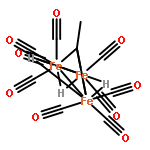
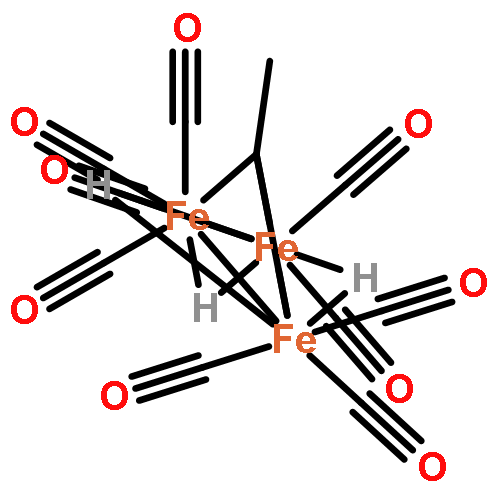
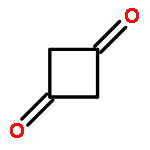
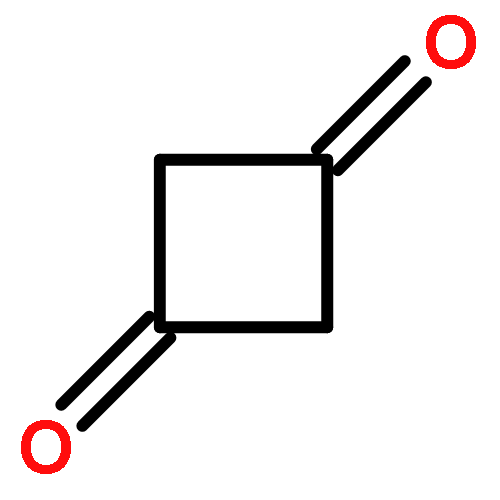
Co-reporter:Daniel M. Chipman
The Journal of Physical Chemistry A 2016 Volume 120(Issue 48) pp:9618-9624
Publication Date(Web):November 10, 2016
DOI:10.1021/acs.jpca.6b09905
The hemibonding interaction in the water dimer cation is studied using coupled cluster electronic structure methods. The hemibonded dimer cation geometry is a local minimum structure characterized by the two participating monomers having both a very short separation and a near parallel relative orientation. It is shown that the vertically ionized dimer at its optimum neutral geometry can convert to the hemibonded dimer cation structure with essentially no energetic hindrance. Direct conversion to the hemibonded structure is therefore an energetically facile alternative to the minimum energy path that connects the vertically ionized neutral water dimer to the global minimum proton-transferred structure. A substantial barrier must be surmounted to convert the hemibonded dimer cation to the proton-transferred structure. The optical absorption spectrum of the hemibonded dimer cation is characterized by three excited near-UV states, two of which have very large oscillator strengths. Relative resonance Raman intensities are estimated for the hemibonded dimer cation vibrational modes, finding the intermolecular stretching mode to be the most strongly enhanced when in near resonance with each of the near-UV excited states, and the anharmonicity and overtones of this mode are estimated. These results provide guidance for the possible observation of hemibonded cations in irradiated liquid water.
Co-reporter:Anna Pomogaeva and Daniel M. Chipman
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 12) pp:3952-3960
Publication Date(Web):October 14, 2011
DOI:10.1021/ct200575c
The performance in describing hydration free energies of a broad class of neutral, cationic, and anionic solutes is tested for the recently proposed FESR (Field-Extremum Short-Range) implicit solvation model for interactions between the solute and nearby water molecules, as taken in conjunction with the previously developed SS(V)PE (Surface and Simulation of Volume Polarization for Electrostatics) dielectric continuum model for long-range interactions with bulk water. The empirical FESR model mainly describes solute–water hydrogen bonding interactions by correlating them with the maximum and minimum values of the electric field produced by the solute at the surface of the cavity that excludes solvent. A preliminary report showed that, with only four adjustable parameters, the FESR model, in conjunction with SS(V)PE, can produce hydration energies comparable to the best analogous efforts in the literature that utilized many more parameters. Here, the performance of the FESR model is more fully documented in several respects. The dependence on the underlying quantum mechanical method used to treat the internal electronic structure of the solute is tested by comparing uncorrelated Hartree–Fock to correlated density functional calculations and by comparing a modest sized to a large basis set. The influence of cavity size is studied in connection with an isodensity contour construction of the cavity. The sensitivity of the results to the parameters in the FESR model is considered, and it is found that the dependence on the electric field strength is quite nonlinear, with an optimum exponent consistently in the range of 3 to 4. Overall, it is concluded that the FESR model shows considerable utility for improving the accuracy of implicit models of aqueous solvation.
Co-reporter:Anna Pomogaeva, Daniel W. Thompson, Daniel M. Chipman
Chemical Physics Letters 2011 Volume 511(1–3) pp:161-165
Publication Date(Web):26 July 2011
DOI:10.1016/j.cplett.2011.05.063
Abstract
A novel model of short-range contributions to hydration free energies is introduced for use with dielectric continuum treatment of long-range bulk electrostatics. The empirical model invokes extrema of the solute normal electric field at the cavity surface that nominally separates solute from solvent. It involves only a small handful of adjustable parameters, which are fitted to experimental data on a large collection of neutral and ionic solutes. The results show mean unsigned errors from experiment of only 0.9 kcal/mol for neutrals and 2.4 kcal/mol for ions, which are comparable to the best analogous previous efforts that utilize many more parameters.
Co-reporter:Daniel M. Chipman
The Journal of Physical Chemistry A 2011 Volume 115(Issue 7) pp:1161-1171
Publication Date(Web):January 27, 2011
DOI:10.1021/jp110238v
The ultraviolet absorption peak commonly used to identify OH radical in liquid water is mainly due to a charge-transfer-from-solvent transition that is prominent when OH is hemibonded, rather than more stable hydrogen bonded, to H2O. This work computationally characterizes the hemibonding interaction and the extent of the geometrical region over which it is significant. Hemibonding is found to be associated with an enlarged energy separation between the two lowest-lying electronic states, which are otherwise always quite close to one another. The lower state, wherein the hemibonding occurs, retains an attractive interaction energy between OH and H2O that can be as much as one-half as strong as the optimum hydrogen-bonding interaction, while the enlarged separation between the states is mainly due to the upper state becoming repulsive over most of the hemibonding region. Hemibonding also leads to a considerable drop in the energy and a considerable increase in the oscillator strength of the characteristic charge-transfer transition. The region of significant hemibonding is found to lie within a moderate range of O−O azimuthal angles and over quite wide ranges of O−O separation distances and hydroxyl OH tilt angles. In particular, significant hemibonding interactions can extend down to surprisingly short O−O distances, where the oscillator strength for the charge-transfer-from-solvent transition becomes very large.
.jpg)
![Methanone, 7-oxabicyclo[2.2.1]hept-2-ene-2,3-diylbis[phenyl-](http://img.cochemist.com/ccimg/74100/74007-32-2.png)
![Methanone, 7-oxabicyclo[2.2.1]hept-2-ene-2,3-diylbis[phenyl-](http://img.cochemist.com/ccimg/74100/74007-32-2_b.png)
![Methanone, 7-oxabicyclo[2.2.1]hepta-2,5-diene-2,3-diylbis[phenyl-](http://img.cochemist.com/ccimg/67000/66933-95-7.png)
![Methanone, 7-oxabicyclo[2.2.1]hepta-2,5-diene-2,3-diylbis[phenyl-](http://img.cochemist.com/ccimg/67000/66933-95-7_b.png)






















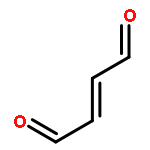





![Methanone, 1,1'-bicyclo[2.2.1]hepta-2,5-diene-2,3-diylbis[1-phenyl-](http://img.cochemist.com/ccimg/1100/1096-50-0.png)
![Methanone, 1,1'-bicyclo[2.2.1]hepta-2,5-diene-2,3-diylbis[1-phenyl-](http://img.cochemist.com/ccimg/1100/1096-50-0_b.png)
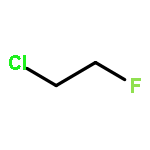
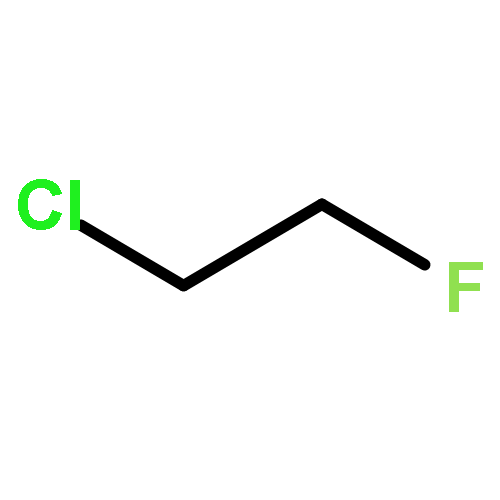




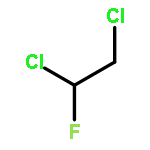

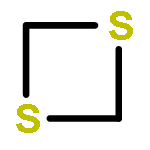
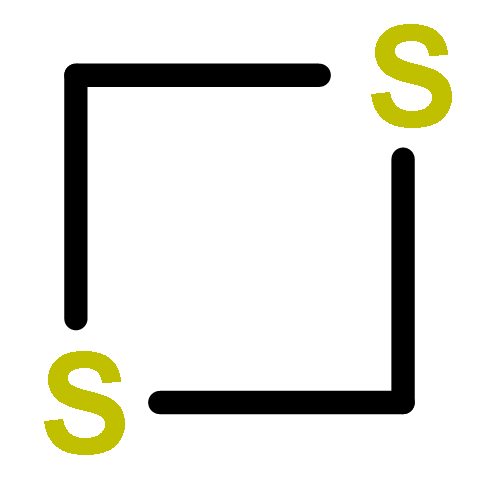


![tricyclo[1.1.0.0~2,4~]butane](http://img.cochemist.com/ccimg/200/157-39-1.png)
![tricyclo[1.1.0.0~2,4~]butane](http://img.cochemist.com/ccimg/200/157-39-1_b.png)