Co-reporter:Benjamin G. Harvey
European Journal of Inorganic Chemistry 2017 Volume 2017(Issue 9) pp:1203-1203
Publication Date(Web):2017/03/03
DOI:10.1002/ejic.201700151
The cover picture shows at the top the first two recognized examples of C–C agostic interactions with a metal center. A stylized arrow demonstrates the progression from the recognition that C–C bonds could donate electron density to a metal center to the understanding that these interactions are present in intermediates of both the Ziegler–Natta and olefin metathesis catalytic cycles. The impact of these interactions is then highlighted by depicting some of the ubiquitous consumer products that are manufactured by use of these catalytic cycles. Details are presented in the Microreview by B. G. Harvey and R. D. Ernst on page 1205 ff (DOI: 10.1002/ejic.201600989). For more on the story behind the cover research, see the Cover Profile (DOI: 10.1002/ejic.201700152).
Co-reporter:Benjamin G. Harvey
European Journal of Inorganic Chemistry 2017 Volume 2017(Issue 9) pp:1204-1204
Publication Date(Web):2017/03/03
DOI:10.1002/ejic.201700152
AbstractInvited for the cover of this issue are Benjamin Harvey at the US Navy, China Lake, California, USA, and Richard Ernst at the University of Utah, Salt Lake City, USA. The cover image shows the first two recognized examples of C–C agostic interactions with a transition metal center and consumer products containing materials prepared by catalytic reactions that rely on these interactions.
Co-reporter:Benjamin G. Harvey
European Journal of Inorganic Chemistry 2017 Volume 2017(Issue 9) pp:1205-1226
Publication Date(Web):2017/03/03
DOI:10.1002/ejic.201600989
The coordination of sigma bonding electron density to a Lewis acid was for some time largely the domain of boron hydride chemistry. Subsequently it was found that C–H bonds could undergo intramolecular coordination to metal centers, forming what are now known as “agostic bonds” and “agostic complexes”. Thereafter, even the H–H bond in molecular hydrogen was found to be capable of coordination, leading to what are commonly referred to as “sigma complexes”. Eventually, coordination by much more shielded C–C bonds was established, and is of particular significance with regard to to the interest in C–C bond activation. This review focuses on the key developments in this field, including the nature of the bonding interactions in these and some related species, as revealed by spectroscopic, structural, and theoretical studies, their involvement in important chemical reactions such as alkene polymerizations and metathesis, and comparisons with the other types of electron-deficient bridge bonding.
Co-reporter:Rehan Basta;Atta M. Arif
Journal of Chemical Crystallography 2013 Volume 43( Issue 2) pp:91-95
Publication Date(Web):2013 February
DOI:10.1007/s10870-012-0389-3
The reactions of the α,ω-diimines (CH2)n[N=CH(C6H5)]2 with Zr(6,6-dmch)2(PMe3)2 (dmch = dimethylcyclohexadienyl) lead to couplings of both imine carbon atoms, each to one of a single dmch ligand’s termini, which thereby also leads to the formation of two Zr–N bonds, and release of the phosphine ligands. The resulting complexes possess 18 electron configurations by virtue of a remaining η5-6,6-dmch ligand, η3-allyl coordination by the coupled 6,6-dmch ligand, and π-amide coordination by each nitrogen donor center. The η5-6,6-dmch ligand displays the distortions expected for a high valent metal pentadienyl complex. The n = 3 complex crystallized in the monoclinic space group P21/c with a = 8.5687(2) Å, b = 27.8768(6) Å, c = 11.7747(3) Å, β = 103.6081(8)°, V = 2,733.64(11) Å3, Dcalc = 1.351 g/cm3 at 150(1) K. For the n = 4 complex, the triclinic space group P\( \overline{1} \) is adopted, with a = 11.1279(3) Å, b = 11.3846(3) Å, c = 14.9154(3) Å, α = 81.6030(13)°, β = 69.8503(13)°, γ = 84.6588(12)°, V = 1,753.05(7) Å3, Dcalc = 1.283 g cm−3 at 150(1) K.
Co-reporter:Yifan Shi;Atta M. Arif
Journal of Chemical Crystallography 2013 Volume 43( Issue 7) pp:360-364
Publication Date(Web):2013 July
DOI:10.1007/s10870-013-0428-8
The new [Co(pydz)6]2+ ion (pydz = pyridazine) has been prepared by a straightforward route in aqueous solution, and isolated as its hexafluorophosphate salt. The salt crystallizes in the monoclinic space group C2/c with a = 18.4796(3) Å, b = 10.6846(2) Å, c = 16.3404(2) Å, β = 92.0518(11)º, V = 3224.30(9) Å3, Dcalc = 1.709 at 150(1) K. The dication has crystallographically imposed 2 symmetry. The analogous, previously reported ruthenium complex ion could be isolated as its tetraphenylborate salt following the reaction of [RuCl2(1,5-COD)]x·(COD = cyclooctadiene) with pyridazine in the presence of hydrogen gas. This salt also crystallizes in the monoclinic space group C2/c, with a = 17.8441(3) Å, b = 19.8693(4) Å, c = 38.4545(7) Å, β = 95.4231(6)º, V = 13573.0(4) Å3, Dcalc = 1.321 at 150(1) K.
Co-reporter:Benjamin G. Harvey;Adam T. Nickel;Atta M. Arif
Journal of Chemical Crystallography 2011 Volume 41( Issue 10) pp:1438-1441
Publication Date(Web):2011 October
DOI:10.1007/s10870-011-0117-4
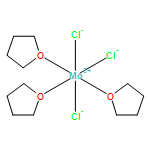
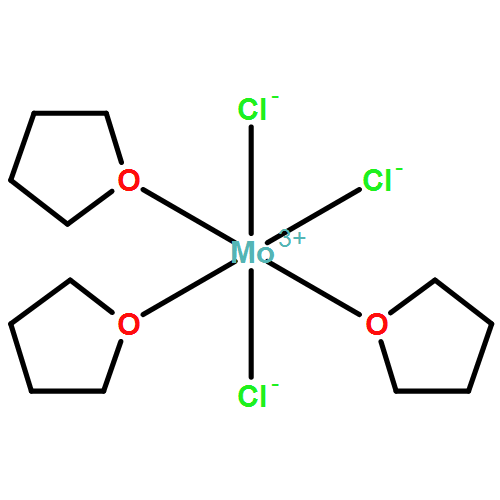
The structure of WCl2(PMe3)4 has been determined in order to allow for a comparison with its molybdenum analogue. The complexes are isomorphous, and exhibit trans-pseudooctahedral coordination, with crystallographically imposed \(\overline{4}2m\) (D2d) symmetry. A significant distortion occurs involving the PMe3 ligands, through which one opposing pair tilts upward toward one axial chloride ligand, while the other pair tilts in an opposite manner, by 9.33°. The complex crystallizes in the tetragonal space group \(I\overline{4}2m\), with a = b = 9.4987(4) Å, c = 12.1058(5) Å, V = 1092.25(8) Å3, Dcalc = 1.700 at 150(1) K. For a better comparison, the structure of the molybdenum complex was redetermined, at a low temperature. Its space group is also \(I\overline{4}2m\) with a = b = 9.5193(3) Å, c = 12.1083(5) Å, V = 1097.22 Å3, Dcalc = 1.426 at 150(1) K. In accord with the larger cell volume, the molybdenum complex possesses slightly longer M–Cl and M–P bonds.
Co-reporter:Rehan Basta;Atta M. Arif
Journal of Chemical Crystallography 2011 Volume 41( Issue 10) pp:
Publication Date(Web):2011 October
DOI:10.1007/s10870-011-0116-5
The reactions of Ti(C5H5)(6,6-dmch)(PMe3) and Zr(C5H5)(6,6-dmch)(PMe3)2 with two equivalents of C6H5C(H)=N(C6H5) have led to couplings of the imines to the 1,5 (terminal) positions of the 6,6-dmch ligands. In both cases the PMe3 ligands are lost, and the metal complexes each attain 18 electron configurations via η5-(C5H5), η3-allyl, and bis(π-amide) coordination. The complexes each crystallize in the monoclinic space group P21/c. For the titanium complex, a = 10.4336(1) Å, b = 9.0407(1) Å, c = 32.5547(6) Å, β = 94.2939(5)°, V = 3062.17(7) Å3, Dcalc = 1.264 at 200(1) K. For the zirconium complex, a = 13.8900(3) Å, b = 10.0873(2) Å, c = 22.4543(6) Å, β = 90.7638(7)°, V = 3145.85(12) Å3, Dcalc = 1.322 at 150(1) K.
Co-reporter:Yifan Shi, Atta M. Arif, Richard D. Ernst
Polyhedron 2011 30(11) pp: 1899-1905
Publication Date(Web):
DOI:10.1016/j.poly.2011.04.023
Co-reporter:Andreas Glöckner, Atta M. Arif, Richard D. Ernst, Thomas Bannenberg, Constantin G. Daniliuc, Peter G. Jones, Matthias Tamm
Inorganica Chimica Acta 2010 Volume 364(Issue 1) pp:23-29
Publication Date(Web):15 December 2010
DOI:10.1016/j.ica.2010.06.049
The reactions of the half-open trozircene [(η7-C7H7)Zr(η5-2,4-C7H11)] (1) with the two-electron donor ligands tert-butyl isocyanide (CN-tBu), 1,2-bis(dimethylphosphino)ethane (dmpe), trimethylphosphine (PMe3) and 1,3,4,5-tetramethylimidazolin-2-ylidene (IMe, :C[N(Me)C(Me)]2) have led to the 1:1 adducts 3, 4, 5 and 6, respectively. The latter three were structurally characterized by X-ray diffraction analysis. Additionally, the stability of the adducts was probed by DFT calculations employing the B3LYP and M05-2X functionals showing that the strongly σ-basic N-heterocyclic carbene forms a thermodynamically much more stable adduct than the other three.The reactivity of the half-open trozircene [(η7-C7H7)Zr(η5-2,4-C7H11)] towards the two-electron donor ligands tert-butyl isocyanide (CN-tBu), 1,2-bis(dimethylphosphino)ethane (dmpe), trimethylphosphine (PMe3) and 1,3,4,5-tetramethylimidazolin-2-ylidene (IMe) has been investigated. The resulting 1:1 adducts are discussed based on experimental and theoretical results.
Co-reporter:Benjamin G. Harvey, Atta M. Arif, Richard D. Ernst
Inorganica Chimica Acta 2010 Volume 363(Issue 1) pp:221-224
Publication Date(Web):4 January 2010
DOI:10.1016/j.ica.2009.08.025
Refluxing WCl4(PMe3)3 under a nitrogen atmosphere in the presence of two equivalents of sodium amalgam leads to a reduction to the W(II) complex [cis,mer-WCl2(PMe3)3]2N2 (1), which can be converted to [mer,trans-WCl3(PMe3)2]2N2 (2) via appropriate oxidation/chlorination. Structural data have been obtained for both complexes, and demonstrate significantly increased steric crowding in 1 due to PMe3/PMe3 interactions. The N–N bond distances in the two compounds are similar, at 1.279(4) and 1.243(18) Å, respectively.The reduction of WCl4(PMe3)3 by sodium under a nitrogen atmosphere leads to [cis,mer-WCl2(PMe3)3]2N2, which can be converted to [mer,trans-WCl3(PMe3)2]2N2. Structural studies reveal respective N–N distances of 1.279(4) and 1.243(18) Å, as well as significant steric crowding in the former complex.
Co-reporter:Brian E. Zaugg;Torsten Kolb;Atta M. Arif
Journal of Chemical Crystallography 2010 Volume 40( Issue 9) pp:778-782
Publication Date(Web):2010 September
DOI:10.1007/s10870-010-9736-4
The structure of the previously reported (py)3ZnFe(CO)4 (py = pyridine) has been determined, confirming the monomeric nature of this species. The complex has average Zn–N and Zn–Fe bond lengths of 2.0970(7) and 2.4017(3) Å, and features a coordination geometry about Fe which is intermediate between trigonal bipyramidal and face monocapped tetrahedral. The space group is P21/c, with a = 8.22080(10) Å, b = 16.1668(3) Å, c = 15.4669(3) Å, β = 102.5869(11)°, V = 2006.21(6) Å3, Dcalc. = 1.558 g/cm3 at 150(1) K. A monomeric cadmium analogue, (pyridine)(neocuproin)CdFe(CO)4, has also been synthesized, and found to possess a similar geometry, with average Cd–N and Cd–Fe bond lengths of 2.352(2) and 2.5380(5) Å. The space group is \( P\overline{1} \) with a = 10.8900(2) Å, b = 11.3042(3) Å, c = 15.5488(4) Å, α = 85.1251(10)°, β = 84.3468(14)°, γ = 72.0377(15)°, V = 1808.93(7) Å3, Dcalc. = 1.478 g/cm3 at 150(1) K.
Co-reporter:Yifan Shi;Atta M. Arif
Journal of Chemical Crystallography 2010 Volume 40( Issue 3) pp:235-240
Publication Date(Web):2010 March
DOI:10.1007/s10870-009-9640-y
The previously reported [Ru(naph)4]2+ complex (naph = 1,8-naphthyridine) has been prepared by a simplified route using [RuCl2(1,5-COD)]x (COD = cyclooctadiene) as starting material and isolated as its tetraphenylborate salt. The salt crystallizes in the monoclinic space group P21/n with a = 13.6531(3) Å, b = 12.5389(4) Å, c = 20.0349(5) Å, β = 96.5884(15)º, V = 3407.22(16) Å, Dcalc = 1.300 at 150(1) K. The dication has crystallographically imposed inversion symmetry. Although the iron analogue has been found to have a coordination number of eight, the ruthenium complex is only six-coordinate, which is achieved by the presence of two monodentate and two bidentate 1,8-naphthyridine ligands. The observation of a higher coordination number for Fe(II) vs. Ru(II) can be explained by the high spin nature of the iron complex. A byproduct complex, [Ru(1,5-COD)(naph)2][B(C6H5)4]2, could also be synthesized, isolated pure, and structurally characterized. The organometallic complex possesses an 18 electron configuration by virtue of the dicationic metal center being coordinated by the diene ligand and all four nitrogen lone pairs. This salt crystallizes in the triclinic space group \( {\text{P}}\bar{1} \) with a = 12.9538(3) Å, b = 14.9485(3) Å, c = 17.4291(3) Å, α = 69.0649(11)º, β = 78.3211(9)º, γ = 78.5629(10)º, V = 3057.50(11) Å3, Dcalc = 1.293 at 150(1) K.
Co-reporter:Benjamin G. Harvey;Atta M. Arif
Journal of Chemical Crystallography 2010 Volume 40( Issue 9) pp:783-787
Publication Date(Web):2010 September
DOI:10.1007/s10870-010-9737-3
The reactions of imines of the formula (C6H5)CH=NR (R = C6H5, i-C3H7) with Ti(C5H5)(2,4-C7H11)(PMe3) (C7H11 = dimethylpentadienyl) lead to expulsion of the PMe3 and coupling between the imine’s carbon atom and a single terminus of the 2,4-C7H11 ligand, resulting in C5H5, “diene,” and π-amide coordination in the 16 electron products. Examination of the Ti–C and C–C bonding parameters for the “diene” ligands reveals that they may be more appropriately regarded as enediyl ligands, leading to a formal +4 oxidation state for titanium. Both complexes crystallize in the triclinic space group \( P\overline{1} \). For the R = C6H5 coupling product, a = 10.4590(2) Å, b = 11.6407(2) Å, c = 17.3729(3) Å, α = 74.7610(7)°, β = 79.8600(6)°, γ = 82.2895(11)°, V = 2000.28(6) Å3, Dcalc = 1.293 g/cm3 at 150(1) K. For the R = i-C3H7 coupling product, a = 8.1039(2) Å, b = 9.4115(2) Å, c = 13.0116(4) Å, α = 88.9906(18)°, β = 73.2780(15)°, γ = 83.3088(16)°, V = 943.82(4) Å3, Dcalc = 1.250 g/cm3 at 150(1) K.
Co-reporter:Stephan Scheins ; Marc Messerschmidt ; Milan Gembicky ; Mateusz Pitak ; Anatoliy Volkov ; Philip Coppens ; Benjamin G. Harvey ; Gregory C. Turpin ; Atta M. Arif
Journal of the American Chemical Society 2009 Volume 131(Issue 17) pp:6154-6160
Publication Date(Web):April 14, 2009
DOI:10.1021/ja807649k
The experimental electron density study of Ti(C5H4Me)2[(CH2)2CMe2] provides direct evidence for the presence of (C−C)→Ti agostic interactions. In accord with the model of Scherer and McGrady, the Cα−Cβ bond densities no longer show cylindrical symmetry in the vicinity of the Ti atom and differ markedly from those of the other C−C bonds. At the points along the Cα−Cβ bond where the deviation is maximal the electron density is elongated toward the metal center. The distortion is supported by parallel theoretical calculations. A calculation on an Mo complex in which the agostic interaction is absent supports the Scherer and McGrady criterion for agostic interactions. Despite the formal d0 electron configuration for this Ti(IV) species, a significant nonzero population is observed for the d orbitals, the d orbital population is largest for the dxy orbital, the lobes of which point toward the two Cα atoms. Of the three different basis sets for the Ti atom used in theoretical calculations with the B3LYP functional, only the 6-311++G** set for Ti agrees well with the experimental charge density distribution in the Ti−(Cα−Cβ)2 plane.
Co-reporter:Anne M. Wilson, Arnold L. Rheingold, Thomas E. Waldman, Michael Klein, Frederick G. West, Richard D. Ernst
Journal of Organometallic Chemistry 2009 694(7–8) pp: 1112-1121
Publication Date(Web):
DOI:10.1016/j.jorganchem.2008.09.008
Co-reporter:Andreas Glöckner, Thomas Bannenberg, Matthias Tamm, Atta M. Arif and Richard D. Ernst
Organometallics 2009 Volume 28(Issue 20) pp:5866-5876
Publication Date(Web):September 23, 2009
DOI:10.1021/om900456s
The reactions of Zr(C7H7)(Cl)(tmeda) (tmeda = tetramethylethylenediamine) with pentadienyl anions lead to formally tetravalent Zr(C7H7)(Pdl) complexes, for Pdl = C5H7, 2,4-C7H11, 6,6-dmch, and c-C7H9 (C7H11 = dimethylpentadienyl, dmch = dimethylcyclohexadienyl, c-C7H9 = cycloheptadienyl). Structural characterizations of the first three have been carried out, revealing much shorter Zr−C distances for the C7H7 ligand and a pattern of Zr−C bond distances for the pentadienyl ligands that is consistent with a formally high (+4) metal oxidation state, which is also supported by DFT calculations. As had been found for the analogous Cp complexes, these 16-electron species are susceptible to Lewis base coordination, and the 2,6-xylyl isocyanide adducts of the 2,4-C7H11 and 6,6-dmch complexes have been isolated and characterized by IR spectroscopy and single-crystal X-ray diffraction studies. The IR spectroscopic studies indicate that the pentadienyl ligands are serving as better net electron donors than Cp ligands, opposite what is typically found for related but lower valent species. At high temperatures the 16-electron Zr(C7H7)(C5H7) complex undergoes slow conversion to the corresponding Cp complex.
Co-reporter:Andreas Glöckner, Matthias Tamm, Atta M. Arif and Richard D. Ernst
Organometallics 2009 Volume 28(Issue 24) pp:7041-7046
Publication Date(Web):November 30, 2009
DOI:10.1021/om900847y
The reactions of Zr(C7H7)(Cl)(tmeda) (tmeda = N,N,N′,N′-tetramethylethylene-1,2-diamine) with cyclopentadienyl and substituted cyclopentadienyl anions have led to the expected Zr(C7H7)(C5H4R) and Zr(C7H7)(C9H7) complexes (R = H, CH3, SiMe3, C3H5 (allyl), PPh2; C9H7 = indenyl). The R = H and PPh2 complexes had previously been reported, but their preparations were accompanied by lower yields, among other drawbacks. The approach reported herein thus appears to offer a fairly general, effective, and convenient method for the preparation of zirconium complexes containing the C7H7 and a variety of monoanionic ligands. Structural data have been obtained for the R = CH3, SiMe3, and allyl species, as well as the indenyl complex. These data are consistent with previous conclusions that the zirconium center is present in a formal +4 oxidation state. Despite the 16-electron configurations for these species, the R = allyl complex showed no coordination of this substituent. As an apparent result of the hard nature of Zr(IV), weak THF coordination to the R = SiMe3 complex was observed to take place, via a structural study. Structural data were also obtained for the previously characterized Zr(C7H7)(C5H5){CN[2,6-C6H3(CH3)2]} complex.
Co-reporter:Andreas Glöckner, Atta M. Arif and Richard D. Ernst
Organometallics 2008 Volume 27(Issue 3) pp:327-333
Publication Date(Web):January 17, 2008
DOI:10.1021/om700437w
The reaction of Zr(C5H5)2Br2 with bromine can be used as an effective route to Zr(C5H5)Br3. Through the slow addition of reactants, Zr(C5H5)Br3 can be isolated in crystalline form. An X-ray diffraction study has revealed it to exist as a one-dimensional polymer, isomorphous with Zr(C5H5)Cl3. In the presence of excess bromine, additional loss of C5H5 occurs, leading to the formation of the salt complex [Zr3(C5H5)3Br3(μ2-Br)3(μ3-Br)2]+[Zr2Br9]−, as well as additional products. However, use of Zr(C5H5)2Br2 that has been exposed to air leads to traces of a water complex, Zr2(C5H5)2Br4(μ2-Br)2(μ2-H2O), which has been revealed through a structural study as having a confacial bioctahedral geometry, with the water serving as one of the three bridging ligands.
Co-reporter:Daesung Chong, William E. Geiger, Nathan A. Davis, Anke Weisbrich, Yifan Shi, Atta M. Arif and Richard D. Ernst
Organometallics 2008 Volume 27(Issue 3) pp:430-436
Publication Date(Web):January 15, 2008
DOI:10.1021/om700886k
The syntheses of the new compounds Fe(3-Me3Si-6,6-dmch)2, 2, and Fe[3-(i-Pr)3Si-6,6-dmch]2, 3, are reported, along with X-ray structural studies of these species, and of the previously reported Fe(1,3,5,6-temch)2 (dmch = dimethylcyclohexadienyl; temch = tetramethylcyclohexadienyl). Each species crystallized in something close to the expected gauche-eclipsed conformation. In accord with previous work on Fe(6,6-dmch)2, but in contrast to results for open ferrocenes such as Fe(2,4-C7H11)2 (C7H11 = dimethylpentadienyl), the three species under study undergo reversible one-electron oxidations at room temperature to stable 17-electron cations, with potentials for oxidation being more favorable than that for ferrocene by 0.51–0.78 V. The edge-bridged open ferrocenes also react in a 1:1 ratio with TCNE (TCNE = tetracyanoethylene), yielding salts that were shown spectroscopically to contain the expected cationic 17-electron metal complexes and the TCNE radical anion.
Co-reporter:Benjamin G. Harvey, Yifan Shi, Bryce K. Peterson, Atta M. Arif, Richard D. Ernst
Inorganica Chimica Acta 2006 Volume 359(Issue 3) pp:839-845
Publication Date(Web):1 February 2006
DOI:10.1016/j.ica.2005.06.016
The reactions of RuCl2[P(C6H5)3]3, RuCl2(tmeda)2, and RuCl2(1,5-COD)(tmeda) with polybasic amines such as pyrazole have been studied. From the phosphine complex, a binuclear complex has been isolated in which one pyrazole has been incorporated, while reactions of the latter two with excess pyrazole lead to the replacement of a tmeda ligand by two pyrazoles.The reaction of RuCl2[P(C6H5)3]3 with one equivalent of pyrazole led to the incorporation of one pyrazole in a di(ruthenium) complex, presumably reflecting an instability of a 1:1 complex. The reaction of pyrazole with RuCl2(tmeda)2 has led to the isolation of RuCl2(tmeda)2(C3N2H4)2. Both species, as well as RuCl2(tmeda)2 and RuCl2(1,5-COD)(tmeda), have been structurally characterized.
Co-reporter:Rehan Basta, Benjamin G. Harvey, Atta M. Arif, Richard D. Ernst
Inorganica Chimica Acta 2004 Volume 357(Issue 13) pp:3883-3888
Publication Date(Web):1 November 2004
DOI:10.1016/j.ica.2004.04.012
The reactions of Zr(C5H5)(6,6-dmch)(PMe3)2 and Zr(6,6-dmch)2(PMe3)2 (dmch=dimethylcyclohexadienyl) with CO lead to the selective replacement of one PMe3 ligand by CO. Both carbonyl complexes have been structurally characterized. Additionally, the reaction of the latter complex with PhC2SiMe3 leads to a similar replacement of one PMe3 ligand, involving simple coordination of the alkyne, rather than any coupling to the dmch ligand.The reactions of Zr(C5H5)(6,6-dmch)(PMe3)2 or Zr(6,6-dmch)2(PMe3)2 (dmch=dimethylcyclohexadienyl) with CO lead to the replacement of one phosphine ligand by CO. Additionally, the reaction of the latter complex with PhC2SiMe3 leads to a similar replacement of one phosphine ligand, initially yielding a complex in which dienyl/alkyne coupling has not taken place.
Co-reporter:Rehan Basta, Atta M. Arif, Richard D. Ernst
Journal of Organometallic Chemistry 2004 Volume 689(Issue 3) pp:685-688
Publication Date(Web):9 February 2004
DOI:10.1016/j.jorganchem.2003.11.010
The reaction of [Ru(C5Me5)Cl2]2 with an excess of 1,3-cyclononadiene in the presence of metallic zinc leads to Ru(C5Me5)(1,2,3,6,7-η5-C9H13), in which the nine-membered ring provides both allyl and olefin coordinations, linked together on each side by C2H4 bridges. The complex has been characterized analytically, spectroscopically, and structurally.The incorporation of 1,3-cyclononadiene into a Ru(C5Me5) complex occurs in conjunction with its deprotonation and subsequent isomerization to an η5-1,2,3,6,7-cyclononadienyl ligand, having isolated allyl and olefin fragments rather than a contiguous η5-dienyl ligand. This formulation has been supported spectroscopically and by a single crystal structural study.
Co-reporter:Rehan Basta, Richard D Ernst, Atta M Arif
Journal of Organometallic Chemistry 2003 Volume 683(Issue 1) pp:64-69
Publication Date(Web):7 October 2003
DOI:10.1016/S0022-328X(03)00408-X
The reaction of ZrCl4 with four equivalents of the 6,6-dimethylcyclohexadienyl anion (6,6-dmch−) in the presence of PMe3 leads to the 18 electron Zr(6,6-dmch)2(PMe3)2. This complex was found to undergo a coupling reaction with two equivalents of PhCHNPh, such that the couplings involved the two termini of the same dienyl ligand, yielding a formal Zr(η5-dienyl)(η3-allyl)(π-amide)2 complex. Both metal complexes have been structurally characterized.An edge-bridged open ziroconocene has been isolated as an 18 electron bis(trimethylphosphine) adduct, Zr(6,6-dmch)2(PMe3)2. This complex reacts with two equivalents of PhCHNPh, resulting in couplings to both ends of one of the dienyl ligands.
Co-reporter:Gregory C. Turpin, Arnold L. Rheingold, Richard D. Ernst
Journal of Organometallic Chemistry 2003 Volume 672(1–2) pp:109-114
Publication Date(Web):14 April 2003
DOI:10.1016/S0022-328X(03)00149-9
Ru(2-methyl-4-phenylpentadienyl)2 has been isolated from the reaction of ruthenium chloride complexes with the appropriate diene and zinc metal in ethanol. The complex exists as a pair of diastereomers, which could be readily separated due to their significantly different solubilities. Structural studies reveal, as expected, that one of the isomers exists in the C1 point group, while the other possesses (noncrystallographic) C2 symmetry.The reaction of RuCl3 hydrate with 2-methyl-4-phenyl pentadienes leads to diastereomeric Ru(2-methyl-4-phenylpentadienyl)2 complexes, which have been found to possess significantly different physical, spectral, and structural properties.
Co-reporter:Rehan Basta, David R Wilson, Huairang Ma, Atta M Arif, Rolfe H Herber, Richard D Ernst
Journal of Organometallic Chemistry 2001 Volumes 637–639() pp:172-181
Publication Date(Web):3 December 2001
DOI:10.1016/S0022-328X(01)00901-9
A 17 electron edge-bridged open metallocene, bis(6,6-dimethylcyclohexadienyl)iron cation, which had previously been observed electrochemically, has been isolated and characterized. Mössbauer spectroscopic data have been obtained for this compound, and for various neutral 18 electron open, half-open, and ‘closed’ ferrocenes, and are in accord with the previous indications that much greater metal–ligand orbital mixing occurs for pentadienyl, as opposed to cyclopentadienyl, ligands. Pertinent structural data have also been obtained for Fe(C5H5)(2,4-C7H11) and Fe(c-C7H9)2 (C7H11=dimethylpentadienyl; c-C7H9=cycloheptadienyl) in order to aid in comparisons between metal–pentadienyl and metal–cyclopentadienyl bonding.Mössbauer and structural comparisons between ferrocene and its open and half-open analogues are reported, and are consistent with earlier proposals of greater metal–ligand orbital mixing for the open pentadienyl ligands. This can be attributed directly to the lower resonance stabilization (i.e. higher energy donor orbitals and lower energy accepting orbitals) of non-aromatic pentadienyl anions.
Co-reporter:Benjamin G. Harvey, Atta M. Arif, Richard D. Ernst
Journal of Molecular Structure (12 November 2008) Volume 890(Issues 1–3) pp:
Publication Date(Web):12 November 2008
DOI:10.1016/j.molstruc.2008.04.001
The reaction of the edge-bridged open zirconocene, Zr(6,6-dmch)2(PMe3)2 (dmch = dimethylcyclohexadienyl) with two equivalents of PhC2SiMe3 leads to a 5 + 2 + 2 ring construction, resulting in a 4.3.1 bicyclodecadienyl skeleton. The resulting complex, with η3-allyl, η4-diene, and η5-dienyl coordination, has a formal 16 electron configuration, and contains close contacts between the metal center and two lengthened C–C bonds, indicative of weak agostic interactions.
.jpg)






![N,N'-bis[(E)-phenylmethylidene]butane-1,4-diamine](http://img.cochemist.com/ccimg/7000/6970-47-4.png)
![N,N'-bis[(E)-phenylmethylidene]butane-1,4-diamine](http://img.cochemist.com/ccimg/7000/6970-47-4_b.png)

