Co-reporter:Pinrao Song, Ming Chen, Xiaodong Ma, Lei Xu, Tao Liu, Yubo Zhou, Yongzhou Hu
Bioorganic & Medicinal Chemistry 2015 23(8) pp: 1858-1868
Publication Date(Web):
DOI:10.1016/j.bmc.2015.02.004
Co-reporter:Suwen Hu, Zhilong Wang, Tingjun Hou, Xiaodong Ma, Jing Li, Tao Liu, Xin Xie, Yongzhou Hu
Bioorganic & Medicinal Chemistry 2015 23(5) pp: 1157-1168
Publication Date(Web):
DOI:10.1016/j.bmc.2014.12.052
Co-reporter:Suwen Hu, Quan Gu, Zhilong Wang, Zhiyong Weng, Yunrui Cai, Xiaowu Dong, Yongzhou Hu, Tao Liu, Xin Xie
European Journal of Medicinal Chemistry 2014 Volume 71() pp:259-266
Publication Date(Web):7 January 2014
DOI:10.1016/j.ejmech.2013.11.013
•We designed and synthesized a series of novel piperidine-4-carboxamide derivatives.•Two compounds showed similar activity to that of the positive control based on calcium mobilization assay.•Two compounds displayed nanomolar activity by HIV-1 single cycle antiviral assay.•The pharmacokinetic properties and cardiovascular safety (hERG) of 16g were evaluated.Based on a putative ‘Y shape’ pharmacophore model of CCR5 inhibitors, a series of novel piperidine-4-carboxamide derivatives were designed and synthesized using a group-reverse strategy. Among synthesized target compounds, 16g (IC50 = 25.73 nM) and 16i (IC50 = 25.53 nM) showed equivalent inhibitory activity against CCR5 to that of the positive control maraviroc (IC50 = 25.43 nM) in calcium mobilization assay. Selected compounds were further tested for their antiviral activity in HIV-1 single cycle assay. Two compounds, 16g and 16i, displayed antiviral activity with IC50 values of 73.01 nM and 94.10 nM, respectively. Additionally, the pharmacokinetic properties and inhibitory potency against hERG of 16g were evaluated, providing a foundation for ongoing optimization.16g and 16i showed equivalent inhibitory activity to that of the positive control maraviroc.
Co-reporter:Wenhu Zhan, Daqiang Li, Jinxin Che, Liangren Zhang, Bo Yang, Yongzhou Hu, Tao Liu, Xiaowu Dong
European Journal of Medicinal Chemistry 2014 Volume 75() pp:11-20
Publication Date(Web):21 March 2014
DOI:10.1016/j.ejmech.2014.01.019
•A novel molecular docking based QSAR model for Akt1 inhibitors was established.•Docking scores, key interaction profiles and molecular descriptors were integrated.•Six 4-aminopyrimidine derivatives were designed, synthesized and biological evaluated.•The application value of MD-SVR model was proved, and potent Akt1 inhibitors were identified.A set of forty-seven Akt1 inhibitors was used for the development of molecular docking based QSAR model by using nonlinear regression. The integration of docking scores, key interaction profiles and molecular descriptors remarkably improved the accuracy of the QSAR models, providing reasonable statistical parameters (Rtrain2 = 0.948, Rtest2 = 0.907 and Qcv2 = 0.794). The established MD-SVR model based structural modification of new 4-amino-pyrimidine derivatives was further performed, and six compounds 56a,b and 60a–d with good prediction activities were synthesized and biologically evaluated. All of these compounds exhibited promising Akt1 inhibitory and antiproliferative activities, suggesting the reliability and good application value of the established MD-SVR model in the development of Akt1 inhibitors.A novel molecular docking based QSAR model (MD-SVR) for Akt1 inhibitor was constructed and applied in the development of 4-amino-pyrimidines as novel Akt1 inhibitors.
Co-reporter:Jiankang Zhang;Mengmeng Han;Xiaodong Ma;Lei Xu;Jiayi Cao;Yubo Zhou;Jia Li;Yongzhou Hu
Chemical Biology & Drug Design 2014 Volume 84( Issue 5) pp:497-504
Publication Date(Web):
DOI:10.1111/cbdd.12342
A series of novel di- and tripeptidyl epoxyketone derivatives composed of β-amino acids were designed, synthesized and evaluated for their proteasome inhibitory activities and anti-proliferation activities against two multiple myeloma cell lines RPMI 8226 and NCI-H929 and normal cells (peripheral blood mononucleated cells). Among these tested compounds, tripeptidyl analogues showed much more potent activities than dipeptides, and four tripeptidyl compounds exhibited proteasome inhibitory activities with IC50 values ranging from 0.97 ± 0.05 to 1.85 ± 0.11 μm. In addition, all the four compounds showed anti-proliferation activities with IC50 values at low micromolar levels against two multiple myeloma cell lines and weak activities against normal cells. Furthermore, Western blot analysis was performed to verify the proteasome inhibition induced by compounds 21d and 21e. All the experimental results validated that the β-amino acid building block has the potential for the development of proteasome inhibitors.
Co-reporter:Pinrao Song, Peng Peng, Mengmeng Han, Xianchao Cao, Xiaodong Ma, Tao Liu, Yubo Zhou, Yongzhou Hu
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 17) pp:4882-4892
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmc.2014.06.044
A series of thienopyridinone derivatives was designed and synthesized as inhibitors of checkpoint kinase 1 (Chk1). Most of them exhibited moderate to good Chk1 inhibitory activities. Among them, compounds 8q, 8t, and 8w with excellent Chk1 inhibitory activities (IC50 values of 4.05, 6.23, and 2.33 nM, respectively) displayed strong synergistic effects with melphalan, a DNA-damaging agent in the cell-based assay. Further kinase profiling indicated that compound 8t was highly selective against CDK2/cyclinA, Aurora A, and PKC.
Co-reporter:Xiaowu Dong, Yanmei Zhao, Xueqin Huang, Kana Lin, Jianzhong Chen, Erqing Wei, Tao Liu, Yongzhou Hu
European Journal of Medicinal Chemistry 2013 Volume 62() pp:754-763
Publication Date(Web):April 2013
DOI:10.1016/j.ejmech.2013.01.041
3D structure of CysLT2 receptor was constructed by using homology modeling and molecular simulations. The binding pocket of CysLT2 receptor and the proposition of the interaction mode between CysLT2 and HAMI3379 were identified. A series of dicarboxylated chalcones was then virtually evaluated through molecular docking studies. A total of six compounds 13a–f with preferable scores was further synthesized and tested for CysLT2 antagonistic activities by determination of the cytosolic free Ca2+ levels in HEK293 cells. Compounds 13e and 13f exhibited potent and selective CysLT2 antagonistic activities with IC50 values being 7.5 and 0.25 μM, respectively.Graphical abstract3D structure of CysLT2 was constructed and applied in the development of dicarboxylated chalcones as novel CysLT2 antagonists. 13e and 13f were identified as potent and selective CysLT2 antagonists.Highlights► 3D structure of CysLT2 receptor was firstly constructed. ► The interaction mode proposed would be useful and valuable. ► A total of six compounds was designed, synthesized and biological evaluated. ► Dicarboxylated chalcone is novel skeleton with CysLT2 antagonistic activities.
Co-reporter:Yang Liu;Yizhe Wu;Haoshu Wu;Li Tang;Peng Wu;Yongzhou Hu
Chemical Biology & Drug Design 2013 Volume 82( Issue 2) pp:140-146
Publication Date(Web):
DOI:10.1111/cbdd.12139
A novel series of (S)-phenylalanine derivatives with a 2-cyanopyrrolidine moiety were designed and synthesized through a rational drug design strategy. Biological evaluation revealed that most tested compounds were potent dipeptidyl peptidase 4 (DPP-4) inhibitors; among them, the cyclopropyl-substituted phenylalanine derivative 11h displayed the most potent DPP-4 inhibitory activity with an IC50 value of 0.247 μm. In addition, molecular docking analysis of the representative compounds 11h, 11k, and 15a were performed, which not only revealed the impact of binding modes on DPP-4 inhibitory activity but also provided additional methodological values for design and optimization.
Co-reporter:Yang Liu, Meimei Si, Li Tang, Shihao Shangguan, Haoshu Wu, Jia Li, Peng Wu, Xiaodong Ma, Tao Liu, Yongzhou Hu
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 18) pp:5679-5687
Publication Date(Web):15 September 2013
DOI:10.1016/j.bmc.2013.07.034
A series of novel benzyl-substituted (S)-phenylalanine derivatives were synthesized and evaluated for their dipeptidyl peptidase 4 (DPP-4) inhibitory activity and selectivity. It was found that most synthesized target compounds were potent DPP-4 inhibitors with IC50 values in 3.79–25.52 nM, which were significantly superior to that of the marketed drug sitagliptin. Furthermore, the 4-fluorobenzyl substituted phenylalanine derivative 6g not only displayed the potent DPP-4 inhibition with an IC50 value of 3.79 nM, but also showed better selectivity against DPP-4 over other related enzymes including DPP-7, DPP-8, and DPP-9. In an oral glucose tolerance test (OGTT) in normal Sprague Dawley rats, compound 6g reduced blood glucose excursion in a dose-dependent manner.Novel benzyl-substituted (S)-phenylalanine derivatives 6a and 6g showed potent DPP-4 inhibitory activity with IC50 values of 9.39 and 3.79 nM, respectively, which was significantly superior to that of lead compound 1.
Co-reporter:Xiaowu Dong, Jingying Yan, Lilin Du, Peng Wu, Shufang Huang, Tao Liu, Yongzhou Hu
Journal of Molecular Graphics and Modelling 2012 Volume 37() pp:77-86
Publication Date(Web):July 2012
DOI:10.1016/j.jmgm.2012.04.003
Pharmacophore models of cyclin-dependent kinase-1 (CDK1) inhibitors were established by using the Catalyst/HypoGen. The best pharmacophore model, Hypo1, consists of one hydrogen bond acceptor (HBA), one hydrogen bond donor (HBD), one hydrophobic (HY) and one ring aromatic (RA) feature. The validation results of Hypo1 through cost analysis, test set prediction, Fisher's cross method and receiver operating characteristic (ROC) study indicated that the Hypo1 was statistically valuable and reliable in identifying structural diverse CDK1 inhibitors. It is further supported by the consistent results from molecular docking studies. Finally, the Hypo1 was used to “in silico” screen the NCI and MayBridge database. The preferable hits obtained were further docked into ATP binding site of CDK1, and nine promising compounds were retrieved as novel potential CDK1 inhibitors for further studies.Graphical abstractPharmacophore model (Hypo1) of CDK1 inhibitors was established and extensively validated. Further “in silico” screening retrieved nine compounds as potential CDK1 inhibitors.Highlights► We established a pharmacophore model of CDK1 inhibitors using HypoGen program. ► Pharmacophore-based virtual screening combined with docking studies were performed. ► Several potential CDK1 inhibitors with different skeleton were identified.
Co-reporter:Zhiyong Weng;Wei Wei;Xiaowu Dong
Monatshefte für Chemie - Chemical Monthly 2012 Volume 143( Issue 2) pp:303-308
Publication Date(Web):2012 February
DOI:10.1007/s00706-011-0645-9
A series of novel piperidin-4-ol derivatives were designed, synthesized, and evaluated for potential treatment of HIV. The compounds were obtained via an efficient synthetic route in excellent yields and have been characterized by 1H NMR, 13C NMR, MS, and elemental analysis. The CCR5 antagonistic activities of the compounds have also been evaluated.

![Spiro[3H-indole-3,4'-piperidine],1,2-dihydro-1-(5-methyl-1H-pyrrolo[2,3-d]pyrimidin-4-yl)-](http://img.cochemist.com/ccimg/908300/908280-59-1.png)
![Spiro[3H-indole-3,4'-piperidine],1,2-dihydro-1-(5-methyl-1H-pyrrolo[2,3-d]pyrimidin-4-yl)-](http://img.cochemist.com/ccimg/908300/908280-59-1_b.png)



![Spiro[3H-indole-3,4'-piperidine],1,2-dihydro-1-(1H-pyrrolo[2,3-d]pyrimidin-4-yl)-](http://img.cochemist.com/ccimg/908300/908280-90-0.png)
![Spiro[3H-indole-3,4'-piperidine],1,2-dihydro-1-(1H-pyrrolo[2,3-d]pyrimidin-4-yl)-](http://img.cochemist.com/ccimg/908300/908280-90-0_b.png)
![2H-THIENO[2,3-D][1,3]OXAZINE-2,4(1H)-DIONE, 6-METHYL-](http://img.cochemist.com/ccimg/877200/877142-62-6.png)
![2H-THIENO[2,3-D][1,3]OXAZINE-2,4(1H)-DIONE, 6-METHYL-](http://img.cochemist.com/ccimg/877200/877142-62-6_b.png)
![3-(carbamoylamino)-5-(3-fluorophenyl)-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide](http://img.cochemist.com/ccimg/860400/860352-01-8.png)
![3-(carbamoylamino)-5-(3-fluorophenyl)-N-[(3S)-piperidin-3-yl]thiophene-2-carboxamide](http://img.cochemist.com/ccimg/860400/860352-01-8_b.png)
![4-[(3-methyl-1-piperazinyl)methyl]-Benzonitrile](http://img.cochemist.com/ccimg/685600/685536-08-7.png)
![4-[(3-methyl-1-piperazinyl)methyl]-Benzonitrile](http://img.cochemist.com/ccimg/685600/685536-08-7_b.png)
![1-[(3-fluorophenyl)methyl]-3-methyl-Piperazine](http://img.cochemist.com/ccimg/685600/685536-00-9.png)
![1-[(3-fluorophenyl)methyl]-3-methyl-Piperazine](http://img.cochemist.com/ccimg/685600/685536-00-9_b.png)






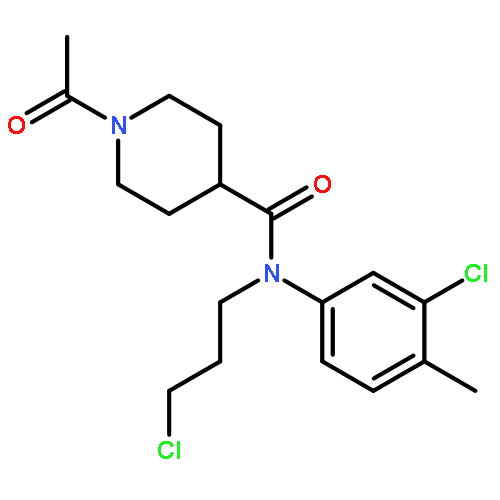
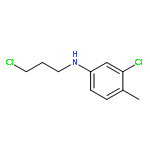
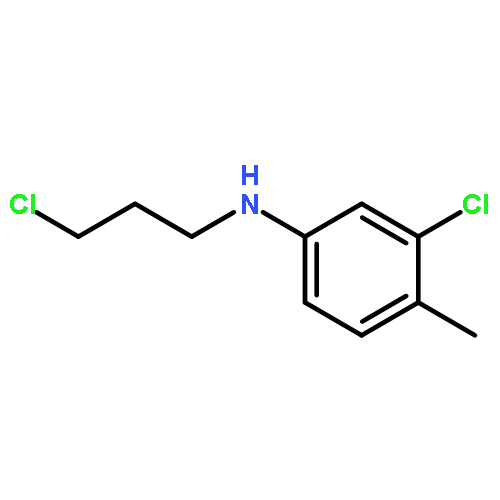
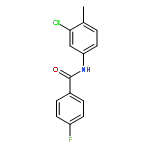
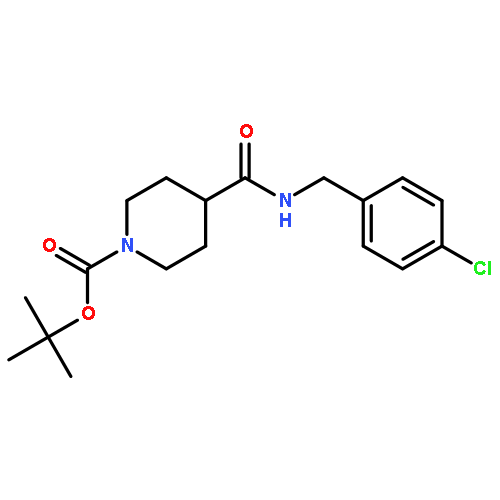
![N-[(4-fluorophenyl)methyl]piperidine-4-carboxamide](http://img.cochemist.com/ccimg/327100/327048-64-6.png)
![N-[(4-fluorophenyl)methyl]piperidine-4-carboxamide](http://img.cochemist.com/ccimg/327100/327048-64-6_b.png)


![1-[(4-fluorophenyl)methyl]-3-methyl-Piperazine](http://img.cochemist.com/ccimg/178700/178624-87-8.png)
![1-[(4-fluorophenyl)methyl]-3-methyl-Piperazine](http://img.cochemist.com/ccimg/178700/178624-87-8_b.png)






![Benzonitrile, 4-[(ethylamino)methyl]-](http://img.cochemist.com/ccimg/133100/133042-86-1.png)
![Benzonitrile, 4-[(ethylamino)methyl]-](http://img.cochemist.com/ccimg/133100/133042-86-1_b.png)
![(1S)-2,5-Diazabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/132400/132339-20-9.png)
![(1S)-2,5-Diazabicyclo[2.2.1]heptane](http://img.cochemist.com/ccimg/132400/132339-20-9_b.png)
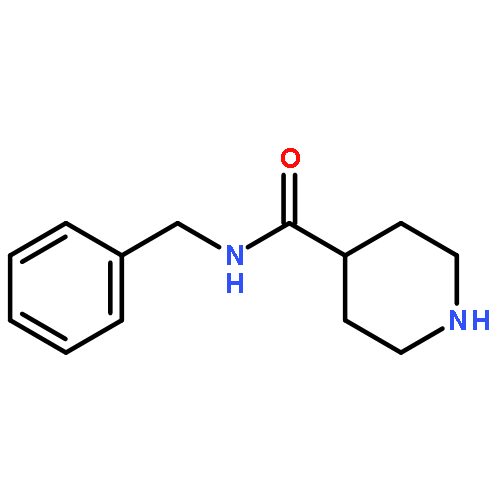


![1,4-DIHYDRO-2H-THIENO[3,2-D][1,3]OXAZINE-2,4-DIONE](http://img.cochemist.com/ccimg/78800/78756-28-2.png)
![1,4-DIHYDRO-2H-THIENO[3,2-D][1,3]OXAZINE-2,4-DIONE](http://img.cochemist.com/ccimg/78800/78756-28-2_b.png)
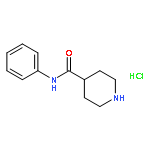
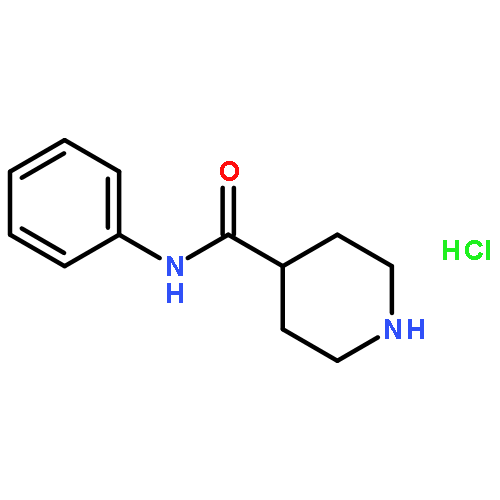
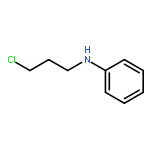













![Tert-butyl N-[(2s)-4-methyl-1-[(2r)-2-methyloxiran-2-yl]-1-oxopentan-2-yl]carbamate](http://img.cochemist.com/ccimg/247100/247068-82-2.png)
![Tert-butyl N-[(2s)-4-methyl-1-[(2r)-2-methyloxiran-2-yl]-1-oxopentan-2-yl]carbamate](http://img.cochemist.com/ccimg/247100/247068-82-2_b.png)