Co-reporter:Qian Huang, Xiao-Fang Mao, Hai-Yun Wu, Hao Liu, ... Yong-Xiang Wang
Brain, Behavior, and Immunity 2017 Volume 62(Volume 62) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.bbi.2017.02.005
•Cynandione A is the principle acetophenone of Cynanchi Wilfordii Radix.•Intrathecal injection of cynandione A produced antinociception in neuropathic rats.•Cynandione A stimulated β-endorphin expression in spinal microglia.•p38β MAPK mediated cynandione A-induced β-endorphin release and antiallodynia.Cynanchi Wilfordii Radix (baishouwu), a medicinal herb, has been widely used in Asia to treat a variety of diseases or illnesses. Cynandione A isolated from C. Wilfordii is the principle acetophenone and exhibits neuroprotective and anti-inflammatory activities. This study aims to evaluate the antihypersensitivity activities of cynandione A in neuropathy and explored its mechanisms of action. Intrathecal injection of cynandione A dose-dependently attenuated spinal nerve ligation-induced mechanical allodynia and thermal hyperalgesia, with maximal possible effects of 57% and 59%, ED50s of 14.9 μg and 6.5 μg, respectively. Intrathecal injection of cynandione A significantly increased β-endorphin levels in spinal cords of neuropathic rats and its treatment concentration-dependently induced β-endorphin expression in cultured primary microglia (but not in neurons or astrocytes), with EC50s of 38.8 and 20.0 μM, respectively. Cynandione A also non-selectively upregulated phosphorylation of mitogen-activated protein kinases (MAPKs), including p38, extracellular signal regulated kinase (ERK1/2), and extracellular signal regulated kinase (JNK) in primary microglial culture; however, cynandione A-stimulated β-endorphin expression was completely inhibited by the specific p38 activation inhibitor SB203580, but not by the ERK1/2 or JNK activation inhibitors. Knockdown of spinal p38β but not p38α using siRNA also completely blocked cynandione A-induced β-endorphin expression in cultured microglial cells. Furthermore, cynandione A-induced antiallodynia in neuropathy was totally inhibited by the microglial inhibitor minocycline, SB203580, anti-β-endorphin antibody, and μ-opioid receptor antagonist CTAP (but not the κ- or δ-opioid receptor antagonist). These results suggest that cynandione A attenuates neuropathic pain through upregulation of spinal microglial expression β-endorphin via p38β MAPK activation.Download high-res image (110KB)Download full-size image
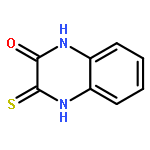
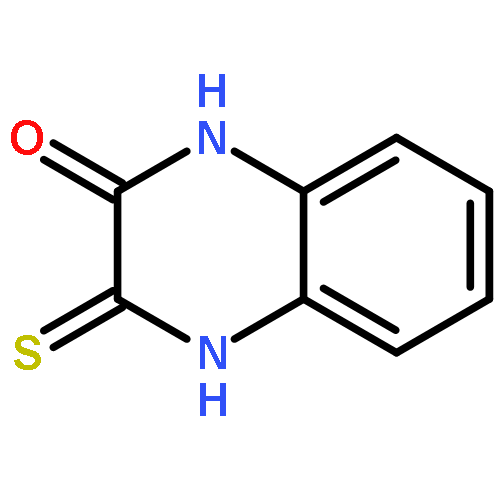
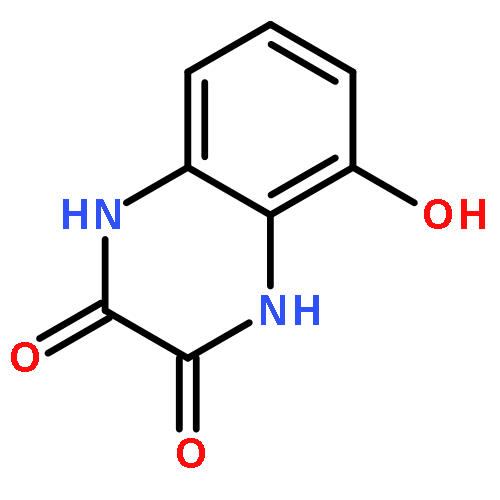
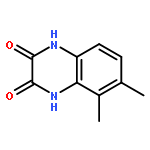
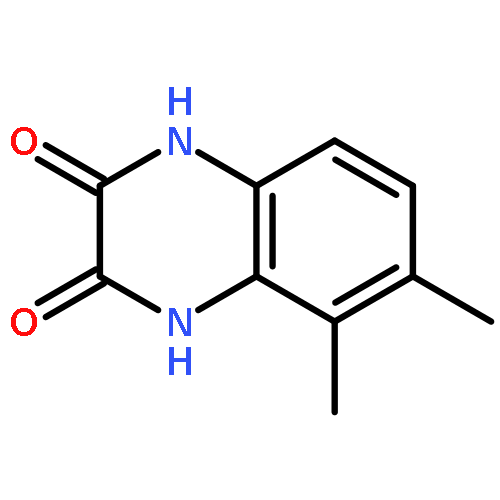
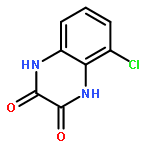
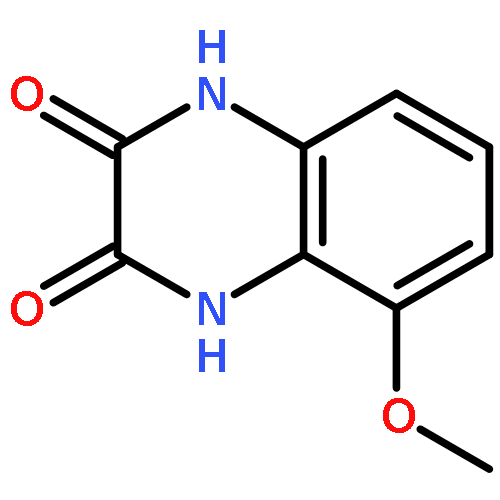
Co-reporter:Dongsheng Xie, Jun Lu, Jin Xie, Junjun Cui, Teng-Fei Li, Yan-Chao Wang, Yuan Chen, Nian Gong, Xin-Yan Li, Lei Fu, Yong-Xiang Wang
European Journal of Medicinal Chemistry 2016 Volume 117() pp:19-32
Publication Date(Web):19 July 2016
DOI:10.1016/j.ejmech.2016.04.017
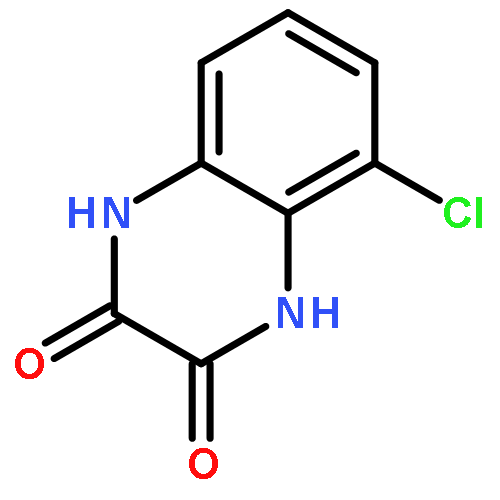
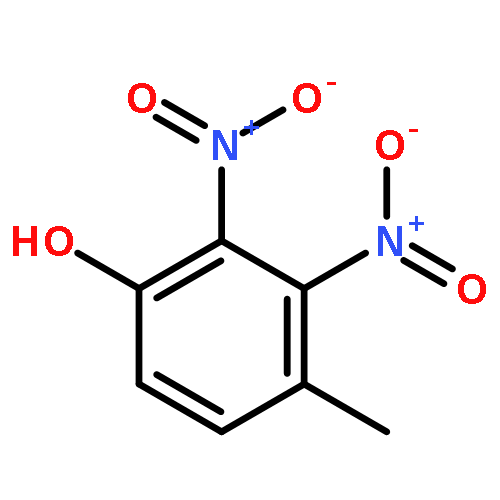
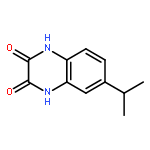
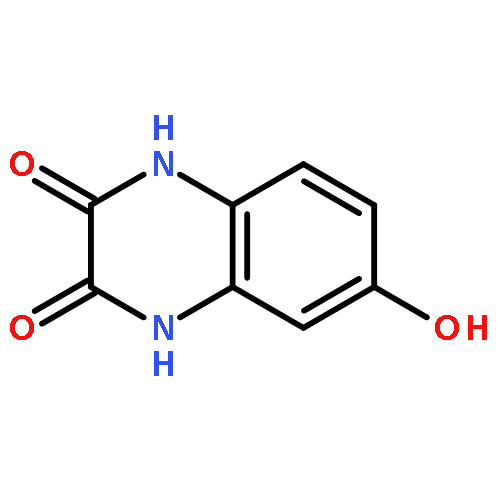
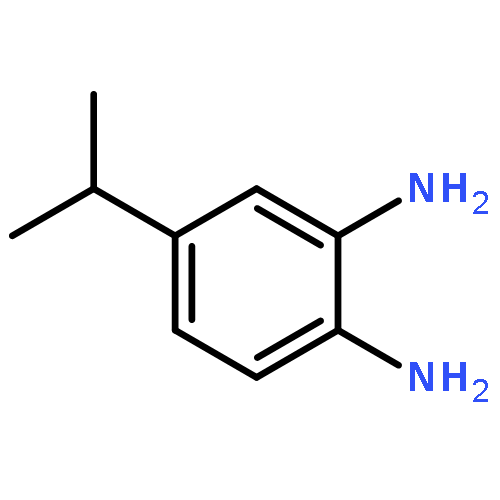
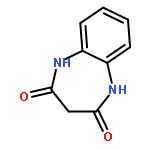
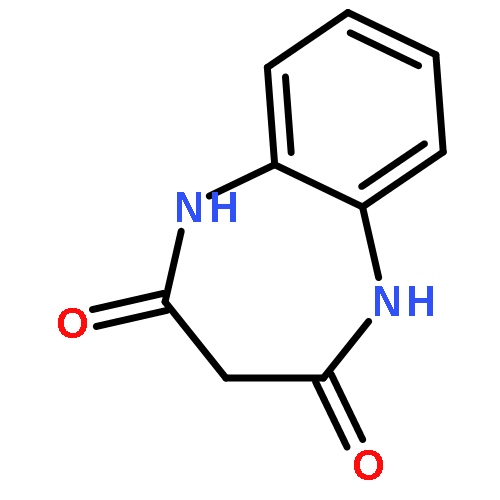
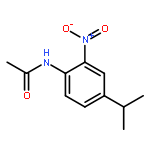
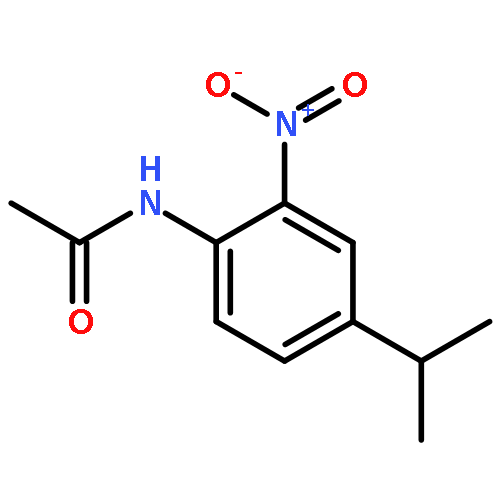
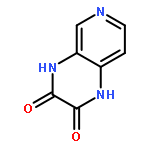
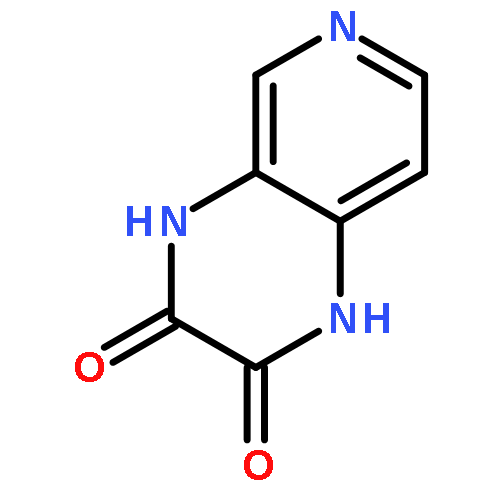
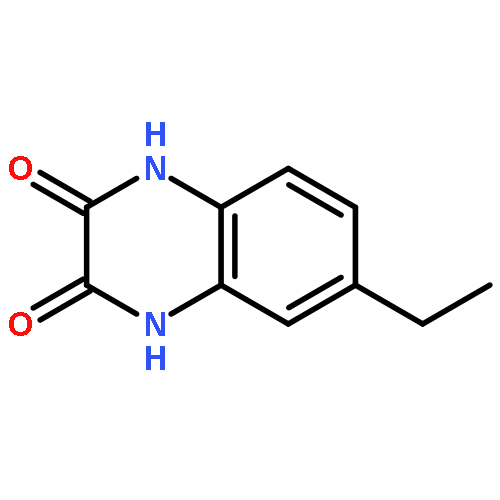
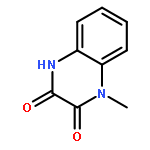
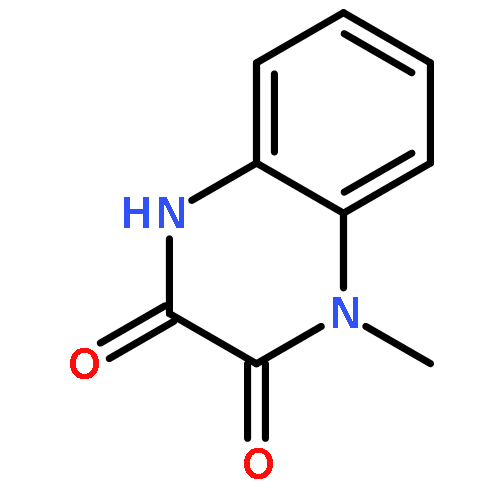
•Disclosed 5-azaquinoxaline-2,3-diones as novel noncarboxylic DAAO inhibitors.•Confirmed the analgesic effects of DAAO inhibitor based on a new scaffold.•Provided a new class of chemical entities for potential analgesic application.A series of 5-azaquinoxaline-2,3-dione derivatives were synthesized and evaluated on d-amino acid oxidase (DAAO) inhibition as potential α-hydroxylactam-based inhibitors. The potent inhibitory activities in vitro suggested that 5-nitrogen could significantly enhance the binding affinity by strengthening relevant hydrogen bond interactions. The analgesic effects of intrathecal and systemic injection of 8-chloro-1,4-dihydropyrido[2,3-b]pyrazine-2,3-dione, a representative molecule of 5-azaquinoxaline-2,3-dione, were investigated in rodents. This research not only confirmed the analgesic effect of the DAAO inhibitors but provided a new class of chemical entities with oral application potential for the treatment of chronic pain and morphine analgesic tolerance.Systemic injection of 8-chloro-1,4-dihydropyrido[2,3-b]pyrazine-2,3-dione (Compound 16), a novel potent d-amino acid oxidase (DAAO) inhibitor, inhibited formalin-induced tonic pain in rodents in a dose-dependent manner.
Co-reporter:Hui Fan;Nian Gong;Teng-Fei Li;Ai-Niu Ma;Xiao-Yan Wu;Ming-Wei Wang;Yong-Xiang Wang
British Journal of Pharmacology 2015 Volume 172( Issue 1) pp:64-79
Publication Date(Web):
DOI:10.1111/bph.12895
Background and Purpose
Two peptide agonists of the glucagon-like peptide-1 (GLP-1) receptor, exenatide and GLP-1 itself, exert anti-hypersensitive effects in neuropathic, cancer and diabetic pain. In this study, we have assessed the anti-allodynic and anti-hyperalgesic effects of the non-peptide agonist WB4-24 in inflammatory nociception and the possible involvement of microglial β-endorphin and pro-inflammatory cytokines.
Experimental Approach
We used rat models of inflammatory nociception induced by formalin, carrageenan or complete Freund's adjuvant (CFA), to test mechanical allodynia and thermal hyperalgesia. Expression of β-endorphin and pro-inflammatory cytokines was measured using real-time quantitative PCR and fluorescent immunoassays.
Key Results
WB4-24 displaced the specific binding of exendin (9–39) in microglia. Single intrathecal injection of WB4-24 (0.3, 1, 3, 10, 30 and 100 μg) exerted dose-dependent, specific, anti-hypersensitive effects in acute and chronic inflammatory nociception induced by formalin, carrageenan and CFA, with a maximal inhibition of 60–80%. Spinal WB4-24 was not effective in altering nociceptive pain. Subcutaneous injection of WB4-24 was also antinociceptive in CFA-treated rats. WB4-24 evoked β-endorphin release but did not inhibit expression of pro-inflammatory cytokines in either the spinal cord of CFA-treated rats or cultured microglia stimulated by LPS. WB4-24 anti-allodynia was prevented by a microglial inhibitor, β-endorphin antiserum and a μ-opioid receptor antagonist.
Conclusions and Implications
Our results suggest that WB4-24 inhibits inflammatory nociception by releasing analgesic β-endorphin rather than inhibiting the expression of proalgesic pro-inflammatory cytokines in spinal microglia, and that the spinal GLP-1 receptor is a potential target molecule for the treatment of pain hypersensitivity including inflammatory nociception.
Co-reporter:Shuai Ma, Xin-Yan Li, Nian Gong, Yong-Xiang Wang
Journal of Pharmaceutical and Biomedical Analysis 2015 Volume 116() pp:131-138
Publication Date(Web):10 December 2015
DOI:10.1016/j.jpba.2015.03.021
•DAAO is a peroxisomal flavoenzyme conversing d-amino acids to hydrogen peroxide.•Multi-daily morphine to mice induced thermal hyperalgesia.•DAAO inhibitor CBIO prevented and reversed chronic morphine-induced hyperalgesia.•PBN and catalase reversed established morphine hyperalgesia.•Spinal DAAO and hydrogen peroxide contribute to morphine-induced hyperalgesia.Spinal d-amino acid oxidase (DAAO) is an FAD-dependent peroxisomal flavoenzyme which mediates the conversion of neutral and polar d-amino acids (including d-serine) to the corresponding α-keto acids, and simultaneously produces hydrogen peroxide and ammonia. This study has aimed to explore the potential contributions of spinal DAAO and its mediated hydrogen peroxide/d-serine metabolism to the development of morphine-induced hyperalgesia. Bi-daily subcutaneous injections of morphine to mice over 7 days induced thermal hyperalgesia as measured by both the hot-plate and tail-immersion tests, and spinal astroglial activation with increased spinal gene expression of DAAO, glial fibrillary acidic protein (GFAP) and pro-inflammatory cytokines (interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α)). Subcutaneous injections of the potent DAAO inhibitor CBIO (5-chloro-benzo[d]isoxazol-3-ol) prevented and reversed the chronic morphine-induced hyperalgesia. CBIO also inhibited both astrocyte activation and the expression of pro-inflammatory cytokines. Intrathecal injection of the hydrogen peroxide scavenger PBN (phenyl-N-tert-butylnitrone) and of catalase completely reversed established morphine hyperalgesia, whereas subcutaneous injections of exogenous d-serine failed to alter chronic morphine-induced hyperalgesia. These results provided evidence that spinal DAAO and its subsequent production of hydrogen peroxide rather than the d-serine metabolism contributed to the development of morphine-induced hyperalgesia.
Co-reporter:Jin-Miao Lu;Nian Gong;Yan-Chao Wang ;Yong-Xiang Wang
British Journal of Pharmacology 2012 Volume 165( Issue 6) pp:
Publication Date(Web):
DOI:10.1111/j.1476-5381.2011.01680.x
BACKGROUND AND PURPOSE Spinal reactive oxygen species (ROS) are critically involved in chronic pain. d-Amino acid oxidase (DAAO) oxidizes d-amino acids such as d-serine to form the byproduct hydrogen peroxide without producing other ROS. DAAO inhibitors are specifically analgesic in tonic pain, neuropathic pain and cancer pain. This study examined the role of spinal hydrogen peroxide in pain and the mechanism of the analgesic effects of DAAO inhibitors.
EXPERIMENTAL APPROACH Formalin-induced pain behaviours and spinal hydrogen peroxide levels were measured in rodents.
KEY RESULTS Formalin injected into the paw increased spinal hydrogen peroxide synchronously with enhanced tonic pain; both were effectively prevented by i.t. fluorocitrate, a selective astrocyte metabolic inhibitor. Given systemically, the potent DAAO inhibitor CBIO (5-chloro-benzo[d]isoxazol-3-ol) blocked spinal DAAO enzymatic activity and specifically prevented formalin-induced tonic pain in a dose-dependent manner. Although CBIO maximally inhibited tonic pain by 62%, it completely prevented the increase in spinal hydrogen peroxide. I.t. catalase, an enzyme specific for decomposition of hydrogen peroxide, completely depleted spinal hydrogen peroxide and prevented formalin-induced tonic pain by 65%. Given systemically, the ROS scavenger PBN (phenyl-N-tert-butylnitrone) also inhibited formalin-induced tonic pain and increase in spinal hydrogen peroxide. Formalin-induced tonic pain was potentiated by i.t. exogenous hydrogen peroxide. CBIO did not increase spinal d-serine level, and i.t. d-serine did not alter either formalin-induced tonic pain or CBIO's analgesic effect.
CONCLUSIONS AND IMPLICATIONS Spinal hydrogen peroxide is specifically and largely responsible for formalin-induced pain, and DAAO inhibitors produce analgesia by blocking spinal hydrogen peroxide production rather than interacting with spinal d-serine.
Co-reporter:Jin-Lu Huang;Xiao-Ling Chen;Cheng Guo;Yong-Xiang Wang
Amino Acids 2012 Volume 43( Issue 5) pp:1905-1918
Publication Date(Web):2012 November
DOI:10.1007/s00726-012-1390-z
d-Amino acid oxidase (DAAO), a FAD-dependent peroxisomal flavoenzyme that catalyzes oxidation of d-amino acids to hydrogen peroxide, is distributed in the spinal cord almost exclusively expressed within astrocytes. The present study aims to explore potential contributions of spinal DAAO to the development of bone cancer pain and morphine tolerance to analgesia. Tibia inoculation of carcinoma cells produced mechanical allodynia (but not heat hyperalgesia), in synchronous with induction of DAAO expression and DAAO enzymatic activity, as well as activation of spinal astrocytes marked by GFAP. Subcutaneous and intrathecal injection of the specific DAAO inhibitor CBIO (5-chloro-benzo[d]isoxazol-3-ol) blocked mechanical allodynia in a dose- and time-dependent manner in tumor-bearing rats, with maximum inhibition of 40–50 %. Multi-daily intrathecal injections of the DAAO gene silencer siRNA/DAAO also yielded anti-allodynic effects by approximately 40 % and the analgesia remained for at least 6 days. Subcutaneous injection of CBIO suppressed the production of spinal hydrogen peroxide and GFAP expression. 7-Day multiple bi-daily injections of CBIO produced anti-allodynia without inducing self-tolerance to analgesia or cross-tolerance to morphine, and concurrent injections of CBIO with morphine produced apparent additive anti-allodynia and completely prevented morphine tolerance in behaviors and spinal expression of μ-opioid receptors. Our results provide the first evidence that spinal DAAO contributes to the development of morphine tolerance to analgesia and bone cancer pain accounting for 40–50 % pain status, probably via production of hydrogen peroxide leading to activation of astrocytes. The unique characterizations of DAAO inhibitors make them a potential for the treatment of cancer pain when they are administered alone or in combination with morphine.
Co-reporter:Nian Gong;Ai-Niu Ma;Li-Jie Zhang;Xiao-Su Luo;Yin-Hui Zhang;Michael Xu;Yong-Xiang Wang
British Journal of Pharmacology 2011 Volume 163( Issue 2) pp:399-412
Publication Date(Web):
DOI:10.1111/j.1476-5381.2011.01227.x
BACKGROUND AND PURPOSE
Exenatide is a 39-amino-acid peptide widely used to manage type 2 diabetes mellitus. However, it has a short plasma half-life and requires a twice daily injection regime. To overcome these drawbacks we used maleimide-polyethylene glycol to induce site-specific PEGylation.
EXPERIMENTAL APPROACH
The analogue PB-105 (ExC39) was produced by replacing cysteine at position 39 of exenatide to provide a free thiol group. PB-105 showed the same glucoregulatory activity as exenatide in mice. Site-specific PEGylation of PB-105 was performed to produce PB-110 (ExC39PEG5kDa), PB-106 (ExC39PEG20kDa), PB-107 (ExC39PEG30kDa) and PB-108 (ExC39PEG40kDa). Their effects on intracellular cAMP, acute glucoregulatory activity and pharmacokinetic profile were compared in mice and rats.
KEY RESULTS
PEGylation shifted the concentration–response curve of PB-105 to the right in a parallel, polyethylene glycol mass-dependent manner but with an inflexion point of at least 20 kDa. The activities of PB-107 and PB-108 but not PB-106 were reduced by 90% and 99%. PEGylation affected in vivo glucoregulatory activity in the same ‘Inflexion-Shift’ fashion at least at 20 kDa, but linearly increased plasma duration and systemic exposure without inflexion. PB-106 had a plasma t1/2 approximately 10-fold that of PB-105, and exhibited superior glucoregulatory activity compared with PB-105 in normal and diabetic mice.
CONCLUSIONS AND IMPLICATIONS
Site-specific PEGylation of exenatide with a permanent amide linkage affects its activity in a new type of ‘Inflexion-Shift’ fashion. PB-106 is a putative new analogue for treating diabetes; it possesses no loss of in vitro activity, prolonged plasma duration and superior, improved in vivo glucoregulatory activity compared with exenatide.
Co-reporter:Wenjuan Zhao;Ryuichi Konno;Xiang-Jun Zhou
Cellular and Molecular Neurobiology 2008 Volume 28( Issue 4) pp:581-591
Publication Date(Web):2008 June
DOI:10.1007/s10571-007-9200-y
(1). We investigated the effects of inhibiting d-amino-acid oxidase (DAO) activity on nociceptive responses through the use of mutant ddY/DAO− mice, which lack DAO activity, and through the application of a selective inhibitor of DAO, sodium benzoate, in the tail flick test, hot-plate test, formalin test, and acetic acid-induced writhing test. (2). Compared with normal ddY/DAO+ mice, ddY/DAO− mice showed significantly prolonged tail withdrawal latency in the tail flick test and licking/jumping latency in the hot-plate test, as well as significantly reduced duration of licking/biting in the late phase of the formalin test and the number of abdominal writhing in the acetic acid-induced writhing test. (3). In addition, we investigated the effects of sodium benzoate in Kunming mice having normal DAO activity. (4). Intravenous administration of sodium benzoate (400 mg/kg) significantly inhibited pain responses of the late phase of the formalin test and abdominal writhing responses in the acetic acid-induced writhing test, with no effects on the early phase flinch responses in the formalin test, nociceptive responses in the tail flick test, or hot-plate test. (5). These results suggest that DAO acts as a pro-nociceptive factor in pain, particularly chronic pain, transmission and modulation, and may be a target for pain treatment.
Co-reporter:Nian Gong, Hui Fan, Ai-Niu Ma, Qi Xiao, Yong-Xiang Wang
Neuropharmacology (September 2014) Volume 84() pp:31-45
Publication Date(Web):1 September 2014
DOI:10.1016/j.neuropharm.2014.04.007
•Geniposide blocked H2O2-induced oxidative damage in an exendin(9-39)-reversible manner.•Subcutaneous and oral geniposide blocked formalin-induced tonic flinching response.•A 7-day subcutaneous geniposide did not induce antinociceptive tolerance.•Geniposide antinociception was prevented by intrathecal exendin(9-39) and siRNA/GLP-1R.•Geniposide iridoid analogs inhibited H2O2-induced oxidative damage and formalin pain.We recently discovered that the activation of the spinal glucagon-like peptide-1 receptors (GLP-1Rs) by the peptidic agonist exenatide produced antinociception in chronic pain. We suggested that the spinal GLP-1Rs are a potential target molecule for the management of chronic pain. This study evaluated the antinociceptive activities of geniposide, a presumed small molecule GLP-1R agonist. Geniposide produced concentration-dependent, complete protection against hydrogen peroxide-induced oxidative damage in PC12 and HEK293 cells expressing rat and human GLP-1Rs, but not in HEK293T cells that do not express GLP-1Rs. The orthosteric GLP-1R antagonist exendin(9-39) right-shifted the concentration–response curve of geniposide without changing the maximal protection, with identical pA2 values in both cell lines. Subcutaneous and oral geniposide dose-dependently blocked the formalin-induced tonic response but not the acute flinching response. Subcutaneous and oral geniposide had maximum inhibition of 72% and 68%, and ED50s of 13.1 and 52.7 mg/kg, respectively. Seven days of multidaily subcutaneous geniposide and exenatide injections did not induce antinociceptive tolerance. Intrathecal geniposide induced dose-dependent antinociception, which was completely prevented by spinal exendin(9-39), siRNA/GLP-1R and cyclic AMP/PKA pathway inhibitors. The geniposide iridoid analogs geniposidic acid, genipin methyl ether, 1,10-anhydrogenipin, loganin and catalpol effectively inhibited hydrogen peroxide-induced oxidative damage and formalin pain in an exendin(9-39)-reversible manner. Our results suggest that geniposide and its iridoid analogs produce antinociception during persistent pain by activating the spinal GLP-1Rs and that the iridoids represented by geniposide are orthosteric agonists of GLP-1Rs that function similarly in humans and rats and presumably act at the same binding site as exendin(9-39).
Co-reporter:Hong Wei, Nian Gong, Jin-Lu Huang, Hui Fan, Ai-Niu Ma, Xin-Yan Li, Yong-Xiang Wang, Antti Pertovaara
Pharmacology Biochemistry and Behavior (October 2013) Volume 111() pp:30-36
Publication Date(Web):1 October 2013
DOI:10.1016/j.pbb.2013.08.003
•d-amino acid oxidase (DAAO) is found in spinal astrocytes.•REM sleep deprivation (REMSD) induces mechanical pain hypersensitivity.•REMSD-induced hypersensitivity was reduced by inhibiting spinal DAAO.•Spinal DAAO contributes to pain hypersensitivity induced by REMSD.We studied the hypothesis that spinal d-amino acid oxidase (DAAO) that is expressed in astrocytes and that has been reported to promote tonic pain in various pathophysiological conditions plays a role in ‘physiological’ pain hypersensitivity induced by rapid eye movement sleep deprivation (REMSD). The experiments were performed in healthy rats with a chronic intrathecal (i.t.) catheter. Pain behavior was assessed by determining limb withdrawal response to repetitive stimulation of the hind paw with a calibrated series of monofilaments. REMSD of 48 h duration produced a significant mechanical hypersensitivity. At 48 h of REMSD, the animals were treated i.t. with a DAAO inhibitor or vehicle. Three structurally different DAAO inhibitors were tested in this study: 6-chlorobenzo[d]isoxazol-3-ol (CBIO), sodium benzoate, and 5-methylpyrazole-3-carboxylic acid (AS-057278). CBIO (1–3 μg), sodium benzoate (30–100 μg) and AS-057278 (3–10 μg) produced dose-related antihypersensitivity effects in sleep-deprived animals. In control animals (with no sleep deprivation), the currently used doses of DAAO inhibitors failed to produce significant changes in mechanically evoked pain behavior. The results indicate that among spinal pain facilitatory mechanisms that contribute to the sleep deprivation-induced mechanical pain hypersensitivity is DAAO, presumably due to production of reactive oxygen species, such as hydrogen peroxide, an endogenous agonist of the pronociceptive TRPA1 ion channel.
Co-reporter:Hong Wei, Hai-Yun Wu, Zuyue Chen, Ai-Niu Ma, Xiao-Fang Mao, Teng-Fei Li, Xin-Yan Li, Yong-Xiang Wang, Antti Pertovaara
Pharmacology Biochemistry and Behavior (November–December 2016) Volumes 150–151() pp:57-67
Publication Date(Web):1 November 2016
DOI:10.1016/j.pbb.2016.09.007
•Prolonged spinal treatment with a TRPA1 antagonist reduced neuropathic pain•Prolonged spinal treatment with a gap junction blocker reduced neuropathic pain•TRPA1 antagonist or gap junction blocker did not prevent development of neuropathy.•TRPA1 & DAAO mRNA were upregulated in dorsal root ganglia of injured nerves.Spinal transient receptor potential ankyrin 1 (TRPA1) channel is associated with various pain hypersensitivity conditions. Spinally, TRPA1 is expressed by central terminals of nociceptive nerve fibers and astrocytes. Among potential endogenous agonists of TRPA1 is H2O2 generated by d-amino acid oxidase (DAAO) in astrocytes. Here we studied whether prolonged block of the spinal TRPA1 or astrocytes starting at time of injury attenuates development and/or maintenance of neuropathic hypersensitivity. Additionally, TRPA1 and DAAO mRNA were determined in the dorsal root ganglion (DRG) and spinal dorsal horn (SDH). Experiments were performed in rats with spared nerve injury (SNI) and chronic intrathecal catheter. Drugs were administered twice daily for the first seven injury days or only once seven days after injury. Mechanical hypersensitivity was assessed with monofilaments. Acute and prolonged treatment with Chembridge-5861528 (a TRPA1 antagonist), carbenoxolone (an inhibitor of activated astrocytes), or gabapentin (a comparison drug) attenuated tactile allodynia-like responses evoked by low (2 g) stimulus. However, antihypersensitivity effect of these compounds was short of significance at a high (15 g) stimulus intensity. No preemptive effects were observed. In healthy controls, carbenoxolone failed to prevent hypersensitivity induced by spinal cinnamaldehyde, a TRPA1 agonist. TRPA1 and DAAO mRNA in the DRG but not SDH were slightly increased in SNI, independent of drug treatment. The results indicate that prolonged peri-injury block of spinal TRPA1 or inhibition of spinal astrocyte activation attenuates maintenance but not development of mechanical (tactile allodynia-like) hypersensitivity after nerve injury.
Co-reporter:Jing-Yang Zhang, Nian Gong, Jin-Lu Huang, Ling-Chen Guo, Yong-Xiang Wang
PAIN® (November 2013) Volume 154(Issue 11) pp:2452-2462
Publication Date(Web):1 November 2013
DOI:10.1016/j.pain.2013.07.027
The present study examined the antinociceptive effects of gelsemine, the principal alkaloid in Gelsemium sempervirens Ait. A single intrathecal injection of gelsemine produced potent and specific antinociception in formalin-induced tonic pain, bone cancer-induced mechanical allodynia, and spinal nerve ligation-induced painful neuropathy. The antinociception was dose-dependent, with maximal inhibition of 50% to 60% and ED50 values of 0.5 to 0.6 μg. Multiple daily intrathecal injections of gelsemine for 7 days induced no tolerance to antinociception in the rat model of bone cancer pain. Spinal gelsemine was not effective in altering contralateral paw withdrawal thresholds, and had only a slight inhibitory effect on formalin-induced acute nociception. The specific antinociception of gelsemine in chronic pain was blocked dose-dependently by the glycine receptor (GlyR) antagonist strychnine with an apparent ID50 value of 3.8 μg. Gelsemine concentration-dependently displaced H3-strychnine binding to the membrane fraction of rat spinal cord homogenates, with a 100% displacement and a Ki of 21.9 μM. Gene ablation of the GlyR α3 subunit (α3 GlyR) but not α1 GlyR, by a 7-day intrathecal injection of small interfering RNA (siRNA) targeting α3 GlyR or α1 GlyR, nearly completely prevented gelsemine-induced antinociception in neuropathic pain. Our results demonstrate that gelsemine produces potent and specific antinociception in chronic pain states without induction of apparent tolerance. The results also suggest that gelsemine produces antinociception by activation of spinal α3 glycine receptors, and support the notion that spinal α3 glycine receptors are a potential therapeutic target molecule for the management of chronic pain.
Co-reporter:Teng-Fei Li, Hui Fan, Yong-Xiang Wang
The Journal of Pain (May 2016) Volume 17(Issue 5) pp:530-548
Publication Date(Web):1 May 2016
DOI:10.1016/j.jpain.2015.12.015
•Bulleyaconitine A (BAA) attenuates neuropathic, bone cancer, and formalin pain.•BAA does not induce antinociceptive tolerance.•Minocycline blocks BAA antiallodynia but not neurotoxicity.•Bulleyaconitine A stimulates dynorphin A expression in spinal microglia.•Dynorphin A antisera and κ-opioid receptor antagonist eliminate BAA antiallodynia.Aconitine and its structurally-related diterpenoid alkaloids have been shown to interact differentially with neuronal voltage-dependent sodium channels, which was suggested to be responsible for their analgesia and toxicity. Bulleyaconitine A (BAA) is an aconitine analogue and has been prescribed for the management of pain. The present study aimed to evaluate the inhibitory effects of BAA on pain hypersensitivity and morphine antinociceptive tolerance, and explore whether the expression of dynorphin A in spinal microglia was responsible for its actions. Single intrathecal or subcutaneous (but not intraventricular or local) injection of BAA blocked spinal nerve ligation-induced painful neuropathy, bone cancer-induced pain, and formalin-induced tonic pain by 60 to 100% with the median effective dose values of 94 to 126 ng per rat (intrathecal) and 42 to 59 μg/kg (subcutaneous), respectively. After chronic treatment, BAA did not induce either self-tolerance to antinociception or cross-tolerance to morphine antinociception, and completely inhibited morphine tolerance. The microglial inhibitor minocycline entirely blocked spinal BAA (but not exogenous dynorphin A) antinociception, but failed to attenuate spinal BAA neurotoxicity. In a minocycline-sensitive and lidocaine- or ropivacaine-insensitive manner, BAA stimulated the expression of dynorphin A in the spinal cord, and primary cultures of microglia but not of neurons or astrocytes. The blockade effects of BAA on nociception and morphine tolerance were totally eliminated by the specific dynorphin A antiserum and/or κ-opioid receptor antagonist. Our results suggest that BAA eliminates pain hypersensitivity and morphine tolerance through directly stimulating dynorphin A expression in spinal microglia, which is not dependent on the interactions with sodium channels.PerspectiveThe newly illustrated mechanisms underlying BAA antinociception help us to better understand and develop novel dynorphin A expression-based painkillers to treat chronic pain.Download high-res image (100KB)Download full-size image
Co-reporter:Hui Fan, Teng-Fei Li, Nian Gong, Yong-Xiang Wang
Neuropharmacology (February 2016) Volume 101() pp:98-109
Publication Date(Web):1 February 2016
DOI:10.1016/j.neuropharm.2015.09.010
•Lamiophlomis rotata is an analgesic herb, and SM is its principle effective iridoid glycoside.•Intrathecal SM produced anti-allodynia in neuropathic rats without inducing tolerance.•SM stimulated β-endorphin expression in spinal microglia and produced anti-allodynia.•p38 MAPK mediated SM-induced β-endorphin expression and anti-allodynia.Lamiophlomis rotata (L. rotata, Duyiwei) is an orally available Tibetan analgesic herb widely prescribed in China. Shanzhiside methylester (SM) is a principle effective iridoid glycoside of L. rotata and serves as a small molecule glucagon-like peptide-1 (GLP-1) receptor agonist. This study aims to evaluate the signal mechanisms underlying SM anti-allodynia, determine the ability of SM to induce anti-allodynic tolerance, and illustrate the interactions between SM and morphine, or SM and β-endorphin, in anti-allodynia and anti-allodynic tolerance. Intrathecal SM exerted dose-dependent and long-lasting (>4 h) anti-allodynic effects in spinal nerve injury-induced neuropathic rats, with a maximal inhibition of 49% and a projected ED50 of 40.4 μg. SM and the peptidic GLP-1 receptor agonist exenatide treatments over 7 days did not induce self-tolerance to anti-allodynia or cross-tolerance to morphine or β-endorphin. In contrast, morphine and β-endorphin induced self-tolerance and cross-tolerance to SM and exenatide. In the spinal dorsal horn and primary microglia, SM significantly evoked β-endorphin expression, which was completely prevented by the microglial inhibitor minocycline and p38 mitogen-activated protein kinase (MAPK) inhibitor SB203580. SM anti-allodynia was totally inhibited by the GLP-1 receptor antagonist exendin(9–39), minocycline, β-endorphin antiserum, μ-opioid receptor antagonist CTAP, and SB203580. SM and exenatide specifically activated spinal p38 MAPK phosphorylation. These results indicate that SM reduces neuropathic pain by activating spinal GLP-1 receptors and subsequently stimulating microglial β-endorphin expression via the p38 MAPK signaling. Stimulation of the endogenous β-endorphin expression may be a novel and effective strategy for the discovery and development of analgesics for the long-term treatment of chronic pain.
Co-reporter:Qian Huang, Yuan Chen, Nian Gong, Yong-Xiang Wang
Metabolism (April 2016) Volume 65(Issue 4) pp:463-474
Publication Date(Web):1 April 2016
DOI:10.1016/j.metabol.2015.12.002
ObjectiveMethylglyoxal is known to be associated with the development of nephropathy, retinopathy, and other complications in diabetes. The present study tested the hypothesis that endogenously increased levels of methylglyoxal in diabetes are causally associated with the induction of neuropathic pain.Materials and methodsStreptozotocin- and methylglyoxal-induced pain models were established in rats, and the anti-nociceptive effects of the methylglyoxal scavenging agents, selective transient receptor potential channel ankyrin 1 (TRPA1) antagonist, and Nav1.8 antagonist were tested.ResultsSystemic injection of streptozotocin in rats induced a prolonged increase in plasma methylglyoxal by approximately 60%, which was correlated with the progressive development of mechanical allodynia and thermal hyperalgesia. Local subcutaneous injection of methylglyoxal into the hindpaw produced dose-dependent and biphasic flinching nociceptive responses, which resembled formaldehyde (formalin)-induced nociception. The local methylglyoxal nociception was significantly blocked by co-injection into the hindpaw of the selective transient receptor potential channel ankyrin 1 (TRPA1) antagonist, A967079, and the Nav1.8 antagonist, A803467. Co-incubation with the methylglyoxal scavengers, aminoguanidine, d-arginine, and metformin, reduced the level of free methylglyoxal by more than 90%, and injection of their incubation solutions into the hindpaw produced negligible (3–17%) nociception. Like the clinically effective anti-diabetic neuropathic pain drug gabapentin, systemic injection of aminoguanidine, d-arginine, and metformin at doses that effectively inhibit paw-injected methylglyoxal-induced nociception significantly blocked streptozotocin-induced mechanical allodynia.ConclusionEndogenously increased methylglyoxal may mediate diabetic neuropathic pain via activation of both TRPA1 and Nav1.8 expressed on primary afferent sensory neurons, and injection of methylglyoxal into the hindpaw may serve as a simple and robust model for testing the anti-diabetic pain drugs.Download high-res image (92KB)Download full-size image
Co-reporter:Hong Wei, Hai-Yun Wu, Hui Fan, Teng-Fei Li, ... Antti Pertovaara
Pharmacological Reports (April 2016) Volume 68(Issue 2) pp:472-475
Publication Date(Web):1 April 2016
DOI:10.1016/j.pharep.2015.11.008
BackgroundProlonged morphine treatment leads to antinociceptive tolerance. Suppression of spinal astrocytes or d-amino acid oxidase (DAAO), an astroglial enzyme catalyzing oxidation of d-amino acids, has reversed morphine antinociceptive tolerance. Since the astrocyte-DAAO pathway generates hydrogen peroxide, an agonist of the TRPA1 channel expressed spinally on nociceptive nerve terminals and astrocytes, we tested a hypothesis that the spinal TRPA1 contributes to antinociceptive tolerance to prolonged spinal morphine treatment.MethodsNociception was assessed using hot-plate test in rats with an intrathecal (it) catheter. Drugs were administered it twice daily from day one to seven in five treatment groups: (i) Saline, (ii) Chembridge-5861528 (a TRPA1 antagonist; 10 μg), (iii) morphine (10 μg), (iv) Chembridge-5861528 (10 μg) + morphine (10 μg), (v) DMSO. Antinociceptive action of morphine was assessed at day one and eight. Additionally, mRNA for DAAO and TRPA1 in the spinal cord was determined on day 8.ResultsMorphine treatment produced antinociceptive tolerance, which was attenuated by co-administration of Chembridge-5861528 that alone had no effect on hot-plate latencies. In animals treated with morphine only, spinal mRNA for DAAO but not TRPA1 was increased. DAAO increase was prevented by co-administration of Chembridge-5861528.ConclusionsAntinociceptive morphine tolerance and up-regulation of spinal DAAO were attenuated in morphine-treated animals by blocking the spinal TRPA1. This finding suggests that spinal TRPA1 may contribute, at least partly, to facilitation of morphine antinociceptive tolerance through mechanisms that possibly involve TRPA1-mediated up-regulation of the astroglial DAAO, a generator of hydrogen peroxide, a pronociceptive compound acting also on TRPA1.
Co-reporter:Wenbin Zhang, Ajith Welihinda, Jordan Mechanic, Haifeng Ding, Liangcheng Zhu, Yuan Lu, Zhongping Deng, Zelin Sheng, Binhua Lv, Yuanwei Chen, Jacques Y. Roberge, Brian Seed, Yong-Xiang Wang
Pharmacological Research (April 2011) Volume 63(Issue 4) pp:284-293
Publication Date(Web):1 April 2011
DOI:10.1016/j.phrs.2011.01.001
Sodium glucose co-transporter 2 (SGLT2) is a renal type III integral membrane protein that co-transports sodium and glucose from filtrate to epithelium in the proximal tubule. Human subjects with homozygous or compound heterozygous mutations in SLC5A2 exhibit glucosuria without hypoglycemia or other obvious morbidity, suggesting that blockade of SGLT2 has the potential to promote normalization of blood glucose without hypoglycemia in the setting of type 2 diabetes. This report presents the in vitro and in vivo pharmacological activities of EGT1442, a recently discovered SGLT2 inhibitor in the C-aryl glucoside class.The inhibitory effects of EGT1442 for human SGLT1 and SGLT2 were evaluated in an AMG uptake assay and the in vivo efficacy of treatment with EGT1442 was investigated in rats and dogs after a single dose and in db/db mice after chronic administration. The effect of EGT1442 on median survival of SHRSP rats was also evaluated.The IC50 values for EGT1442 against human SGLT1 and SGLT2 are 5.6 μM and 2 nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED50 of 0.38 and 0.09 mg/kg, respectively. Following chronic administration to db/db mice, EGT1442 dose-dependently reduced HbA1c and blood glucose concentration without affecting body mass or insulin level. Additionally, EGT1442 significantly prolonged the median survival of SHRSP rats.EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.Download full-size image
Co-reporter:Qian Huang, Ming-Li Sun, Yuan Chen, Xin-Yan Li, Yong-Xiang Wang
Journal of Ethnopharmacology (20 January 2017) Volume 196() pp:151-159
Publication Date(Web):20 January 2017
DOI:10.1016/j.jep.2016.12.027
•Acute bullatine A and dynorphin A additively enhance morphine antinociception.•Chronic bullatine A and dynorphin A induce no antinociceptive tolerance.•Concurrent bullatine A and dynorphin A inhibit morphine antinociceptive tolerance.•Bullatine A antinociception/anti-tolerance are mediated by spinal microglial dynorphin secretion.•Our data provide basis for concurrent use of aconitines and morphine in analgesia.Ethnopharmacological relevanceBullatine A, a C20-diterpenoid alkaloid and one of the major effective ingredients in Aconiti brachypodi Radix (Xue-shang-yi-zhi-hao), can block pain hypersensitivity in a variety of rodent models through expression of spinal microglial dynorphin A.Aim of the studyTo assess the interaction between bullatine A and morphine on antinociception in acute nociception and pain hypersensitivity states, with the exogenous synthetic dynorphin A as a comparisonMaterials and methodsSpinal nerve ligation-induced neuropathic rats and naïve mice were used for assessing the acute and chronic interactions of bullatine A/dynorphin A with morphine.ResultsSingle subcutaneous injection of bullatine A or dynorphin A(1−17) did not either alter formalin- and thermally (hot-plate and water immersion tests)-induced acute nociception or potentiate morphine antinociception in naïve mice. In contrast, bullatine A dose-dependently inhibited formalin-induced tonic pain with the efficacy of 54% inhibition and the half-effective dose of 0.9 mg/kg. Concurrent bullatine A additively enhanced morphine antinociception. In neuropathic rats, the antinociceptive effects of multiple bidaily intrathecal injections of bullatine A and dynorphin A remained consistent over 13 days, whereas morphine produced progressive and complete tolerance to antinociception, which was completely inhibited by concurrent bullatine A and dynorphin A. A single intrathecal injection of bullatine A and dynorphin A immediately reversed established morphine tolerance in neuropathic rats, although the blockade was a less degree in the thermally induced mouse acute nociceptive tests. The inhibitory effects of bullatine A and dynorphin A on morphine tolerance were immediately and completely attenuated by intrathecal dynorphin A antibody and/or selective κ-opioid receptor antagonist GNTI.ConclusionThese results suggest that bullatine A produces antinociception without induction of tolerance and inhibits morphine antinociceptive tolerance, and provide pharmacological basis for concurrent bullatine A and morphine treatment for chronic pain and morphine analgesic tolerance.Download high-res image (245KB)Download full-size image

![Pyrido[2,3-b]pyrazine-2,3-dione,1,4-dihydro-8-methyl-](http://img.cochemist.com/ccimg/144500/144435-01-8.png)
![Pyrido[2,3-b]pyrazine-2,3-dione,1,4-dihydro-8-methyl-](http://img.cochemist.com/ccimg/144500/144435-01-8_b.png)









![3-Hydroxy-2H-benzo[b][1,4]oxazin-2-one](http://img.cochemist.com/ccimg/3600/3597-63-5.png)
![3-Hydroxy-2H-benzo[b][1,4]oxazin-2-one](http://img.cochemist.com/ccimg/3600/3597-63-5_b.png)