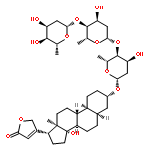
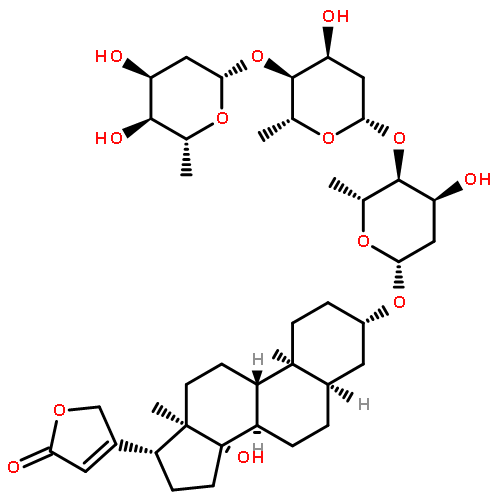
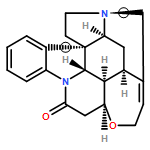
Co-reporter:Guoting Jiang, Hongyan Kang, Yunqiu Yu
Journal of Chromatography B 2017 Volume 1060(Volume 1060) pp:
Publication Date(Web):15 August 2017
DOI:10.1016/j.jchromb.2017.05.023
•Dermal toxicology of phenanthrene was firstly researched using cell metabolomics.•A cross-platform approach was employed to increase the coverage of the metabolome.•48 phenanthrene-regulated metabolites were identified involved with 4 pathways.•Phenanthrene exposure caused a reduced amino pool and a reduced antioxidant status.Phenanthrene (Phe) is one of the most abundant Polycyclic aromatic hydrocarbons (PAHs) contamination from various ambient sources, which has a tremendous impact on public health. However, our knowledge regarding its effects on skin remains limited. In this study, we investigated the metabolite profiling of the human keratinocytes HaCaT cells after Phe exposure to understand the toxic effects of Phe exposure on skin. To obtain a broad picture of metabolome with various hydrophilicity, a cross-platform approach with GC–MS and UHPLC–QTOF-MS has been employed. Data were analyzed by multivariate statistical analysis and samples were separated successfully using supervised PLS-DA models. It was shown that the impacts of Phe exposure on HaCaT cells were both dose-related and time-related. A total of 48 Phe-regulated metabolites were identified and among which 19 were confirmed by reference standards. By pathway analysis, amino acid metabolism, glutathione metabolism and glycerophospholipid metabolism were highlighted as the major metabolic pathways disturbed by Phe. Furthermore, it was found that the mechanisms included a reduced amino pool and a reduced antioxidant status. Overall, these results aid in improving understanding of the dermal toxicology related to Phe, and demonstrate this cross-platform approach is suitable for metabolomics researches on HaCaT cells.Download high-res image (276KB)Download full-size image
Co-reporter:Xia Li, Yu Wang, Qingyuan Zhou, Yunqiu Yu, Lihong Chen, Jie Zheng
Journal of Chromatography B 2015 Volumes 978–979() pp:138-144
Publication Date(Web):26 January 2015
DOI:10.1016/j.jchromb.2014.11.023
•It is a more sensitive LC/MS/MS method firstly reported to determine digoxin with the formate-adduct ion as the precursor ion.•The intensity of digoxin formate-adduct was proved to be 16–18 times stronger than the deprotonated ion.•The new method is rapid, sensitive, less sample volume and injection volume needed.•Steroid saponins with a conjugate lactonic ring in their structure were found to be easier to form stable formate-adduct ion.A sensitive and rapid method based on formate-adduct ion detection was developed and fully validated for digoxin determination in rat plasma. For LC/MS/MS detection with formate-adducts as precursor ions, transitions of m/z 825.5 → 779.9 for digoxin and m/z 809.5 → 763.4 for the internal standard (digitoxin) were monitored in negative mode. To investigate the impact of formic acid on the mass response and method sensitivity, a formic acid concentration range of 0–0.1% (0, 0.0005%, 0.002%, 0.01%, 0.1%, v/v) was evaluated. A concentration of 0.002% gave the highest sensitivity, which was 16- to 18-fold higher than deprotonated ions, and was designated as the contribution giving the strongest ionization enhancement and adduction. A number of parameters were then varied in order to optimize the method, and a limit of quantitation (LOQ) at 0.2 ng/mL was reached with an injection volume of 5 μL, a total run time of 3 min, and 0.1 mL of rat plasma. A calibration curve was plotted over the range 0.2–50 ng/mL (R2 = 0.9998), and the method was successfully applied to study pharmacokinetics in rat following a single oral administration of digoxin (0.05 mg/kg). Four additional steroid saponins (digitoxin, deslanoside, ginsenoside Rg1 and Rb1) were investigated to assess the impact of formic acid on the mass response of steroid saponins. Compounds with a conjugated lactonic ring in their structures such as digoxin, digitoxin and deslanoside tended to form stable formate-adduct ions more easily. The LC/MS/MS method developed here is therefore well suited for the quantification of steroid saponins that are difficult to deprotonate using other MS approaches.
Co-reporter:Xiaoyun Liu;Shuiqing Zheng;Zheng Jiang;Chen Liang;Rong Wang;Zhe Zhou;Yurong Zhang
Journal of Separation Science 2014 Volume 37( Issue 7) pp:764-774
Publication Date(Web):
DOI:10.1002/jssc.201301055
An in vivo study of Strychnos alkaloids metabolites in rats by ultra high performance liquid chromatography with linear ion trap Orbitrap MS is reported for the first time. Two major Strychnos alkaloids compounds including strychnine and brucine were investigated. To obtain optimal extraction efficiency, samples were pretreated by using an SPE plate. The structures of metabolites and their fragment ions were characterized based on the accurate mass and MSn data. Forty-seven metabolites were identified in rat urine, of which 25 were reported for the first time. Four new metabolism pathways were proposed on the basis of the identified metabolites. This study provides a practical approach for rapidly identifying complicated metabolites, a methodology that could be widely applied not only in forensic and clinically toxicological relevant cases, but also for the structural characterization of metabolites of other compounds.
Co-reporter:Qixin Dong;Jiajun Zhu;Qiang Sui;Chao Tang;Xiaomei Wang
Journal of Separation Science 2013 Volume 36( Issue 7) pp:1200-1208
Publication Date(Web):
DOI:10.1002/jssc.201201114
In this study, the objective was to investigate the degradation behavior of Esomeprazole under different recommended stress conditions according to International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use [1] by HPLC. Our research showed that the effect of mobile phase species on separation was significant for the determination of Esomeprazole and its related compounds. Successful separation of the drug from its related impurities and degradation products formed under different stress conditions was achieved using ammonium acetate buffer/ACN by a gradient elution. Compared with phosphate buffer/ACN, ammonium acetate buffer/ACN under same pH and gradient showed a great improvement in resolution due to the change of elution order. The drug was subjected to stress conditions including acidic, alkaline, oxidative, photolytic, and thermal conditions. Extensive degradation occurred in acidic and oxidative conditions, while mild degradation was observed in alkaline and photolytic conditions. Besides, it turned out the drug was extremely stable under thermal condition. The stability-indicating LC–UV method was validated with respect to linearity, precision, accuracy, specificity, and robustness. The LC–MS method was also adopted for the characterization of degradation products. Based on the m/z values and fragmentation patterns, the degradation pathway of the drug has been proposed.
Co-reporter:Zhuhong Yu;Zhongping Wu;Feijun Gong;Rong Wong;Chen Liang;Yurong Zhang
Journal of Separation Science 2012 Volume 35( Issue 20) pp:2773-2780
Publication Date(Web):
DOI:10.1002/jssc.201200263
A novel capillary zone electrophoresis separation coupled to electro spray ionization time-of-flight mass spectrometry method was developed for the simultaneous analysis of six toxic alkaloids: brucine, strychnine, atropine sulfate, anisodamine hydrobromide, scopolamine hydrobromide and anisodine hydrobromide in human plasma and urine. To obtain optimal sensitivity, a solid-phase extraction method using Oasis MCX cartridges (1 mL, 30 mg; Waters, USA) for the pretreatment of samples was used. All compounds were separated by capillary zone electrophoresis at 25 kV within 12 min in an uncoated fused-silica capillary of 75 μm id × 100 cm and were detected by time-of-flight mass spectrometry. This method was validated with regard to precision, accuracy, sensitivity, linear range, limit of detection (LOD), and limit of quantification (LOQ). In the plasma and urine samples, the linear calibration curves were obtained over the range of 0.50–100 ng/mL. The LOD and LOQ were 0.2–0.5 ng/mL and 0.5–1.0 ng/mL, respectively. The intra- and interday precision was better than 12% and 13%, respectively. Electrophoretic peaks could be identified by mass analysis.
Co-reporter:Zhi-Yan Lin, Xiaoyun Liu, Fan Yang, Yun-Qiu Yu
International Journal of Mass Spectrometry 2012 Volumes 328–329() pp:43-66
Publication Date(Web):1 September 2012
DOI:10.1016/j.ijms.2012.07.022
In this work, we reported the use of high-performance liquid chromatography (HPLC) with electrospray ionization multi-stage tandem mass spectrometry (ESI-MSn) to study the characterization of five triterpenoid saponins from Momordica cochinchinensis extracts in positive ion mode and negative ion mode, revealed the fragmentation behavior of these five M. cochinchinensis saponins in different ion modes. Results showed that in negative ion mode, fragmentation on the M. cochinchinensis triterpenoid saponins at positions C28 and O bond were the primary pathways. In positive ion mode, the predominant diagnostic ions were a series of ions resulting from the rupture of the saccharic chain on C28. Comparison of different fragmentation patterns showed that the results from positive and negative ion ESI MSn were complementary. Both modes can yield structurally different information for the characterization of triterpenoid saponins. This study provides a powerful method for the online structural identification of five triterpenoid saponins from M. cochinchinensis, and some of which have not been reported.Graphical abstractHighlights► An LC–MSn method was used to study the characterization of triterpenoid saponins. ► The fragmentation behavior of triterpenoid saponins was different in two ion modes. ► Five saponins from Momordica cochinchinensis was deduced by this method. ► A rapid approach was applied to online structural deduced of triterpenoid saponins.
Co-reporter:Le Pan;Yanping Guo;Zhongdong Li;Jun Chen;Tian Jiang
Chromatographia 2010 Volume 72( Issue 7-8) pp:627-633
Publication Date(Web):2010 October
DOI:10.1365/s10337-010-1683-x
For the first time a sensitive, specific and rapid LC–MS–MS assay is presented for the simultaneous determination of levodopa (L-DP), 3-O-methyldopa (3-OMD) and benserazide (BSZ) in human serum. The three compounds were extracted from human serum by protein precipitation followed by dilution of the supernatant with aqueous formic acid. In serum, linearity was observed between 50 and 1,000 ng mL−1 of L-DP, 3-OMD and BSZ, respectively. Intra-day and inter-day RSD values were below 10.56 and 6.22% at concentrations of 120, 360 and 720 ng mL−1. The presented method showed excellent specificity and sensitivity compared with other methods reported. It was applied to a pharmacokinetic study and demonstrated its applicability to pre-clinical and clinical pharmacological research.
Co-reporter:Le Pan;Longwei Sun
Chromatographia 2010 Volume 72( Issue 5-6) pp:425-430
Publication Date(Web):2010 September
DOI:10.1365/s10337-010-1672-0
A reliable and validated LC–MS method was established for a hexokinase inhibitor study based on adenosine 5′-triphosphate (ATP) determination. By adding 5 mM ammonium acetate in the aqueous phase, this method enabled the determination of ATP by LC–MS and greatly increased the MS signal of ATP. This method was used in the study of the anticancer mechanism of Momordica cochinchinensis, an exact ingredient which had exhibited certain hexokinase inhibitor activity. This might reveal the anticancer mechanism of Momordica cochinchinensis.
Co-reporter:Shengmin Su, Yunqiu Yu
Journal of Chromatography A 2009 Volume 1216(Issue 9) pp:1490-1495
Publication Date(Web):27 February 2009
DOI:10.1016/j.chroma.2008.12.050
A sensitive and effort-saving method was established and validated for the quantitative determination of recombinant Arg-Gly-Asp-hirudin (rRGD-hirudin) in human urine samples. The assay was performed on a uncoated fused silica capillary of 70 cm × 50 μm I.D. and a positive voltage of 30 kV was applied. The sample was injected under pressure of 50 mbar for 300 s and the temperature of capillary was kept 25 °C. Sheath liquid consisting of 30% methanol and 70% of 0.1% formic acid aqueous solution flowing at 7 μL/min was supplied to the CE-electrospray interface. Utilizing the dynamic pH junction technique, a lower limit of quantitation of approximately 35 nM was achieved (concentration coefficiency was about 100-fold) without complex sample preprocessing procedure. CE-MS conditions and parameters were also optimized to obtain better performance. The method has been successfully applied in clinical research of rRGD-hirudin.
Co-reporter:Shengmin Su, Yunqiu Yu, Wei Mo, Yanling Zhang, Houyan Song, Qinfen Chen, Yi Xie
Journal of Chromatography B 2008 Volume 870(Issue 1) pp:27-31
Publication Date(Web):1 July 2008
DOI:10.1016/j.jchromb.2008.05.032
A reliable and validated LC–MS method was established for determination of r-RGD-Hirudin in human serum. Ultrafiltration was used instead of liquid–liquid extraction or solid phase extraction for water solubility drug r-RGD-Hirudin extraction. Freeze drying was used for concentration. The experiment conditions, including pre-processing procedure and LC–MS, have been investigated and optimized. Comparing with reported assays, the current method showed significant improvement in specificity, linearity, precision and sensitivity. This method has been successfully applied in clinical research of r-RGD-Hirudin.
Co-reporter:Zheng Jiang;Yuan Lu;Weilun Ke;Xinbi Li
Chromatographia 2008 Volume 68( Issue 1-2) pp:111-116
Publication Date(Web):2008 July
DOI:10.1365/s10337-008-0654-y
Rhodamine 123 has been frequently used to evaluate the functional activity of P-glycoprotein, assess the related drug interactions, analyze mitochondrial distribution and function, sort functionally distinct cell subpopulations and measure mitochondrial or cellular membrane potential. We developed a sensitive and rapid liquid chromatographic method with fluorescence detection after simple sample preparation procedure for the determination of Rhodamine 123 in P-glycoprotein efflux studies. The mobile phase consisted of methanol and 15 mM dibasic sodium phosphate buffer (pH 6.0) (80:20,v/v), delivered at a rate of 1.0 mL min−1. 15 μL of the samples were injected into a reversed-phase C18 column with a fixed excitation wavelength at 505 nm and altered emission wavelengths. The whole LC analysis was accomplished within 6 min. The established linearity range from 1 to 100 ng mL−1, with the inter-day and intra-day RSD below 7.57 and 4.89% at concentrations of 4.4, 44 and 88 ng mL−1. All the calibration standards and quality controls were prepared in cell lysate and were stable for three freeze-thaw cycles, for 12 h at 4 °C and for 6 h at room temperature. This rapid LC method has been applied to the quantification of Rhodamine 123 in cell lysate obtained from P-glycoprotein efflux study conducted in rat brain capillary endothelial cells.
Co-reporter:Li Zhang, Rong Wang, Yunqiu Yu, Yurong Zhang
Journal of Chromatography B 2007 Volume 857(Issue 1) pp:130-135
Publication Date(Web):15 September 2007
DOI:10.1016/j.jchromb.2007.07.009
In the current paper, we report the development of a new capillary electrophoresis method using pre-column derivatization and laser-induced fluorescence detection for the determination of ephedrine and amphetamine drugs. Our new method allows for the identification and quantification of six commonly used illicit drugs namely pseudoephedrine, ephedrine, amphetamine, methamphetamine, 3,4-methylenedioxyamphetamine, and 3,4-methylenedioxymethylamphetamine, respectively, as well as propafenone (internal standard). Following derivatization with fluorescein isothiocyanate, a total of six amphetamine drugs and the internal standard could readily be separated using a fused-silica 75 μm ID × 60 cm length (effective length: 50.2 cm) capillary column. The mobile phase consisted of buffer containing 20 mM borate (pH 12, adjusted with sodium hydroxide). Samples were injected in pressure mode with the capillary being operated at 25 kV/25 °C, and the detection of the derivatized compounds was sought using a laser-induced fluorescence (LIF) detector (λex = 488 nm and λem = 520 nm), with a run-time of 20 min. The current method was validated with regard to precision (relative standard deviation, RSD), accuracy, sensitivity, linear range, limit of detection (LOD) and limit of quantification (LOQ). In human blood and urine samples, detection limits were 0.2 ng mL−1, and the linear range of the calibration curves was 0.5–100 ng mL−1. The intra-day and inter-day precisions were both less than 13.22%.
Co-reporter:Ni Li;Jianming Huang;Weiyu Weng;Zhaochang Huang
Annali di Chimica 2005 Volume 95(Issue 6) pp:
Publication Date(Web):1 JUN 2005
DOI:10.1002/adic.200590054