5-Pyrrolidinedione, 1-[(4-methoxybenzoyl)oxy]-](http://img.cochemist.com/ccimg/30400/30364-57-9.png)
![2,5-Pyrrolidinedione, 1-[(4-methoxybenzoyl)oxy]-](http://img.cochemist.com/ccimg/30400/30364-57-9_b.png)


![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)

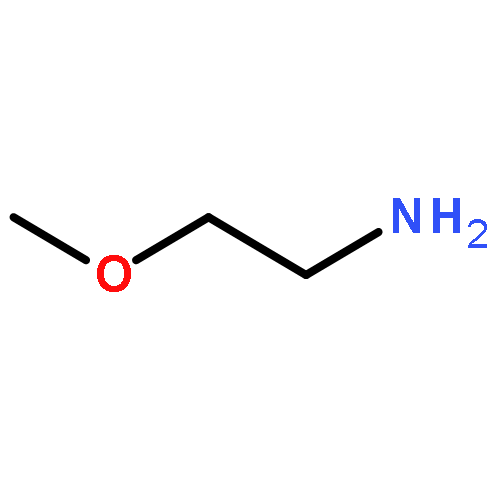
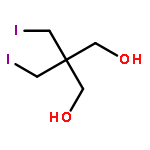
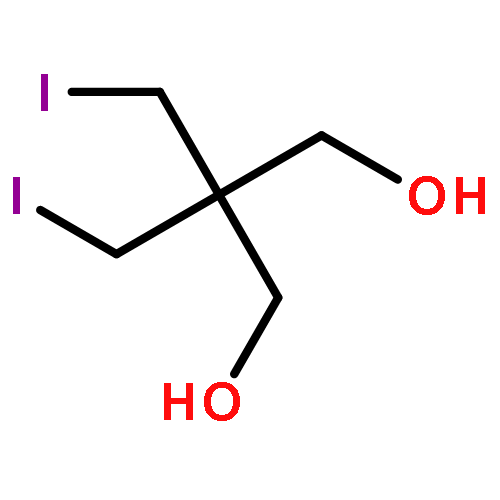


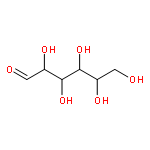
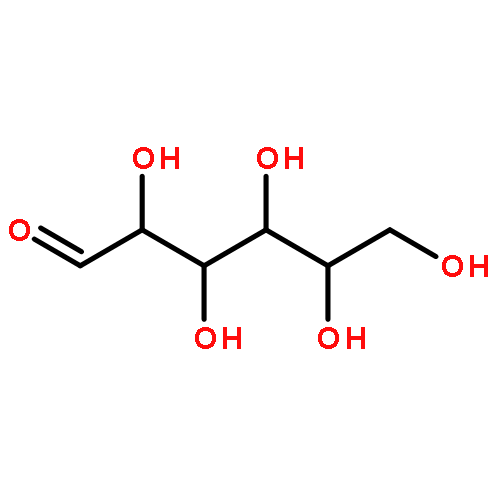


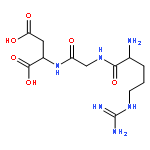

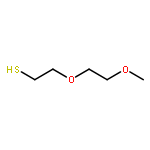


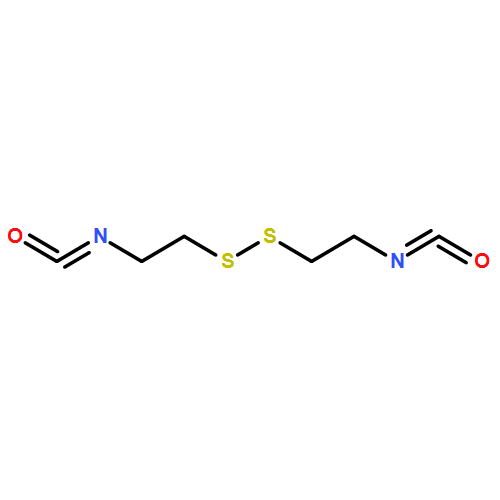
![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7.png)
![Ethanol, 2-[2-(2-azidoethoxy)ethoxy]-](/data/chemimg/964400/86520-52-7_b.png)
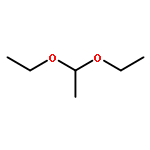
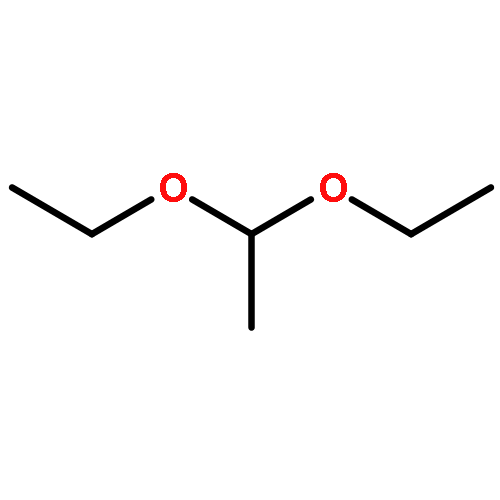
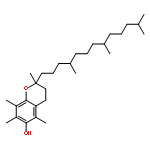
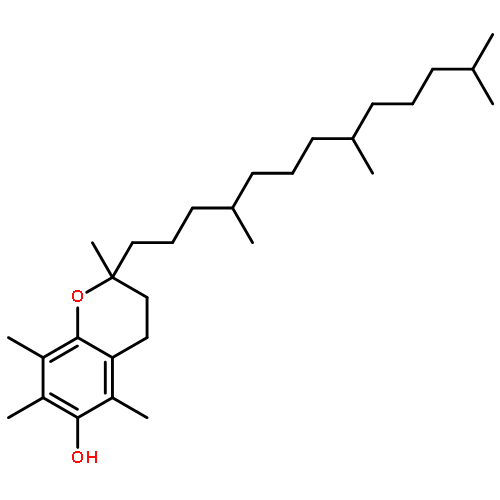
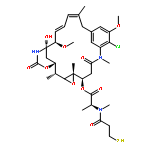











![3H-Indolium, 2-[5-[1-[6-[(2,5-dioxo-1-pyrrolidinyl)oxy]-6-oxohexyl]-1,3-dihydro-3,3-dimethyl-5-sulfo-2H-indol-2-ylidene]-1,3-pentadien-1-yl]-1-ethyl-3,3-](/data/chemimg/105700/146368-14-1.png)
![3H-Indolium, 2-[5-[1-[6-[(2,5-dioxo-1-pyrrolidinyl)oxy]-6-oxohexyl]-1,3-dihydro-3,3-dimethyl-5-sulfo-2H-indol-2-ylidene]-1,3-pentadien-1-yl]-1-ethyl-3,3-](/data/chemimg/105700/146368-14-1_b.png)






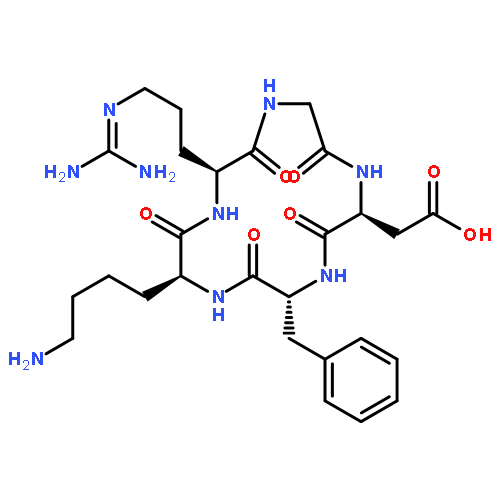





![Carbamic acid, N-[2-[(2-aminoethyl)dithio]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/105100/485800-26-8.png)
![Carbamic acid, N-[2-[(2-aminoethyl)dithio]ethyl]-, 1,1-dimethylethyl ester](/data/chemimg/105100/485800-26-8_b.png)