Co-reporter:Yu-Chen Lo, Silvia Senese, Robert Damoiseaux, and Jorge Z. Torres
ACS Chemical Biology 2016 Volume 11(Issue 8) pp:2244
Publication Date(Web):June 10, 2016
DOI:10.1021/acschembio.6b00253
Target identification remains a major challenge for modern drug discovery programs aimed at understanding the molecular mechanisms of drugs. Computational target prediction approaches like 2D chemical similarity searches have been widely used but are limited to structures sharing high chemical similarity. Here, we present a new computational approach called chemical similarity network analysis pull-down 3D (CSNAP3D) that combines 3D chemical similarity metrics and network algorithms for structure-based drug target profiling, ligand deorphanization, and automated identification of scaffold hopping compounds. In conjunction with 2D chemical similarity fingerprints, CSNAP3D achieved a >95% success rate in correctly predicting the drug targets of 206 known drugs. Significant improvement in target prediction was observed for HIV reverse transcriptase (HIVRT) compounds, which consist of diverse scaffold hopping compounds targeting the nucleotidyltransferase binding site. CSNAP3D was further applied to a set of antimitotic compounds identified in a cell-based chemical screen and identified novel small molecules that share a pharmacophore with Taxol and display a Taxol-like mechanism of action, which were validated experimentally using in vitro microtubule polymerization assays and cell-based assays.
Co-reporter:S Senese, Y C Lo, D Huang, T A Zangle, A A Gholkar, L Robert, B Homet, A Ribas, M K Summers, M A Teitell, R Damoiseaux and J Z Torres
Cell Death & Disease 2014 5(10) pp:e1462
Publication Date(Web):2014-10-01
DOI:10.1038/cddis.2014.420
Cancer cell proliferation relies on the ability of cancer cells to grow, transition through the cell cycle, and divide. To identify novel chemical probes for dissecting the mechanisms governing cell cycle progression and cell division, and for developing new anti-cancer therapeutics, we developed and performed a novel cancer cell-based high-throughput chemical screen for cell cycle modulators. This approach identified novel G1, S, G2, and M-phase specific inhibitors with drug-like properties and diverse chemotypes likely targeting a broad array of processes. We further characterized the M-phase inhibitors and highlight the most potent M-phase inhibitor MI-181, which targets tubulin, inhibits tubulin polymerization, activates the spindle assembly checkpoint, arrests cells in mitosis, and triggers a fast apoptotic cell death. Importantly, MI-181 has broad anti-cancer activity, especially against BRAFV600E melanomas.
Co-reporter:Sugunadevi Sakkiah;Mahreen Arooj;Keun Woo Lee
Medicinal Chemistry Research 2014 Volume 23( Issue 9) pp:3998-4010
Publication Date(Web):2014 September
DOI:10.1007/s00044-014-0983-3
Bayesian and pharmacophore modeling approaches were utilized to identify the fragments and critical chemical features of small molecules that enhance sirtuin1 (SIRT1) activity. Initially, 48 Bayesian models (BMs) were developed by exploring 12 different fingerprints (ECFC, ECFP, EPFC, EPFP, FPFC, FPFP, FCFC, FCFP, LCFC, LCFP, LPFC, and LPLP) with diameters of 4, 6, 8, and 10. Among them the BM1 model was selected as the best model based on its good statistical parameters including total accuracy: 0.98 and positive recalls: 0.95. Additionally, BM1 showed good predictive power for the test set (total accuracy: 0.87 and positive recall: 0.87). In addition, 10 qualitative pharmacophore models were generated using 6 well-known SIRT1 activators. Hypothesis2 (Hypo2) was selected as best hypothesis, among 10 Hypos, based on its discriminant ability between the highly active and least/moderately active SIRT1 activators. The best models, BM1 and Hypo2 were used as a query in virtual screens of a drug-like database and the hit molecules were sorted based on Bayesian score and fit value, respectively. In addition, the highest occupied molecular orbital, lowest unoccupied molecular orbital, and energy gap values were calculated for the selected virtual screening hits using density functional theory. Finally, 16 compounds were selected as leads based on their energy gap values, which represent the high reactivity of molecules. Thus, our results indicated that the combination of two-dimensional (2D) and 3D approaches are useful for the discovery and development of specific and potent SIRT1 activators, and will benefit medicinal chemists focused on designing novel lead
compounds that activate SIRT1.
.jpg)
![N-(2-(3-(Piperazin-1-ylmethyl)imidazo[2,1-b]thiazol-6-yl)phenyl)quinoxaline-2-carboxamide](http://img.cochemist.com/ccimg/925500/925434-55-5.png)
![N-(2-(3-(Piperazin-1-ylmethyl)imidazo[2,1-b]thiazol-6-yl)phenyl)quinoxaline-2-carboxamide](http://img.cochemist.com/ccimg/925500/925434-55-5_b.png)




![Benzamide, N-[2-(1H-benzimidazol-2-yl)phenyl]-3,4-dimethoxy-](http://img.cochemist.com/ccimg/925500/925432-19-5.png)
![Benzamide, N-[2-(1H-benzimidazol-2-yl)phenyl]-3,4-dimethoxy-](http://img.cochemist.com/ccimg/925500/925432-19-5_b.png)
![Benzamide, 3,4-dimethoxy-N-(2-oxazolo[4,5-b]pyridin-2-ylphenyl)-](http://img.cochemist.com/ccimg/925500/925432-07-1.png)
![Benzamide, 3,4-dimethoxy-N-(2-oxazolo[4,5-b]pyridin-2-ylphenyl)-](http://img.cochemist.com/ccimg/925500/925432-07-1_b.png)






















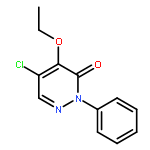
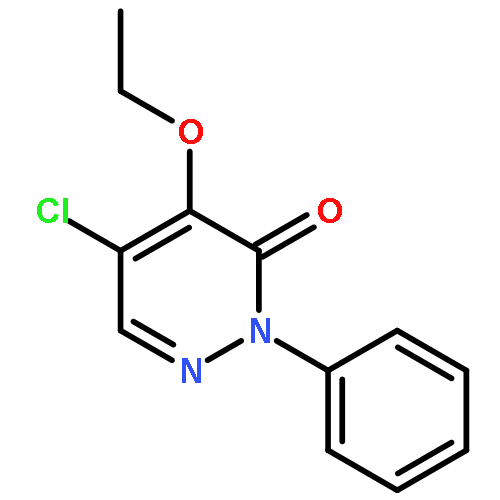


![3,6-Dimethylimidazo[2,1-b]thiazole](http://img.cochemist.com/ccimg/26000/25944-60-9.png)
![3,6-Dimethylimidazo[2,1-b]thiazole](http://img.cochemist.com/ccimg/26000/25944-60-9_b.png)



