Co-reporter:Hanwool Yoon, Vesselin Kolev, and Arieh Warshel
The Journal of Physical Chemistry B October 12, 2017 Volume 121(Issue 40) pp:9358-9358
Publication Date(Web):September 14, 2017
DOI:10.1021/acs.jpcb.7b07726
The study of the function of proteins on a quantitative level requires consideration of the water molecules in and around the protein. This requirement presents a major computational challenge due to the fact that the insertion of water molecules can have a very high activation barrier and would require a long simulation time. Recently, we developed a water flooding (WF) approach which is based on a postprocessing Monte Carlo ranking of possible water configurations. This approach appears to provide a very effective way for assessing the insertion free energies and determining the most likely configurations of the internal water molecules. Although the WF approach was used effectively in modeling challenging systems that have not been addressed reliably by other microscopic approaches, it was not validated by a comparison to the more rigorous grand canonical Monte Carlo (GCMC) method. Here we validate the WF approach by comparing its performance to that of the GCMC method. It is found that the WF approach reproduces the GCMC results in well-defined test cases but does so much faster. This established the WF approach as a useful strategy for finding correct water configurations in proteins and thus to provide a powerful way for studies of the functions of proteins.
Co-reporter:Garima Jindal, Balajee Ramachandran, Ram Prasad Bora, and Arieh Warshel
ACS Catalysis May 5, 2017 Volume 7(Issue 5) pp:3301-3301
Publication Date(Web):March 30, 2017
DOI:10.1021/acscatal.7b00171
Computer-aided enzyme design presents a major challenge since in most cases it has not resulted in an impressive catalytic power. The reasons for the problems with computational design include the use of nonquantitative approaches, but they may also reflect other difficulties that are not completely obvious. Thus, it is very useful to try to learn from the trend in directed evolution experiments. Here we explore the nature of the refinement of Kemp eliminases by directed evolution, trying to gain an understanding of related requirements from computational design. The observed trend in the directed evolution refinement of KE07 and HG3 are reproduced, showing that in the case of KE07 the directed evolution leads to ground-state destabilization, whereas in the case of HG3 the directed evolution leads to transition-state stabilization. The nature of the different paths of the directed evolution is examined and discussed. The present study seems to indicate that computer-aided enzyme design may require more than calculations of the effect of single mutations and should be extended to calculations of the effect of simultaneous multiple mutations (that make a few residues preorganized effectively). However, the analysis of two known evolution paths can still be accomplished using the relevant sequences and structures. Thus, by comparing two directed evolution paths of Kemp eliminases we reached the important conclusion that the more effective path leads to transition-state stabilization.Keywords: directed evolution; enzyme design; EVB; Kemp eliminase; transition-state stabilization;
Co-reporter:Shayantani Mukherjee
Photosynthesis Research 2017 Volume 134( Issue 1) pp:1-15
Publication Date(Web):03 July 2017
DOI:10.1007/s11120-017-0411-x
Molecular motors are multi-subunit complexes that are indispensable for accomplishing various tasks of the living cells. One such molecular motor is the FOF1 ATP synthase that synthesizes ATP at the expense of the membrane proton gradient. Elucidating the molecular origin of the motor function is challenging despite significant advances in various experimental fields. Currently atomic simulations of whole motor complexes cannot reach to functionally relevant time scales that extend beyond the millisecond regime. Moreover, to reveal the underlying molecular origin of the function, one must model the coupled chemical and conformational events using physically and chemically meaningful multiscaling techniques. In this review, we discuss our approach to model the action of the F1 and FO molecular motors, where emphasis is laid on elucidating the molecular origin of the driving force that leads to directional rotation at the expense of ATP hydrolysis or proton gradients. We have used atomic structures of the motors and used hierarchical multiscaling techniques to generate low dimensional functional free energy surfaces of the complete mechano-chemical process. These free energy surfaces were studied further to calculate important characteristics of the motors, such as, rotational torque, temporal dynamics, occurrence of intermittent dwell states, etc. We also studied the result of mutating various parts of the motor domains and our observations correspond very well with the experimental findings. Overall, our studies have generated a cumulative understanding of the motor action, and especially highlight the crucial role of electrostatics in establishing the mechano-chemical coupling.
Co-reporter:Ilsoo Kim
PNAS 2016 Volume 113 (Issue 28 ) pp:7810-7815
Publication Date(Web):2016-07-12
DOI:10.1073/pnas.1608118113
Measurements of voltage changes in response to charge separation within membrane proteins can offer fundamental information
on spectroscopically “invisible” steps. For example, results from studies of voltage changes associated with electron and
proton transfer in cytochrome c oxidase could, in principle, be used to discriminate between different theoretical models describing the molecular mechanism
of proton pumping. Earlier analyses of data from these measurements have been based on macroscopic considerations that may
not allow for exploring the actual molecular mechanisms. Here, we have used a coarse-grained model describing the relation
between observed voltage changes and specific charge-transfer reactions, which includes an explicit description of the membrane,
the electrolytes, and the electrodes. The results from these calculations offer mechanistic insights at the molecular level.
Our main conclusion is that previously assumed mechanistic evidence that was based on electrogenic measurements is not unique.
However, the ability of our calculations to obtain reliable voltage changes means that we have a tool that can be used to
describe a wide range of electrogenic charge transfers in channels and transporters, by combining voltage measurements with
other experiments and simulations to analyze new mechanistic proposals.
Co-reporter:Garima Jindal and Arieh Warshel
The Journal of Physical Chemistry B 2016 Volume 120(Issue 37) pp:9913-9921
Publication Date(Web):August 23, 2016
DOI:10.1021/acs.jpcb.6b07203
Although QM/MM calculations are the primary current tool for modeling enzymatic reactions, the reliability of such calculations can be limited by the size of the QM region. Thus, we examine in this work the dependence of QM/MM calculations on the size of the QM region, using the reaction of catechol-O-methyl transferase (COMT) as a test case. Our study focuses on the effect of adding residues to the QM region on the activation free energy, obtained with extensive QM/MM sampling. It is found that the sensitivity of the activation barrier to the size of the QM is rather limited, while the dependence of the reaction free energy is somewhat larger. Of course, the results depend on the inclusion of the first solvation shell in the QM regions. For example, the inclusion of the Mg2+ ion can change the activation barrier due to charge transfer effects. However, such effects can easily be included in semiempirical approaches by proper parametrization. Overall, we establish that QM/MM calculations of activation barriers of enzymatic reactions are not highly sensitive to the size of the QM region, beyond the immediate region that describes the reacting atoms.
Co-reporter:Raphael Alhadeff and Arieh Warshel
The Journal of Physical Chemistry B 2016 Volume 120(Issue 42) pp:10951-10958
Publication Date(Web):September 21, 2016
DOI:10.1021/acs.jpcb.6b08126
The structures of transport proteins have been steadily revealed in the last few decades, and yet the conversion of this information into molecular-level understanding of their function is still lagging behind. In this study, we try to elucidate how the action of the archaeal sodium/proton antiporter MjNhaP1 depends on its structure-energy relationship. To this end, we calculate the binding energies of its substrates and evaluate the conformational change barrier, focusing on the rotation of the catalytic residue D161. We find that sodium ions and protons compete against a common binding site and that the accessibility of this binding site is restricted to either the inside or outside of the cell. We suggest that the rotation of D161 χ1 angle correlates with the conformational change and is energetically unfavorable when D161 does not bind any substrate. This restriction ensures coupling between the sodium ions and the protons, allowing MjNhaP1 and probably other similar transporters to exchange substrates with minimal leak. Using Monte Carlo simulations we demonstrate the feasibility of our model. Overall we present a complete picture that reproduces the electroneutral (at 1:1 substrate ratio) and coupled transport activity of MjNhaP1 including the energetic basis for the criteria provided by Jardetzky half a century ago.
Co-reporter:Ilsoo Kim and Arieh Warshel
The Journal of Physical Chemistry B 2016 Volume 120(Issue 3) pp:418-432
Publication Date(Web):December 30, 2015
DOI:10.1021/acs.jpcb.5b10956
The voltage sensitivity of membrane proteins is reflected in the response of the voltage sensing domains (VSDs) to the changes in membrane potential. This response is implicated in the displacement of positively charged residues, associated with the conformational changes of VSDs. The displaced charges generate nonlinear (i.e., voltage-dependent) capacitance current called the gating current (and its corresponding gating charge), which is a key experimental quantity that characterizes voltage activation in VSMP. However, the relevant theoretical/computational approaches, aimed to correlate the structural information on VSMP to electrophysiological measurements, have been rather limited, posing a broad challenge in computer simulations of VSMP. Concomitant with the development of our coarse-graining (CG) model of voltage coupling, we apply our theoretical framework for the treatments of voltage effects in membrane proteins to modeling the VSMP activation, taking the VSDs (Ci-VSD) derived from the Ciona intestinalis voltage sensitive phosphatase (Ci-VSP) as a model system. Our CG model reproduces the observed gating charge of Ci-VSD activation in several different perspectives. In particular, a new closed-form expression of the gating charge is evaluated in both nonequilibrium and equilibrium ways, while considering the fluctuation–dissipation relation that connects a nonequilibrium measurement of the gating charge to an equilibrium measurement of charge fluctuations (i.e., the voltage-independent linear component of membrane capacitance). In turn, the expression uncovers a novel link that connects an equilibrium measurement of the voltage-independent linear capacitance to a nonequilibrium measurement of the voltage-dependent nonlinear capacitance (whose integral over voltage is equal to the gating charge). In addition, our CG model yields capacitor-like voltage dependent free energy parabolas, resulting in the free energy difference and the free energy barrier for the Ci-VSD activation at “zero” (depolarization) membrane potential. Significantly, the resultant voltage dependent energetics enables a direct evaluation of capacitance–voltage relationship (C–V curve) as well as charge–voltage relationship (Q–V curve) that is in a good agreement with the observed measurement of Ci-VSD voltage activation. Importantly, an extension of our kinetic/thermodynamic model of voltage dependent activation in VSMP allows for novel derivations of voltage-dependent rate constants, whose parameters are expressed by the intrinsic properties of VSMP. These novel closed-form expressions offer a physicochemical foundation for the semiempirical Eyring-type voltage dependent rate equations that have been the cornerstone for the phenomenological (kinetic) descriptions of gating and membrane currents in the mechanistic study of ion channels and transporters. Our extended theoretical framework developed in the present study has potential implications on the roles played by gating charge fluctuations for the spike generations in nerve cells within the framework of the Hodgkin–Huxley-type model.
Co-reporter:Jerônimo Lameira, Ilya Kupchencko, and Arieh Warshel
The Journal of Physical Chemistry B 2016 Volume 120(Issue 9) pp:2155-2164
Publication Date(Web):February 11, 2016
DOI:10.1021/acs.jpcb.5b11966
Despite the enormous increase in computer power, it is still extremely challenging to obtain computationally converging sampling of ab initio QM/MM (QM(ai)/MM) free energy surfaces in condensed phases. The sampling problem can be significantly reduced by the use of the reference potential paradynamics (PD) approach, but even this approach still requires major computer time in studies of enzymatic reactions. To further reduce the sampling problem we developed here a new PD version where we use an empirical valence bond reference potential that has a minimum rather than a maximum at the transition state region of the target potential (this is accomplished conveniently by shifting the EVB of the product state). Hence, we can map the TS region in a more efficient way. Here, we introduce and validate the inverted EVB PD approach. The validation involves the study of the SN2 step of the reaction catalyzed by haloakene dehalogenase (DhlA) and the GTP hydrolysis in the RasGAP system. In addition, we have also studied the corresponding reaction in water for each of the systems described here and the reaction involving trimethylsulfonium and dimethylamine in solution. The results are encouraging and the new strategy appears to provide a powerful way of evaluating QM(ai)/MM activation free energies.
Co-reporter:Ram Prasad Bora, Matthew J. L. Mills, Maria P. Frushicheva, and Arieh Warshel
The Journal of Physical Chemistry B 2015 Volume 119(Issue 8) pp:3434-3445
Publication Date(Web):January 23, 2015
DOI:10.1021/jp5124025
The ability to design effective enzymes presents a fundamental challenge in biotechnology and also in biochemistry. Unfortunately, most of the progress on this field has been accomplished by bringing the reactants to a reasonable orientation relative to each other, rather than by rational optimization of the polar preorganization of the environment, which is the most important catalytic factor. True computer based enzyme design would require the ability to evaluate the catalytic power of designed active sites. This work considers the evolution from a phosphotriesterase (with the paraoxon substrate) to arylesterase (with the 2-naphthylhexanoate (2NH) substrate) catalysis. Both the original and the evolved enzymes involve two zinc ions and their ligands, making it hard to obtain a reliable quantum mechanical description and then to obtain an effective free energy sampling. Furthermore, the options for the reaction path are quite complicated. To progress in this direction we started with DFT calculations of the energetics of different mechanistic options of cluster models and then used the results to calibrate empirical valence bond (EVB) models and to generate properly sampled free energy surfaces for different mechanisms in the enzyme. Interestingly, it is found that the catalytic effect depends on the Zn–Zn distance making the mechanistic analysis somewhat complicated. Comparing the activation barriers of paraoxon and the 2NH ester at the beginning and end of the evolutionary path reproduced the observed evolutionary trend. However, although our findings provide an advance in exploring the nature of promiscuous enzymes, they also indicate that modeling the reaction mechanism in the case of enzymes with a binuclear zinc center is far from trivial and presents a challenge for computer-aided enzyme design.
Co-reporter:Arieh Warshel;Shayantani Mukherjee
PNAS 2015 Volume 112 (Issue 46 ) pp:14121-14122
Publication Date(Web):2015-11-17
DOI:10.1073/pnas.1519066112
Co-reporter:Raphael Alhadeff
PNAS 2015 Volume 112 (Issue 40 ) pp:12378-12383
Publication Date(Web):2015-10-06
DOI:10.1073/pnas.1516881112
The molecular basis of the function of transporters is a problem of significant importance, and the emerging structural information
has not yet been converted to a full understanding of the corresponding function. This work explores the molecular origin
of the function of the bacterial Na+/H+ antiporter NhaA by evaluating the energetics of the Na+ and H+ movement and then using the resulting landscape in Monte Carlo simulations that examine two transport models and explore
which model can reproduce the relevant experimental results. The simulations reproduce the observed transport features by
a relatively simple model that relates the protein structure to its transporting function. Focusing on the two key aspartic
acid residues of NhaA, D163 and D164, shows that the fully charged state acts as an Na+ trap and that the fully protonated one poses an energetic barrier that blocks the transport of Na+. By alternating between the former and latter states, mediated by the partially protonated protein, protons, and Na+ can be exchanged across the membrane at 2:1 stoichiometry. Our study provides a numerical validation of the need of large
conformational changes for effective transport. Furthermore, we also yield a reasonable explanation for the observation that
some mammalian transporters have 1:1 stoichiometry. The present coarse-grained model can provide a general way for exploring
the function of transporters on a molecular level.
Co-reporter:Patrick Schopf;Matthew J. L. Mills
PNAS 2015 Volume 112 (Issue 14 ) pp:4328-4333
Publication Date(Web):2015-04-07
DOI:10.1073/pnas.1503828112
The catalytic power of enzymes containing coenzyme B12 has been, in some respects, the “last bastion” for the strain hypothesis. Our previous study of this system established by
a careful sampling that the major part of the catalytic effect is due to the electrostatic interaction between the ribose
of the ado group and the protein and that the strain contribution is very small. This finding has not been sufficiently appreciated
due to misunderstandings of the power of the empirical valence bond (EVB) calculations and the need of sufficient sampling.
Furthermore, some interesting new experiments point toward entropic effects as the source of the catalytic power, casting
doubt on the validity of the electrostatic idea, at least, in the case of B12 enzymes. Here, we focus on the observation of the entropic effects and on analyzing their origin. We clarify that our EVB
approach evaluates free energies rather than enthalpies and demonstrate by using the restraint release (RR) approach that
the observed entropic contribution to the activation barrier is of electrostatic origin. Our study illustrates the power of
the RR approach by evaluating the entropic contributions to catalysis and provides further support to our paradigm for the
origin of the catalytic power of B12 enzymes. Overall, our study provides major support to our electrostatic preorganization idea and also highlights the basic
requirements from ab initio quantum mechanics/molecular mechanics calculations of activation free energies of enzymatic reactions.
Co-reporter:Shayantani Mukherjee
PNAS 2015 Volume 112 (Issue 9 ) pp:2746-2751
Publication Date(Web):2015-03-03
DOI:10.1073/pnas.1500979112
Unraveling the molecular nature of the conversion of chemical energy (ATP hydrolysis in the α/β-subunits) to mechanical energy
and torque (rotation of the γ-subunit) in F1-ATPase is very challenging. A major part of the challenge involves understanding the rotary–chemical coupling by a nonphenomenological
structure–energy description, while accounting for the observed torque generated on the γ-subunit and its change due to mutation
of this unit. Here we extend our previous study that used a coarse-grained model of the F1-ATPase to generate a structure-based free energy landscape of the rotary–chemical process. Our quantitative analysis of the
landscape reproduced the observed torque for the wild-type enzyme. In doing so, we found that there are several possibilities
of torque generation from landscapes with various shapes and demonstrated that a downhill slope along the chemical coordinate
could still result in negligible torque, due to ineffective coupling of the chemistry to the γ-subunit rotation. We then explored
the relationship between the functionality and the underlying sequence through systematic examination of the effect of various
parts of the γ-subunit on free energy surfaces of F1-ATPase. Furthermore, by constructing several types of γ-deletion systems and calculating the corresponding torque generation,
we gained previously unknown insights into the molecular nature of the F1-ATPase rotary motor. Significantly, our results are in excellent agreement with recent experimental findings and indicate
that the rotary–chemical coupling is primarily established through electrostatic effects, although specific contacts through
γ-ionizable residue side chains are not essential for establishing the basic features of the coupling.
Co-reporter:Maria P Frushicheva, Matthew JL Mills, Patrick Schopf, Manoj K Singh, Ram B Prasad, Arieh Warshel
Current Opinion in Chemical Biology 2014 Volume 21() pp:56-62
Publication Date(Web):August 2014
DOI:10.1016/j.cbpa.2014.03.022
•The origin of enzyme catalysis can only be determined by computational approaches.•Enzyme catalysis is due to preorganization, and not to other exotic factors.•Computer aided enzyme design must be able to produce known catalytic effects.•The EVB model allows one to produce the trend of directed evolution.•At present the main advances in enzyme design are due to directed evolution.Gaining a deeper understanding of enzyme catalysis is of great practical and fundamental importance. Over the years it has become clear that despite advances made in experimental mutational studies, a quantitative understanding of enzyme catalysis will not be possible without the use of computer modeling approaches. While we believe that electrostatic preorganization is by far the most important catalytic factor, convincing the wider scientific community of this may require the demonstration of effective rational enzyme design. Here we make the point that the main current advances in enzyme design are basically advances in directed evolution and that computer aided enzyme design must involve approaches that can reproduce catalysis in well-defined test cases. Such an approach is provided by the empirical valence bond method.
Co-reporter:Manoj Kumar Singh, Zhen T. Chu, and Arieh Warshel
The Journal of Physical Chemistry B 2014 Volume 118(Issue 42) pp:12146-12152
Publication Date(Web):September 18, 2014
DOI:10.1021/jp507592g
One of the greatest challenges in biotechnology and in biochemistry is the ability to design efficient enzymes. In fact, such an ability would be one of the most convincing manifestations of a full understanding of the origin of enzyme catalysis. Despite some progress on this front, most of the advances have been made by placing the reacting fragments in the proper places rather than by optimizing the preorganization of the environment, which is the key factor in enzyme catalysis. A rational improvement of the preorganization and a consistent assessment of the effectiveness of different design options require approaches capable of evaluating reliably the actual catalytic effect. In this work we examine the ability of the empirical valence bond (EVB) to reproduce the results of directed evolution improvements of the catalysis of diethyl 7-hydroxycoumarinyl by a designed mononuclear zinc metalloenzyme. Encouragingly, our study reproduced the catalytic effect obtained by directed evolution and offers a good start for further studies of this system.
Co-reporter:Ilsoo Kim;Suman Chakrabarty;Peter Brzezinski
PNAS 2014 Volume 111 (Issue 31 ) pp:11353-11358
Publication Date(Web):2014-08-05
DOI:10.1073/pnas.1411573111
Measurements of voltage changes in response to charge separation within membrane proteins can offer fundamental information
on mechanisms of charge transport and displacement processes. A recent example is provided by studies of cytochrome c oxidase. However, the interpretation of the observed voltage changes in terms of the number of charge equivalents and transfer
distances is far from being trivial or unique. Using continuum approaches to describe the voltage generation may involve significant
uncertainties and reliable microscopic simulations are not yet available. Here, we attempt to solve this problem by using
a coarse-grained model of membrane proteins, which includes an explicit description of the membrane, the electrolytes, and
the electrodes. The model evaluates the gating charges and the electrode potentials (c.f. measured voltage) upon charge transfer
within the protein. The accuracy of the model is evaluated by a comparison of measured voltage changes associated with electron
and proton transfer in bacterial photosynthetic reaction centers to those calculated using our coarse-grained model. The calculations
reproduce the experimental observations and thus indicate that the method is of general use. Interestingly, it is found that
charge-separation processes with different spatial directions (but the same distance perpendicular to the membrane) can give
similar observed voltage changes, which indicates that caution should be exercised when using simplified interpretation of
the relationship between charge displacement and voltage changes.
Co-reporter:Shayantani Mukherjee
PNAS 2014 Volume 111 (Issue 20 ) pp:E2077
Publication Date(Web):2014-05-20
DOI:10.1073/pnas.1404542111
Co-reporter:Ilsoo Kim
PNAS 2014 Volume 111 (Issue 6 ) pp:2128-2133
Publication Date(Web):2014-02-11
DOI:10.1073/pnas.1324014111
Quantitative structure-based modeling of voltage activation of ion channels is very challenging. For example, it is very hard
to reach converging results, by microscopic simulations while macroscopic treatments involve major uncertainties regarding
key features. The current work overcomes some of the above challenges by using our recently developed coarse-grained (CG)
model in simulating the activation of the Kv1.2 channel. The CG model has allowed us to explore problems that cannot be fully
addressed at present by microscopic simulations, while providing insights on some features that are not usually considered
in continuum models, including the distribution of the electrolytes between the membrane and the electrodes during the activation
process and thus the physical nature of the gating current. Here, we demonstrate that the CG model yields realistic gating
charges and free energy landscapes that allow us to simulate the fluctuating gating current in the activation processes. Our
ability to simulate the time dependence of the fast gating current allows us to reproduce the observed trend and provides
a clear description of its relationship to the landscape involved in the activation process.
Co-reporter:Anna Rychkova;Shayantani Mukherjee;Ram Prasad Bora
PNAS 2013 110 (25 ) pp:10195-10200
Publication Date(Web):2013-06-18
DOI:10.1073/pnas.1307869110
The nature of the coupling between the stalling of the elongated nascent peptide chain in the ribosome and its insertion through
the translocon is analyzed, focusing on the recently discovered biphasic force that overcomes the stalling barrier. The origin
of this long-range coupling is explored by coarse-grained simulations that combine the translocon (TR) insertion profile and
the effective chemical barrier for the extension of the nascent chain in the ribosome. Our simulation determined that the
inserted H segment is unlikely to climb the TR barrier in parallel with the peptide synthesis chemical step and that the nascent
chain should first overcome the chemical barriers and move into the ribosome–TR gap region before the insertion into the TR
tunnel. Furthermore, the simulations indicate that the coupled TR-chemistry free energy profile accounts for the biphasic
force. Apparently, although the overall elongation/insertion process can be depicted as a tug-of-war between the forces of
the TR and the ribosome, it is actually a reflection of the combined free-energy landscape. Most importantly, the present
study helps to relate the experimental observation of the biphasic force to crucial information about the elusive path and
barriers of the TR insertion process.
Co-reporter:Shayantani Mukherjee
PNAS 2013 Volume 110 (Issue 43 ) pp:E2076E2077-17331
Publication Date(Web):2013-10-22
DOI:10.1073/pnas.1317641110
Understanding the basis for the action of myosin motors and related molecular machines requires a quantitative energy-based
description of the overall functional cycle. Previous theoretical attempts to do so have provided interesting insights on
parts of the cycle but could not generate a structure-based free energy landscape for the complete cycle of myosin. In particular,
a nonphenomenological structure/energy-based understanding of the unidirectional motion is still missing. Here we use a coarse-grained
model of myosin V and generate a structure-based free energy surface of the largest conformational change, namely the transition
from the post- to prepowerstroke movement. We also couple the observed energetics of ligand binding/hydrolysis and product
release to that of the conformational surface and reproduce the energetics of the complete mechanochemical cycle. It is found
that the release in electrostatic free energy upon changing the conformation of the lever arm and the convertor domain from
its post- to prepowerstroke state provides the necessary energy to bias the system towards the unidirectional movement of
myosin V on the actin filament. The free energy change of 11 kcal is also in the range of ∼2–3 pN, which is consistent with
the experimentally observed stalling force required to stop the motor completely on its track. The conformational-chemical
coupling generating a successful powerstroke cycle is believed to be conserved among most members of the myosin family, thus
highlighting the importance of the previously unknown role of electrostatics free energy in guiding the functional cycle in
other actin-based myosin motors.
Co-reporter:Anna Rychkova
PNAS 2013 Volume 110 (Issue 2 ) pp:495-500
Publication Date(Web):2013-01-08
DOI:10.1073/pnas.1220361110
The elucidation of the molecular nature of the translocon-assisted protein insertion is a challenging problem due to the complexity
of this process. Furthermore, the limited availability of crucial structural information makes it hard to interpret the hints
about the insertion mechanism provided by biochemical studies. At present, it is not practical to explore the insertion process
by brute force simulation approaches due to the extremely lengthy process and very complex landscape. Thus, this work uses
our previously developed coarse-grained model and explores the energetics of the membrane insertion and translocation paths.
The trend in the calculated free-energy profiles is verified by evaluating the correlation between the calculated and observed
effect of mutations as well as the effect of inverting the signal peptide that reflects the “positive-inside” rule. Furthermore,
the effect of the tentative opening induced by the ribosome is found to reduce the kinetic barrier. Significantly, the trend
of the forward and backward energy barriers provides a powerful way to analyze key energetics information. Thus, it is concluded
that the insertion process is most likely a nonequilibrium process. Moreover, we provided a general formulation for the analysis
of the elusive apparent membrane insertion energy, ΔGapp, and conclude that this important parameter is unlikely to correspond to the free-energy difference between the translocon
and membrane. Our formulation seems to resolve the controversy about ΔGapp for Arg.
Co-reporter:Nikolay V. Plotnikov, B. Ram Prasad, Suman Chakrabarty, Zhen T. Chu, and Arieh Warshel
The Journal of Physical Chemistry B 2013 Volume 117(Issue 42) pp:12807-12819
Publication Date(Web):April 21, 2013
DOI:10.1021/jp4020146
Understanding the nature of the free-energy surfaces for phosphate hydrolysis is a prerequisite for understanding the corresponding key chemical reactions in biology. Here, the challenge has been to move to careful ab initio QM/MM (QM(ai)/MM) free-energy calculations, where obtaining converging results is very demanding and computationally expensive. This work describes such calculations, focusing on the free-energy surface for the hydrolysis of phosphate monoesters, paying special attention to the comparison between the one water (1W) and two water (2W) paths for the proton-transfer (PT) step. This issue has been explored before by energy minimization with implicit solvent models and by nonsystematic QM/MM energy minimization, as well as by nonsystematic free-energy mapping. However, no study has provided the needed reliable 2D (3D) surfaces that are necessary for reaching concrete conclusions. Here we report a systematic evaluation of the 2D (3D) free-energy maps for several relevant systems, comparing the results of QM(ai)/MM and QM(ai)/implicit solvent surfaces, and provide an advanced description of the relevant energetics. It is found that the 1W path for the hydrolysis of the methyl diphosphate (MDP) trianion is 6–9 kcal/mol higher than that the 2W path. This difference becomes slightly larger in the presence of the Mg2+ ion because this ion reduces the pKa of the conjugated acid form of the phosphate oxygen that accepts the proton. Interestingly, the BLYP approach (which has been used extensively in some studies) gives a much smaller difference between the 1W and 2W activation barriers. At any rate, it is worth pointing out that the 2W transition state for the PT is not much higher that the common plateau that serves as the starting point of both the 1W and 2W PT paths. Thus, the calculated catalytic effects of proteins based on the 2W PT mechanistic model are not expected to be different from the catalytic effects predicted using the 1W PT mechanistic model, which was calibrated on the observed barrier in solution and in which the TS charge distribution was similar to the that of the plateau (as was done in all of our previous EVB studies).
Co-reporter:B. Ram Prasad, Nikolay V. Plotnikov, and Arieh Warshel
The Journal of Physical Chemistry B 2013 Volume 117(Issue 1) pp:153-163
Publication Date(Web):December 1, 2012
DOI:10.1021/jp309778n
The nature and mechanism of phosphate hydrolysis reactions are of great interest in view of the crucial role of these reactions in key biological processes. Although it is becoming clearer that the ultimate way of resolving mechanistic controversies must involve reliable theoretical studies, it is not widely realized that such studies cannot be performed at present by using most existing automated ways and that only careful systematic studies can lead to meaningful conclusions. The present work clarifies the above point by considering the hydrolysis of phosphate monoesters. The clarification starts by defining the actual issues that should be addressed in careful studies and by highlighting the problems with studies that ignore the need for unique mechanistic definitions (e.g., works that confuse associative and dissociative pathways). We then focus on the analysis of the proton transfer (PT) pathways in phosphate hydrolysis and on recent suggestions that PT involves more than one water molecule. Here we point out that most of the studies that found a proton transfer through several water molecules have not involved a sufficient systematic search of the relevant reaction coordinates. This includes both energy minimization approaches as well as a recent metadynamics (MTD) simulation study. To illustrate the crucial need of exploring the potential surfaces reliably, rather than relying on automated approaches, we present here a very careful study of the free energy landscape along a 3D reaction coordinate (RC) exploring both the standard 2D RC, comprised of the attacking and leaving group reaction coordinates, as well as of the proton transfer (PT) coordinate. Our study points out that QM/MM minimization or MTD studies that concluded that the hydrolysis of phosphate monoesters involves a PT through several water molecules, have not explored carefully the single water (1W) path (that involves a direct PT form the attacking water molecule to the phosphate oxygen). Furthermore, we identified the most likely reason for the difficulty in finding the 1W path by QM/MM minimization methods, as well as by the current MTD simulations. We also discuss the problems with current studies that challenge the phosphate as a base mechanism and emphasize that all recent studies found associative/concerted paths (although many have not realized the meaning of their results). Finally, although we clearly do not have the last word about the 1W versus 2W paths we believe that we illustrated that the crucial mechanistic problems with alternative pathways should not be resolved by just running black box search approaches.
Co-reporter:Anna Rychkova and Arieh Warshel
The Journal of Physical Chemistry B 2013 Volume 117(Issue 44) pp:13748-13754
Publication Date(Web):October 2, 2013
DOI:10.1021/jp406925y
The nature of the biological free energy scale (ΔGapp), obtained from translocon mediated insertion studies, has been a major puzzle and the subject of major controversies. Part of the problem has been the complexity of the insertion process that discouraged workers from considering the feasible kinetics schemes and left the possible impression that ΔGapp presents some simple partition. Here we extend and clarify our recent analysis of the insertion problem using well-defined kinetics schemes and a free energy profile. We point out that although the rate constants of some steps are far from being obvious, it is essential to consider explicitly such schemes in order to advance in analyzing the meaning of ΔGapp. It is then shown that under some equilibrium conditions the kinetics scheme leads to a simple formula that allows one to relate ΔGapp to the actual free energy of partitioning between the water, the membrane, and the translocon. Other options are also considered (including limits with irreversible transitions that can be described by linear free energy relationships (LFERs)). It is concluded that it is unlikely that a kinetics plus thermodynamic based analysis can lead to a result that identifies ΔGapp with the partition between the membrane and the translocon. Thus, we argue that unless such analysis is presented, it is unjustified to assume that ΔGapp corresponds to the membrane translocon equilibrium or to some other arbitrary definition. Furthermore, we point out that the presumption that it is sufficient to just calculate the PMF for going from the translocon (TR) to the membrane and then to assume irreversible diffusive motion to water and for further entrance to the membrane is not a valid analysis. Overall, we point out that it is important to try to relate ΔGapp to a well-defined kinetics scheme (regardless of the complication of the system) in order to determine whether the energies of inserting positively charged residues to the membrane are related to the corresponding ΔGapp. It is also suggested that deviations from our simple formula for equilibrium conditions can help in identifying and analyzing kinetics barriers.
Co-reporter:Ram Prasad B;Nikolay V. Plotnikov;Jeronimo Lameira
PNAS 2013 Volume 110 (Issue 51 ) pp:20509-20514
Publication Date(Web):2013-12-17
DOI:10.1073/pnas.1319854110
GTPases play a major role in cellular processes, and gaining quantitative understanding of their activation demands reliable
free energy surfaces of the relevant mechanistic paths in solution, as well as the interpolation of this information to GTPases.
Recently, we generated ab initio quantum mechanical/molecular mechanical free energy surfaces for the hydrolysis of phosphate
monoesters in solution, establishing quantitatively that the barrier for the reactions with a proton transfer (PT) step from
a single attacking water (1W) is higher than the one where the PT is assisted by a second water (2W). The implication of this
finding on the activation of GTPases is quantified here, by using the ab initio solution surfaces to calibrate empirical valence
bond surfaces and then exploring the origin of the activation effect. It is found that, although the 2W PT path is a new element,
this step is not rate determining, and the catalytic effect is actually due to the electrostatic stabilization of the pre-PT
transition state and the subsequent plateau. Thus, the electrostatic catalytic effect found in our previous studies of the
Ras GTPase activating protein (RasGAP) and the elongation factor-Tu (EF-Tu) with a 1W mechanism is still valid for the 2W
path. Furthermore, as found before, the corresponding activation appears to involve a major allosteric effect. Overall, we
believe that our finding is general to both GTPases and ATPases. In addition to the biologically relevant finding, we also
provide a critical discussion of the requirements from reliable surfaces for enzymatic reactions.
Co-reporter:Edina Rosta and Arieh Warshel
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 10) pp:3574-3585
Publication Date(Web):March 29, 2012
DOI:10.1021/ct2009329
Understanding the relationship between the adiabatic free energy profiles of chemical reactions and the underlining diabatic states is central to the description of chemical reactivity. The diabatic states form the theoretical basis of linear free energy relationships (LFERs) and thus play a major role in physical organic chemistry and related fields. However, the theoretical justification for some of the implicit LFER assumptions has not been fully established by quantum mechanical studies. This study follows our earlier works(1, 2) and uses the ab initio frozen density functional theory (FDFT) method(3) to evaluate both the diabatic and the adiabatic free energy surfaces and to determine the corresponding off-diagonal coupling matrix elements for a series of SN2 reactions. It is found that the off-diagonal coupling matrix elements are almost the same regardless of the nucleophile and the leaving group but change upon changing the central group. Furthermore, it is also found that the off-diagonal elements are basically the same in gas phase and in solution, even when the solvent is explicitly included in the ab initio calculations. Furthermore, our study establishes that the FDFT diabatic profiles are parabolic to a good approximation, thus providing a first-principles support to the origin of LFER. These findings further support the basic approximation of the empirical valence bond treatment.
Co-reporter:Shayantani Mukherjee
PNAS 2012 Volume 109 (Issue 37 ) pp:
Publication Date(Web):2012-09-11
DOI:10.1073/pnas.1212841109
The molecular origin of the action of the F0 proton gradient-driven rotor presents a major puzzle despite significant structural advances. Although important conceptual
models have provided guidelines of how such systems should work, it has been challenging to generate a structure-based molecular
model using physical principles that will consistently lead to the unidirectional proton-driven rotational motion during ATP
synthesis. This work uses a coarse-grained (CG) model to simulate the energetics of the F0-ATPase system in the combined space defined by the rotational coordinate and the proton transport (PTR) from the periplasmic
side (P) to the cytoplasmic side (N). The model establishes the molecular origin of the rotation, showing that this effect
is due to asymmetry in the energetics of the proton path rather than only the asymmetry of the interaction of the Asp on the
c-ring helices and Arg on the subunit-a. The simulation provides a clear conceptual background for further exploration of
the electrostatic basis of proton-driven mechanochemical systems.
Co-reporter:Anatoly Dryga;Suman Chakrabarty;Spyridon Vicatos
PNAS 2012 Volume 109 (Issue 9 ) pp:
Publication Date(Web):2012-02-28
DOI:10.1073/pnas.1121094109
Understanding the detailed mechanism of the activation of voltage-gated ion channels has been a problem of great current interest.
Reliable molecular simulations of voltage effects present a major challenge because meaningful converging microscopic simulations
are not yet available and macroscopic treatments involve major uncertainties regarding the dielectric constant used and other
key features. The current work has overcome some of the above challenges by using our recently developed coarse-grained (CG)
model in simulating the activation of the Kv1.2 channel. The CG model has allowed us to explore problems that cannot be addressed
at present by fully microscopic simulations, while providing insights on some features that are not usually considered in
continuum models, including the distribution of the electrolytes between the membrane and the electrodes during the activation
process and thus the nature of the gating current. Furthermore, the clear connection to microscopic descriptions combined
with the power of CG modeling offers a powerful tool for exploring the energy balance between the protein conformational energy
and the interaction with the external potential in voltage-activated channels. Our simulations have reproduced the observed
experimental trend of the gating charge and, most significantly, the correct trend in the free energies, where the closed
channel is more stable at negative potential and the open channel is more stable at positive potential. Moreover, we provide
a unique view of the activation landscape and the time dependence of the activation process.
Co-reporter:Nikolay V. Plotnikov and Arieh Warshel
The Journal of Physical Chemistry B 2012 Volume 116(Issue 34) pp:10342-10356
Publication Date(Web):August 1, 2012
DOI:10.1021/jp304678d
The performance of the paradynamics (PD) reference potential approach in QM/MM calculations is examined. It is also clarified that, in contrast to some possible misunderstandings, this approach provides a rigorous strategy for QM/MM free energy calculations. In particular, the PD approach provides a gradual and controlled way of improving the evaluation of the free energy perturbation associated with moving from the EVB reference potential to the target QM/MM surface. This is achieved by moving from the linear response approximation to the full free energy perturbation approach in evaluating the free energy changes. We also present a systematic way of improving the reference potential by using Gaussian-based correction potentials along a reaction coordinate. In parallel, we review other recent adaptations of the reference potential approach, emphasizing and demonstrating the advantage of using the EVB potential as a reference potential, relative to semiempirical QM/MM molecular orbital potentials. We also compare the PD results to those obtained by direct calculations of the potentials of the mean force (PMF). Additionally, we propose a way of accelerating the PMF calculations by using Gaussian-based negative potentials along the reaction coordinate (which are also used in the PD refinement). Finally, we discuss performance of the PD and the metadynamics approaches in ab initio QM/MM calculations and emphasize the advantage of using the PD approach.
Co-reporter:Maria P. Frushicheva, Shayantani Mukherjee, and Arieh Warshel
The Journal of Physical Chemistry B 2012 Volume 116(Issue 45) pp:13353-13360
Publication Date(Web):October 22, 2012
DOI:10.1021/jp3084327
The development of enzyme mimetic catalysts as well as the analysis of the catalytic effects of such catalysts has been a major challenge for synthetic chemists. One of the impressive examples of artificial catalysts has been the development of a highly charged host compound that provides a significant acceleration to the hydrolysis of orthoformates and other systems. However, the origin of the catalytic effect has not been quantified, and its origin remains somewhat unclear. The understanding of the corresponding supramolecular catalysis has thus become a major challenge, both in terms of computational modeling and in terms of the analysis of the corresponding acid-catalyzed reaction. Here we present a computer simulation study and kinetic analyses that reproduce the experimentally observed catalytic effect, establishing that this effect is due to electrostatic stabilization of the positively charged transition state (relative to the uncharged bound complex). Our study illustrates the crucial need for careful analysis of the complex kinetics of the catalytic effect and the host system, as well as the need for computational modeling in analyzing the catalytic effect and in the potential design of better catalysts. Finally, our finding of the large stabilization of the bound H3O+ points out the very low “local pH” inside the host system even when the solvent is kept at a high pH.
Co-reporter:Maria P. Frushicheva ; Dr. Arieh Warshel
ChemBioChem 2012 Volume 13( Issue 2) pp:215-223
Publication Date(Web):
DOI:10.1002/cbic.201100600
Abstract
The prospect for consistent computer-aided refinement of stereoselective enzymes is explored by simulating the hydrolysis of enantiomers of an α-substituted ester by wild-type and mutant Candida antarctica lipase A, using several strategies. In particular, we focused on the use of the empirical valence bond (EVB) method in a quantitative screening for enantioselectivity, and evaluate both kcat and kcat/KM of the R and S stereoisomers. We found that an extensive sampling is essential for obtaining converging results. This requirement points towards possible problems with approaches that use a limited conformational sampling. However, performing the proper sampling appears to give encouraging results and to offer a powerful tool for the computer-aided design of enantioselective enzymes. We also explore faster strategies for identifying mutations that will help in augmenting directed-evolution experiments, but these approaches require further refinement.
Co-reporter:Shina Caroline Lynn Kamerlin and Arieh Warshel
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 22) pp:10401-10411
Publication Date(Web):27 Apr 2011
DOI:10.1039/C0CP02823A
Recent years have witnessed a tremendous explosion in computational power, which in turn has resulted in great progress in the complexity of the biological and chemical problems that can be addressed by means of all-atom simulations. Despite this, however, our computational time is not infinite, and in fact many of the key problems of the field were resolved long before the existence of the current levels of computational power. This review will start by presenting a brief historical overview of the use of multiscale simulations in biology, and then present some key developments in the field, highlighting several cases where the use of a physically sound simplification is clearly superior to a brute-force approach. Finally, some potential future directions will be discussed.
Co-reporter:Maria P. Frushicheva, Jie Cao, and Arieh Warshel
Biochemistry 2011 Volume 50(Issue 18) pp:
Publication Date(Web):March 28, 2011
DOI:10.1021/bi200063a
One of the fundamental challenges in biotechnology and biochemistry is the ability to design effective enzymes. Despite recent progress, most of the advances on this front have been made by placing the reacting fragments in the proper places, rather than by optimizing the preorganization of the environment, which is the key factor in enzyme catalysis. Thus, rational improvement of the preorganization would require approaches capable of evaluating reliably the actual catalytic effect. This work considers the catalytic effects in different Kemp eliminases as a benchmark for a computer-aided enzyme design. It is shown that the empirical valence bond provides a powerful screening tool, with significant advantages over current alternative strategies. The insights provided by the empirical valence bond calculations are discussed with an emphasis on the ability to analyze the difference between the linear free energy relationships obtained in solution and those found in the enzymes. We also point out the trade-off between the reliability and speed of the calculations and try to determine what it takes to realize reliable computer-aided screening.
Co-reporter:Andrew J. Adamczyk
PNAS 2011 Volume 108 (Issue 24 ) pp:9827-9832
Publication Date(Web):2011-06-14
DOI:10.1073/pnas.1105714108
The crucial process of aminoacyl-tRNA delivery to the ribosome is energized by the GTPase reaction of the elongation factor
Tu (EF-Tu). Advances in the elucidation of the structure of the EF-Tu/ribosome complex provide the rare opportunity of gaining
a detailed understanding of the activation process of this system. Here, we use quantitative simulation approaches and reproduce
the energetics of the GTPase reaction of EF-Tu with and without the ribosome and with several key mutants. Our study provides
a novel insight into the activation process. It is found that the critical H84 residue is not likely to behave as a general
base but rather contributes to an allosteric effect, which includes a major transition state stabilization by the electrostatic
effect of the P loop and other regions of the protein. Our findings have general relevance to GTPase activation, including
the processes that control signal transduction.
Co-reporter:Nikolay V. Plotnikov, Shina C. L. Kamerlin, and Arieh Warshel
The Journal of Physical Chemistry B 2011 Volume 115(Issue 24) pp:7950-7962
Publication Date(Web):May 27, 2011
DOI:10.1021/jp201217b
Recent years have seen tremendous effort in the development of approaches with which to obtain quantum mechanics/molecular mechanics (QM/MM) free energies for reactions in the condensed phase. Nevertheless, there remain significant challenges to address, particularly, the high computational cost involved in performing proper configurational sampling and, in particular, in obtaining ab initio QM/MM (QM(ai)/MM) free-energy surfaces. One increasingly popular approach that seems to offer an ideal way to progress in this direction is the elegant metadynamics (MTD) approach. However, in the current work, we point out the subtle efficiency problems associated with this approach and illustrate that we have at hand what is arguably a more powerful approach. More specifically, we demonstrate the effectiveness of an updated version of our original idea of using a classical reference potential for QM(ai)/MM calculations [ J. Phys. Chem. 1995, 99, 17516)], which we refer to as paradynamics (PD). This approach is based on the use of an empirical valence bond (EVB) reference potential, which is already similar to the real ab initio potential. The reference potential is fitted to the ab initio potential by an iterative and, to a great degree, automated refinement procedure. The corresponding free-energy profile is then constructed using the refined EVB potential, and the linear response approximation (LRA) is used to evaluate the QM(ai)/MM activation free-energy barrier. The automated refinement of the EVB surface (and thus the reduction of the difference between the reference and ab initio potentials) is a key factor in accelerating the convergence of the LRA approach. We apply our PD approach to a test reaction, namely, the SN2 reaction between a chloride ion and methyl chloride, and demonstrate that, at present, this approach is far more powerful and cost-effective than the metadynamics approach (at least in its current implementation). We also discuss the general features of the PD approach in terms of its ability to explore complex systems and clarify that it is not a specialized approach limited to only accelerating QM(ai)/MM calculations with proper sampling, but rather can be used in a wide variety of applications. In fact, we point out that the use of a reference (CG) potential coupled with its PD refinement, as well as our renormalization approach, provides very general and powerful strategies that can be used very effectively to explore any property that has been studied by the MTD approach.
Co-reporter:Shayantani Mukherjee
PNAS 2011 108 (51 ) pp:
Publication Date(Web):2011-12-20
DOI:10.1073/pnas.1117024108
Understanding the nature of energy transduction in life processes requires a quantitative description of the energetics of
the conversion of ATP to ADP by ATPases. Previous attempts to do so have provided an interesting insight but could not account
for the rotary mechanism by a nonphenomenological structure/energy description. In particular it has been very challenging
to account for the observations of the 80° and 40° rotational substates, without any prior information about such states in
the simulation procedure. Here we use a coarse-grained model of F1-ATPase and generate, without the adjustment of phenomenological
parameters, a structure-based free energy landscape that reproduces the energetics of the mechanochemical process. It is found
that the landscape along the relevant rotary path is determined by the electrostatic free energy and not by steric effects.
Furthermore, the generated surface and the corresponding Langevin dynamics simulations identify a hidden conformational barrier
that provides a new fundamental interpretation of the catalytic dwell and illuminate the nature of the energy conversion process.
Co-reporter:Andrew J. Adamczyk;Jie Cao;Shina C. L. Kamerlin
PNAS 2011 108 (34 ) pp:
Publication Date(Web):2011-08-23
DOI:10.1073/pnas.1111252108
The proposal that enzymatic catalysis is due to conformational fluctuations has been previously promoted by means of indirect considerations. However, recent works have focused on cases where the relevant motions have components toward distinct conformational
regions, whose population could be manipulated by mutations. In particular, a recent work has claimed to provide direct experimental evidence for a dynamical contribution to catalysis in dihydrofolate reductase, where blocking a relevant conformational
coordinate was related to the suppression of the motion toward the occluded conformation. The present work utilizes computer
simulations to elucidate the true molecular basis for the experimentally observed effect. We start by reproducing the trend
in the measured change in catalysis upon mutations (which was assumed to arise as a result of a “dynamical knockout” caused
by the mutations). This analysis is performed by calculating the change in the corresponding activation barriers without the
need to invoke dynamical effects. We then generate the catalytic landscape of the enzyme and demonstrate that motions in the
conformational space do not help drive catalysis. We also discuss the role of flexibility and conformational dynamics in catalysis,
once again demonstrating that their role is negligible and that the largest contribution to catalysis arises from electrostatic
preorganization. Finally, we point out that the changes in the reaction potential surface modify the reorganization free energy
(which includes entropic effects), and such changes in the surface also alter the corresponding motion. However, this motion
is never the reason for catalysis, but rather simply a reflection of the shape of the reaction potential surface.
Co-reporter:Shina C. L. Kamerlin
Journal of Physical Organic Chemistry 2010 Volume 23( Issue 7) pp:677-684
Publication Date(Web):
DOI:10.1002/poc.1620
Abstract
Enzymatic reactions are crucial toward controlling and performing most life processes, and, as such, understanding how they really work has both fundamental and practical importance. Thus, one of the major current challenges of biophysics involves understanding the origin of the enormous catalytic power of enzymes, an issue that is still not widely understood and remains controversial within the scientific community. Several proposals have been put forth to try to explain the origin of enzyme catalysis, one of which is the idea that enzyme catalysis involves special factors such as nuclear quantum mechanical (NQM) effects, and, in particular, nuclear tunneling. Here, we will discuss both the factors for and against this proposition, and demonstrate that an analysis of all the relevant facts and arguments seems to establish that enzyme catalysis does not involve large contributions from nuclear tunneling. Copyright © 2010 John Wiley & Sons, Ltd.
Co-reporter:Arieh Warshel;Anna Rychkova;Spyridon Vicatos
PNAS 2010 Volume 107 (Issue 41 ) pp:17598-17603
Publication Date(Web):2010-10-12
DOI:10.1073/pnas.1012207107
The understanding of the mechanism of insertion of transmembrane (TM) helixes through the translocon presents a major open
challenge. Although the experimental information about the partition of the inserted helices between the membrane and the
solution contains crucial information about this process, it is not clear how to extract this information. In particular,
it is not clear how to rationalize the small apparent insertion energy, ΔGapp, of an ionized residue in the center of a TM helix. Here we explore the nature of the insertion energies, asking what should
be the value of these parameters if their measurements represent equilibrium conditions. This is done using a coarse-grained
model with advanced electrostatic treatment. Estimating the energetics of ionized arginine of a TM helix in the presence of
neighboring helixes or the translocon provides a rationale for the observed ΔGapp of ionized residues. It is concluded that the apparent insertion free energy of TM with charged residues reflects probably
more than just the free energy of moving the isolate single helix from water into the membrane. The present approach should
be effective not only in exploring the mechanism of the operation of the translocon but also for studies of other membrane
proteins.
Co-reporter:Arieh Warshel;Shina C. L. Kamerlin
PNAS 2010 Volume 107 (Issue 17 ) pp:E72
Publication Date(Web):2010-04-27
DOI:10.1073/pnas.1002658107
Co-reporter:Shina C.L. Kamerlin;Pankaz K. Sharma;Zhen T. Chu
PNAS 2010 Volume 107 (Issue 9 ) pp:4075-4080
Publication Date(Web):2010-03-02
DOI:10.1073/pnas.0914579107
One of the best systems for exploring the origin of enzyme catalysis has been the reaction of ketosteroid isomerase (KSI).
Studies of the binding of phenolates to KSI have been taken as proof that the electrostatic preorganization effect only makes
a minor contribution to the binding of the real, multiring, transition state (TS). However, our simulation study has determined
that the difference between the phenolates and the TS arises from the fact that the nonpolar state of the phenolate can rotate freely relative to the oxyanion hole and thus loses the preorganization contribution. A
recent study explored the reactivity of both small and multiring systems and concluded that their similar reactivity contradicts
our preorganization idea. Herein, we establish that the available experiments in fact provide what is perhaps the best proof
and clarification of the preorganization idea and its crucial role in enzyme catalysis. First, we analyze the binding energy
and the pKa of equilenin and identify direct experimental evidence for our prediction about the differential electrostatic stabilization
of the large TS and the small phenolates. Subsequently, we show that the similar reactivity of the small and large systems
is also due to an electrostatic preorganization effect but that this effect only appears in the intermediate state because
the TS is not free to rotate. This establishes the electrostatic origin of enzyme catalysis. We also clarify the crucial importance
of having a well-defined physical concept when examining catalytic effects and the need for quantitative tools for analyzing
such effects.
Co-reporter:Anatoly Dryga and Arieh Warshel
The Journal of Physical Chemistry B 2010 Volume 114(Issue 39) pp:12720-12728
Publication Date(Web):September 13, 2010
DOI:10.1021/jp1056122
Simulations of long time process in condensed phases, in general, and in biomolecules, in particular, present a major challenge that cannot be overcome at present by brute force molecular dynamics (MD) approaches. This work takes the renormalization method, intruded by us sometime ago, and establishes its reliability and potential in extending the time scale of molecular simulations. The validation involves a truncated gramicidin system in the gas phase. This system is small enough to allow for very long explicit simulations and sufficiently complex to present the physics of realistic ion channels. The renormalization approach is found to be reliable and arguably presents the first approach that allows one to exploit the otherwise problematic steered molecular dynamics (SMD) treatments in quantitative and meaningful studies. It is established that we can reproduce the long time behavior of large systems by using Langevin dynamics (LD) simulations of a renormalized implicit model. This is done without spending the enormous time needed to obtain such trajectories in the explicit system. The present study also provides a promising advance in accelerated evaluation of free energy barriers. This is done by adjusting the effective potential in the implicit model to reproduce the same passage time as that obtained in the explicit model under the influence of an external force. Here having a reasonable effective friction provides a way to extract the potential of mean force (PMF) without investing the time needed for regular PMF calculations. The renormalization approach, which is illustrated here in realistic calculations, is expected to provide a major help in studies of complex landscapes and in exploring long time dynamics of biomolecules.
Co-reporter:Maria P. Frushicheva;Zhen T. Chu;Jie Cao
PNAS 2010 Volume 107 (Issue 39 ) pp:16869-16874
Publication Date(Web):2010-09-28
DOI:10.1073/pnas.1010381107
One of the fundamental challenges in biotechnology and in biochemistry is the ability to design effective enzymes. Doing so
would be a convincing manifestation of a full understanding of the origin of enzyme catalysis. Despite an impressive progress,
most of the advances on this front have been made by placing the reacting fragments in the proper places, rather than by optimizing
the environment preorganization, which is the key factor in enzyme catalysis. Rational improvement of the preorganization
would require approaches capable of evaluating reliably the actual catalytic effect. This work takes apreviously designed
kemp eliminases as a benchmark for a computer aided enzyme design, using the empirical valence bond as the main screening
tool. The observed absolute catalytic effect and the effect of directed evolution are reproduced and analyzed (assuming that
the substrate is in the designed site). It is found that, in the case of kemp eliminases, the transition state charge distribution
makes it hard to exploit the active site polarity, even with the ability to quantify the effect of different mutations. Unexpectedly,
it is found that the directed evolution mutants lead to the reduction of solvation of the reactant state by water molecules
rather that to the more common mode of transition state stabilization used by naturally evolved enzymes. Finally it is pointed
out that our difficulties in improving Kemp eliminase are not due to overlooking exotic effect, but to the challenge in designing
a preorganized environment that would exploit the small change it charge distribution during the formation of the transition
state.
Co-reporter:Maite Roca, Alexandra Vardi-Kilshtain and Arieh Warshel
Biochemistry 2009 Volume 48(Issue 14) pp:
Publication Date(Web):January 22, 2009
DOI:10.1021/bi802191b
The ability to design effective enzymes is one of the most fundamental challenges in biotechnology and in some respects in biochemistry. In fact, such ability would be one of the most convincing manifestations of a full understanding of the origin of enzyme catalysis. In this work, we explore the reliability of different simulation approaches, in terms of their ability to rank different possible active site constructs. This validation is done by comparing the ability of different approaches to evaluate the catalytic contributions of various residues in chorismate mutase. It is demonstrated that the empirical valence bond (EVB) model can serve as a practical yet accurate tool in the final stages of computer-aided enzyme design (CAED). Other approaches for fast screening are also examined and found to be less accurate and mainly useful for qualitative screening of ionized residues. It is pointed out that accurate ranking of different options for enzyme design cannot be accomplished by approaches that cannot capture the electrostatic preorganization effect. This is in particular true with regard to current design approaches that use gas phase or small cluster calculations and then estimate the interaction between the enzyme and the transition state (TS) model rather than the TS binding free energy or the relevant activation free energy. The ability of the EVB model to provide a tool for quantitative ranking in the final stage of CAED may help in progressing toward the design of enzymes whose catalytic power is closer to that of native enzymes than to that of the current generation of designer enzymes.
Co-reporter:Shina C. L. Kamerlin, Charles E. McKenna, Myron F. Goodman and A. Warshel
Biochemistry 2009 Volume 48(Issue 25) pp:
Publication Date(Web):April 24, 2009
DOI:10.1021/bi900140c
DNA polymerases make up a family of enzymes responsible for regulating DNA replication and repair, which in turn maintains the integrity of the genome. However, despite intensive kinetic, crystallographic, and computational studies, elucidation of the detailed enzymatic mechanism still presents a significant challenge. We recently developed an alternative strategy for exploring the fidelity and mechanism of DNA polymerases, by probing leaving group effects on nucleotidyl transfer using a series of dGTP bisphosphonate analogues in which the β,γ-bridging oxygen was replaced by a series of substituted methylene groups (X = CYZ, where Y and Z = H, halogen, or another substituent). Pre-steady state kinetic measurements of DNA polymerase-catalyzed incorporation of correctly base paired (R) and mispaired (W) analogues demonstrated a strong linear free energy relationship (LFER) between the polymerase rate constant (kpol) and the highest pKa of the free bisphosphonic acid corresponding to the leaving group. However, unexpectedly, the data segregated into two distinctly different linear correlations depending on the nature of the substituent. The discrepancy between the two lines was considerably greater when the dGTP analogue formed an incorrect (G·T) rather than a correct (G·C) base pair, although the reason for this phenomenon remains unexplained. Here, we have evaluated the complete free energy surfaces for bisphosphonate hydrolysis in aqueous solution and evaluated the corresponding LFER. Our study, which employs several alternative solvation models, finds a split of the calculated LFER for the mono- and dihalogen compounds into two parallel lines, reflecting their behavior in the polymerase-catalyzed condensation reaction. We suggest that the division into two linear subsets may be a generalized solvation phenomenon involving the overall electrostatic interaction between the substrates and their surroundings and would also be observed in polar solvents in the absence of the enzyme, if the reaction in solvent is in fact identical to that of the enzyme. However, the amplified differences between the LFER lines for the incorporation of matched and mismatched deoxynucleotides probably reflects the differences in the electrostatic interaction between the TS charges in the polymerase active site. An understanding of the mechanism of this reaction in solution could thereby provide a steppingstone for understanding the factors governing the fidelity of DNA polymerases.
Co-reporter:Shina C. L. Kamerlin and Arieh Warshel
The Journal of Physical Chemistry B 2009 Volume 113(Issue 47) pp:15692-15698
Publication Date(Web):November 4, 2009
DOI:10.1021/jp907223t
ATP hydrolysis is the driving force of many life processes, yet the exact nature of and contributions to the energetics of this reaction are far from being clear. In particular, it is unclear how much of the driving force of this reaction is due to the separation of the already dissociated ADP + Pi moieties rather than to the chemical event. This fundamental issue is explored here by ab initio calculations that use different solvation models, and it is found that, while the calculations are sensitive to the theoretical approach used, it is quite likely that the dissociation of the charged fragments makes a significant contribution to the energetics of ATP hydrolysis.
Co-reporter:Shina C. L. Kamerlin Dr.;Maciej Haranczyk Dr.
ChemPhysChem 2009 Volume 10( Issue 7) pp:1125-1134
Publication Date(Web):
DOI:10.1002/cphc.200800753
Co-reporter:Xiaojiang S. Chen;Hanbin Liu;Yemin Shi
PNAS 2009 Volume 106 (Issue 18 ) pp:7449-7454
Publication Date(Web):2009-05-05
DOI:10.1073/pnas.0900532106
The molecular origin of the action of helicases is explored, starting with a model built based on the different X-ray structures
of the large tumor antigen (LTag) hexameric helicase and a simplified model containing the ionized phosphate backbones of
a single-strand DNA. The coupling between the protein structural changes and the translocation process is quantified using
an effective electrostatic free-energy surface for the protein/DNA complex. This surface is then used in Langevin dynamics
simulations of the time dependence of the translocation process. Remarkably, the simulated motion along the free-energy surface
results in a vectorial translocation of the DNA, consistent with the biological process. The electrostatic energy of the system
appears to reproduce the directionality of this process. Thus, we are able to provide a consistent structure-based molecular
description of the energetic and dynamics of the translocation process. This analysis may have general implications for relating
structural models to translocation directionality in helicases and other DNA translocases.
Co-reporter:Shina C. L. Kamerlin, Jie Cao, Edina Rosta and Arieh Warshel
The Journal of Physical Chemistry B 2009 Volume 113(Issue 31) pp:10905-10915
Publication Date(Web):July 16, 2009
DOI:10.1021/jp901709f
In recent years, the EVB has become a widely used tool in the QM/MM modeling of reactions in condensed phases and in biological systems, with ever increasing popularity. However, despite the fact that its power and validity have been repeatedly established since 1980, a recent work (Valero, R.; et al. J. Chem. Theory Comput. 2009, 5, 1) has strongly criticized this approach, while not discussing the fact that one of the authors is effectively using it himself for both gas-phase and solution studies. Here, we have responded to the most serious unjustified assertions of that paper, covering both the more problematic aspects of that work and the more complex scientific aspects. Additionally, we have demonstrated that the poor EVB results shown in Valero et al. which where presented as verification of the unreliability of the EVB model were in fact obtained by the use of incorrect parameters, without comparing to the correct surface obtained by our program.
Co-reporter:Andrei V. Pisliakov;Jie Cao;Shina C. L. Kamerlin
PNAS 2009 Volume 106 (Issue 41 ) pp:17359-17364
Publication Date(Web):2009-10-13
DOI:10.1073/pnas.0909150106
The idea that enzymes catalyze reactions by dynamical coupling between the conformational motions and the chemical coordinates
has recently attracted major experimental and theoretical interest. However, experimental studies have not directly established
that the conformational motions transfer energy to the chemical coordinate, and simulating enzyme catalysis on the relevant
timescales has been impractical. Here, we introduce a renormalization approach that transforms the energetics and dynamics
of the enzyme to an equivalent low-dimensional system, and allows us to simulate the dynamical coupling on a ms timescale.
The simulations establish, by means of several independent approaches, that the conformational dynamics is not remembered
during the chemical step and does not contribute significantly to catalysis. Nevertheless, the precise nature of this coupling
is a question of great importance.
Co-reporter:Nidhi Singh and Arieh Warshel
The Journal of Physical Chemistry B 2009 Volume 113(Issue 20) pp:7372-7382
Publication Date(Web):April 29, 2009
DOI:10.1021/jp811063v
The evaluation of the solvation entropies is a major conceptual and practical challenge. On the one hand, it is interesting to quantify the factors that are responsible for the solvation entropies in solutions, whereas on the other, it is essential to be able to assess the contributions of the solvation entropies to the binding free energies and related properties. In fact, the solvation entropies are neglected in almost all of the studies of the binding entropies. The main problem is that widely used approaches, such as the quasiharmonic (QH) approximation, do not provide reliable results particularly in cases of shallow potential and multidimensional surfaces while brute force evaluations of the entropic effects by simulating temperature dependence of the free energy converges very slowly. This paper addresses the above issue by starting with an analysis of the factors that are responsible for the negative solvation entropy of ions, showing that it is not due to the change in the solvent vibration modes or to the solvent force constant but to the changes in the solvent configurational space upon change in the solute charges. We begin by clarifying that when one deals with aqueous solutions, it is easy to evaluate the corresponding entropic effect by the Langevin dipole (LD) treatment. However, in this work we are interested in developing a general microscopic tool that can be used to study similar effects in the proteins. To this end, we explore the ability of our restraint release (RR) approach to evaluate the solvation entropy. We start this analysis by reviewing the foundation of this approach and in particular, the requirements of minimizing the enthalpy contribution to the RR free energy. We then establish that our approach is not a specialized harmonic treatment but a rather powerful general approach. Moving to the main topic of this work, we demonstrate that the RR approach provides quantitative results for the solvation entropies of monovalent and divalent ions and effectively captures the physics of these entropic effects. The success of the current approach indicates that it should be applicable to the studies of the solvation entropies in the proteins and also, in examining hydrophobic effects. Thus, we believe that the RR approach provides a powerful tool for evaluating the corresponding contributions to the binding entropies and, eventually, to the binding free energies. This holds promise for extending the information theory modeling to proteins and protein−ligand complexes in aqueous solutions and consequently, facilitating computer-aided drug design.
Co-reporter:Shina C. L. Kamerlin, Maciej Haranczyk and Arieh Warshel
The Journal of Physical Chemistry B 2009 Volume 113(Issue 5) pp:1253-1272
Publication Date(Web):December 4, 2008
DOI:10.1021/jp8071712
Hybrid quantum mechanical/molecular mechanical (QM/MM) approaches have been used to provide a general scheme for chemical reactions in proteins. However, such approaches still present a major challenge to computational chemists, not only because of the need for very large computer time in order to evaluate the QM energy but also because of the need for proper computational sampling. This review focuses on the sampling issue in QM/MM evaluations of electrostatic energies in proteins. We chose this example since electrostatic energies play a major role in controlling the function of proteins and are key to the structure−function correlation of biological molecules. Thus, the correct treatment of electrostatics is essential for the accurate simulation of biological systems. Although we will be presenting different types of QM/MM calculations of electrostatic energies (and related properties) here, our focus will be on pKa calculations. This reflects the fact that pKa’s of ionizable groups in proteins provide one of the most direct benchmarks for the accuracy of electrostatic models of macromolecules. While pKa calculations by semimacroscopic models have given reasonable results in many cases, existing attempts to perform pKa calculations using QM/MM-FEP have led to discrepancies between calculated and experimental values. In this work, we accelerate our QM/MM calculations using an updated mean charge distribution and a classical reference potential. We examine both a surface residue (Asp3) of the bovine pancreatic trypsin inhibitor and a residue buried in a hydrophobic pocket (Lys102) of the T4-lysozyme mutant. We demonstrate that, by using this approach, we are able to reproduce the relevant side chain pKa’s with an accuracy of 3 kcal/mol. This is well within the 7 kcal/mol energy difference observed in studies of enzymatic catalysis, and is thus sufficient accuracy to determine the main contributions to the catalytic energies of enzymes. We also provide an overall perspective of the potential of QM/MM calculations in general evaluations of electrostatic free energies, pointing out that our approach should provide a very powerful and accurate tool to predict the electrostatics of not only solution but also enzymatic reactions, as well as the solvation free energies of even larger systems, such as nucleic acid bases incorporated into DNA.
Co-reporter:Edina Rosta, Shina C. L. Kamerlin and Arieh Warshel
Biochemistry 2008 Volume 47(Issue 12) pp:
Publication Date(Web):February 29, 2008
DOI:10.1021/bi702106m
The hydrolysis of phosphate esters is crucially important to biological systems, being involved in, among other things, signaling, energy transduction, biosynthesis, and the regulation of protein function. Despite this, there are many questions that remain unanswered in this important field, particularly with regard to the preferred mechanism of hydrolysis of phosphate esters, which can proceed through any of multiple pathways that are either associative or dissociative in nature. Previous comparisons of calculated and observed linear free energy relationships (LFERs) for phosphate monoester dianions with different leaving groups showed that the TS character gradually changes from associative to dissociative with the increasing acidity of the leaving group, while reproducing the experimental LFER. Here, we have generated ab initio potential energy surfaces for the hydrolysis of phosphate diesters in solution, with a variety of leaving groups. Once again, the reaction changes from a compact concerted pathway to one that is more expansive in character when the acidity of the leaving group increases. When such systems are examined in solution, it is essential to take into consideration the contribution of solute to the overall activation entropy, which remains a major computational challenge. The popular method of calculating the entropy using a quasi-harmonic approximation appears to markedly overestimate the configurational entropy for systems with multiple occupied energy wells. We introduce an improved restraint release approach for evaluating configurational entropies and apply this approach to our systems. We demonstrate that when this factor is taken into account, it is possible to reproduce the experimental LFER for this system with reasonable accuracy.
Co-reporter:Hiroshi Ishikita Dr. Dr.
Angewandte Chemie 2008 Volume 120( Issue 4) pp:709-712
Publication Date(Web):
DOI:10.1002/ange.200704178
Co-reporter:Hiroshi Ishikita Dr. Dr.
Angewandte Chemie International Edition 2008 Volume 47( Issue 4) pp:697-700
Publication Date(Web):
DOI:10.1002/anie.200704178
Co-reporter:Maite Roca;Donald Hilvert;Benjamin Messer
PNAS 2008 Volume 105 (Issue 37 ) pp:13877-13882
Publication Date(Web):2008-09-16
DOI:10.1073/pnas.0803405105
Elucidating the relationship between the folding landscape of enzymes and their catalytic power has been one of the challenges
of modern enzymology. The present work explores this issue by using a simplified folding model to generate the free-energy
landscape of an enzyme and then to evaluate the activation barriers for the chemical step in different regions of the landscape.
This approach is used to investigate the recent finding that an engineered monomeric chorismate mutase exhibits catalytic
efficiency similar to the naturally occurring dimer even though it exhibits the properties of an intrinsically disordered
molten globule. It is found that the monomer becomes more confined than its native-like counterpart upon ligand binding but
still retains a wider catalytic region. Although the overall rate acceleration is still determined by reduction of the reorganization
energy, the detailed contribution of different barriers yields a more complex picture for the chemical process than that of
a single path. This work provides insight into the relationship between folding landscapes and catalysis. The computational
approach used here may also provide a powerful strategy for modeling single-molecule experiments and designing enzymes.
Co-reporter:Pankaz K. Sharma;Zhen T. Chu;Andrei V. Pisliakov;Maciej Haranczyk
PNAS 2008 Volume 105 (Issue 22 ) pp:7726-7731
Publication Date(Web):2008-06-03
DOI:10.1073/pnas.0800580105
Gaining detailed understanding of the energetics of the proton-pumping process in cytochrome c oxidase (CcO) is one of the challenges of modern biophysics. Despite promising mechanistic proposals, most works have not
related the activation barriers of the different assumed steps to the protein structure, and there has not been a physically
consistent model that reproduced the barriers needed to create a working pump. This work reevaluates the activation barriers
for the primary proton transfer (PT) steps by calculations that reflect all relevant free energy contributions, including
the electrostatic energies of the generated charges, the energies of water insertion, and large structural rearrangements
of the donor and acceptor. The calculations have reproduced barriers that account for the directionality and sequence of events
in the primary PT in CcO. It has also been found that the PT from Glu-286 (E) to the propionate of heme a3 (Prd) provides a gate for an initial back leakage from the high pH side of the membrane. Interestingly, the rotation of E
that brings it closer to Prd appears to provide a way for blocking competing pathways in the primary PT. Our study elucidates
and quantifies the nature of the control of the directionality in the primary PT in CcO and provides instructive insight into
the role of the water molecules in biological PT, showing that “bridges” of several water molecules in hydrophobic regions
present a problem (rather than a solution) that is minimized in the primary PT.
Co-reporter:Pankaz K. Sharma;Zhen T. Chu;Mats H. M. Olsson;
Proceedings of the National Academy of Sciences 2007 104(23) pp:9661-9666
Publication Date(Web):May 21, 2007
DOI:10.1073/pnas.0702238104
The catalytic power of enzymes containing coenzyme B12 cofactor has been, in some respects, the “last bastion” for the strain hypothesis. The present work explores the origin of
this effect by using simulation methods that overcome the sampling difficulties of previous energy minimization studies. It
is found that the major part of the catalytic effect is due to the electrostatic interaction between the ribose and the protein,
and that the strain contribution is very small. Remarkably, enzymes can use electrostatic effects even in a radical process,
when the charge distribution of the reacting fragments does not change significantly during the reaction. Electrostatic catalysis
can, in such cases, be obtained by attaching a polar group to the leaving fragment and designing an active site that interacts
more strongly with this group in the product state than in the reactant state. The finding that evolution had to use this
trick provides further evidence to the observation that it is extremely hard to catalyze enzymatic reactions by nonelectrostatic
factors. The trick used by B12 enzymes may, in fact, be a very powerful new strategy in enzyme design.
Co-reporter:Arieh Warshel, Pankaz K. Sharma, Mitsunori Kato, William W. Parson
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2006 Volume 1764(Issue 11) pp:1647-1676
Publication Date(Web):November 2006
DOI:10.1016/j.bbapap.2006.08.007
Electrostatic energies provide what is perhaps the most effective tool for structure–function correlation of biological molecules. This review considers the current state of simulations of electrostatic energies in macromolecules as well as the early developments of this field. We focus on the relationship between microscopic and macroscopic models, considering the convergence problems of the microscopic models and the fact that the dielectric ‘constants’ in semimacroscopic models depend on the definition and the specific treatment. The advances and the challenges in the field are illustrated considering a wide range of functional properties including pKa's, redox potentials, ion and proton channels, enzyme catalysis, ligand binding and protein stability. We conclude by pointing out that, despite the current problems and the significant misunderstandings in the field, there is an overall progress that should lead eventually to quantitative descriptions of electrostatic effects in proteins and thus to quantitative descriptions of the function of proteins.
Co-reporter:Arieh Warshel
PNAS 2005 Volume 102 (Issue 6 ) pp:1813-1814
Publication Date(Web):2005-02-08
DOI:10.1073/pnas.0409788102
Co-reporter:William W. Parson, Arieh Warshel
Chemical Physics 2004 Volume 296(2–3) pp:201-216
Publication Date(Web):26 January 2004
DOI:10.1016/j.chemphys.2003.10.006
Abstract
The dispersed-polaron (spin-boson) model is reviewed briefly and then used to develop a density-matrix model for studies of electron transfer in condensed phases. The frequencies and Franck–Condon factors for solvent vibrational modes that are coupled to electron transfer are obtained from molecular dynamics (MD) simulations by the dispersed-polaron treatment. Microscopic rate constants for vibrational relaxations, dephasing and coherence transfer between the solvent modes are obtained by fitting the time dependence of the solvent coordinates in the density-matrix treatment to the corresponding time dependence obtained from molecular-dynamics simulations with a classical linear-response approximation. This is done by adjusting a single parameter, the time constant for thermal equilibration of the two lowest levels of a solvent mode (T10). The model thus focuses on the coupling between solvent modes, rather than on the more widely studied coupling of solute modes by the thermal bath. The resulting density-matrix model is used to examine vibronic coupling in the initial electron-transfer step in photosynthetic bacterial reaction centers. Values of T10 in the range of 1–2 ps are consistent with molecular-dynamics simulations of the time-dependent energy gap between the reactant and product states (P* and P+B−), and also with the damping of coherent vibrational motions that are seen experimentally after excitation of reaction centers with a short pulse of light. In both the density-matrix model and the MD simulations, the autocorrelation function of the energy gap also has a decay component with a time constant of about 50 fs, which we ascribe to the group dephasing of oscillatory motions at many different frequencies. This component is insensitive to vibrational relaxations and is largely irrelevant to the electron-transfer dynamics. Using values of T10 in the range of 1–2 ps, a model with five vibrational modes reproduces the main features of electron transfer from P* to B, including stepwise formation of the product during the period when the system retains vibrational coherences. Although the rate does not depend strongly on whether P* is prepared coherently or incoherently, speeding up vibrational relaxations decreases the rate. At least part of the adverse effect of rapid relaxations can be viewed as a manifestation of the quantum Zeno paradox, which arises when off-diagonal elements of the density matrix decay very rapidly.
Co-reporter:Montserrat Barbany;Hugo Gutiérrez-de-Terán;Ferran Sanz ;Jordi Villà-Freixa Dr.
ChemBioChem 2003 Volume 4(Issue 4) pp:
Publication Date(Web):26 MAR 2003
DOI:10.1002/cbic.200390048
The effective design of catalytic antibodies represents a major conceptual and practical challenge. It is implicitly assumed that a proper transition state analogue (TSA) can elicit a catalytic antibody (CA) that will catalyze the given reaction in a similar way to an enzyme that would evolve (or was evolved) to catalyze this reaction. However, in most cases it was found that the TSA used produced CAs with relatively low rate enhancement as compared to the corresponding enzymes, when these exist. The present work explores the origin of this problem, by developing two approaches that examine the similarity of the TSA and the corresponding transition state (TS). These analyses are used to assess the proficiency of the CA generated by the given TSA. Both approaches focus on electrostatic effects that have been found to play a major role in enzymatic reactions. The first method uses molecular interaction potentials to look for the similarity between the TSA and the TS and, in principle, to help in designing new haptens by using 3D quantitative struture–activity relationships. The second and more quantitative approach generates a grid of Langevin dipoles, which are polarized by the TSA, and then uses the grid to bind the TS. Comparison of the resulting binding energy with the binding energy of the TS to the grid that was polarized by the TS provides an estimate of the proficiency of the given CA. Our methods are used in examining the origin of the difference between the catalytic power of the 1F7 CA and chorismate mutase. It is demonstrated that the relatively small changes in charge and structure between the TS and TSA are sufficient to account for the difference in proficiency between the CA and the enzyme. Apparently the environment that was preorganized to stabilize the TSA charge distribution does not provide a sufficient stabilization to the TS. The general implications of our findings and the difficulties in designing a perfect TSA are discussed. Finally, the possible use of our approach in screening for an optimal TSA is pointed out.
Co-reporter:Jan Florián;Myron F. Goodman
Biopolymers 2003 Volume 68(Issue 3) pp:
Publication Date(Web):30 JAN 2003
DOI:10.1002/bip.10244
Computer simulations can provide in principle quantitative correlation between the structures of DNA polymerases and the replication fidelity. This paper describes our progress in this direction. Using several theoretical approaches, including the free energy perturbation (FEP), linear response approximation (LRA), and the empirical valence bond (EVB) methods, we examined the stability of several mismatched base pairs in DNA duplex in aqueous solution, the contribution of binding energy to the fidelity of DNA polymerases β and T7, and the mechanism and energetics of the polymerization reaction catalyzed by T7 DNA polymerase. © 2003 Wiley Periodicals, Inc. Biopolymers 68: 286–299, 2003
Co-reporter:Arieh Warshel ;Jan Florián;Marek Štrajbl;Jordi Villà
ChemBioChem 2001 Volume 2(Issue 2) pp:
Publication Date(Web):26 JAN 2001
DOI:10.1002/1439-7633(20010202)2:2<109::AID-CBIC109>3.0.CO;2-9
The enormous enhancement of the transformation of orotate into uracil in the active site of orotidine monophosphate decarboxylase (ODCase) has been recently attributed to a ground-state destabilization (GSD) mechanism. These proposals are scrutinized here by reconsidering the relevant energetics on the basis of the crystal structure of this enzyme (see picture of the active site with bound substrate). The present analysis leads to the conclusion that ODCase owes its catalytic power to a transition-state stabilization mechanism rather than to a GSD mechanism.
Co-reporter:J. Villà;M. Štrajbl;T. M. Glennon;Y. Y. Sham;Z. T. Chu;A. Warshel
PNAS 2000 Volume 97 (Issue 22 ) pp:11899-11904
Publication Date(Web):2000-10-24
DOI:10.1073/pnas.97.22.11899
The idea that enzymes accelerate their reactions by entropic
effects has played a major role in many prominent proposals about the
origin of enzyme catalysis. This idea implies that the binding to an
enzyme active site freezes the motion of the reacting fragments and
eliminates their entropic contributions,
(ΔScat‡)′, to the
activation energy. It is also implied that the binding entropy is equal
to the activation entropy,
(ΔSw‡)′, of the
corresponding solution reaction. It is, however, difficult to examine
this idea by experimental approaches. The present paper defines the
entropic proposal in a rigorous way and develops a computer simulation
approach that determines (ΔS‡)′. This
approach allows us to evaluate the differences between
(ΔS‡)′ of an enzymatic reaction and of the
corresponding reference reaction in solution. Our approach is used in a
study of the entropic contribution to the catalytic reaction of
subtilisin. It is found that this contribution is much smaller than
previously thought. This result is due to the following:
(i) Many of the motions that are free in the
reactants state of the reference solution reaction are also free at the
transition state. (ii) The binding to the enzyme
does not completely freeze the motion of the reacting fragments so that
(ΔS‡)′ in the enzymes is not zero.
(iii) The binding entropy is not necessarily equal
to (ΔSw‡)′.
Co-reporter:Suman Chakrabarty, Ida Namslauer, Peter Brzezinski, Arieh Warshel
Biochimica et Biophysica Acta (BBA) - Bioenergetics (April 2011) Volume 1807(Issue 4) pp:413-426
Publication Date(Web):April 2011
DOI:10.1016/j.bbabio.2011.01.004
Co-reporter:Shina Caroline Lynn Kamerlin, Janez Mavri, A. Warshel
FEBS Letters (2 July 2010) Volume 584(Issue 13) pp:2759-2766
Publication Date(Web):2 July 2010
DOI:10.1016/j.febslet.2010.04.062
The idea that tunneling is enhanced by the compression of the donor–acceptor distance has attracted significant interest. In particular, recent studies argued that this proposal is consistent with pressure effects on enzymatic reactions, and that the observed pressure effects support the idea of vibrationally enhanced catalysis. However, a careful analysis of the current works reveals serious inconsistencies in the evidence presented to support these hypotheses. Apparently, tunneling decreases upon compression, and external pressure does not lead to the applicable compression of the free energy surface. Additionally, pressure experiments do not provide actual evidence for vibrationally enhanced catalysis. Finally, the temperature dependence of the entropy change in hydride transfer reactions is shown to reflect simple electrostatic effects.
Co-reporter:Peter Oelschlaeger, Marco Klahn, William A. Beard, Samuel H. Wilson, Arieh Warshel
Journal of Molecular Biology (16 February 2007) Volume 366(Issue 2) pp:687-701
Publication Date(Web):16 February 2007
DOI:10.1016/j.jmb.2006.10.095
Human DNA polymerase β (pol β) fills gaps in DNA as part of base excision DNA repair. Due to its small size it is a convenient model enzyme for other DNA polymerases. Its active site contains two Mg2+ ions, of which one binds an incoming dNTP and one catalyzes its condensation with the DNA primer strand. Simulating such binuclear metalloenzymes accurately but computationally efficiently is a challenging task. Here, we present a magnesium-cationic dummy atom approach that can easily be implemented in molecular mechanical force fields such as the ENZYMIX or the AMBER force fields. All properties investigated here, namely, structure and energetics of both Michaelis complexes and transition state (TS) complexes were represented more accurately using the magnesium-cationic dummy atom model than using the traditional one-atom representation for Mg2+ ions. The improved agreement between calculated free energies of binding of TS models to different pol β variants and the experimentally determined activation free energies indicates that this model will be useful in studying mutational effects on catalytic efficiency and fidelity of DNA polymerases. The model should also have broad applicability to the modeling of other magnesium-containing proteins.
Co-reporter:Maite Roca, Benjamin Messer, Arieh Warshel
FEBS Letters (15 May 2007) Volume 581(Issue 10) pp:2065-2071
Publication Date(Web):15 May 2007
DOI:10.1016/j.febslet.2007.04.025
The ability to predict the thermal stability of proteins based on their corresponding sequence is a problem of great fundamental and practical importance. Here we report an approach for calculating the electrostatic contribution to protein stability based on the use of the semimacroscopic protein dipole Langevin dipole (PDLD/S) in its linear response approximation version for self-energy with a dielectric constant, (εp) and an effective dielectric for charge–charge interactions (εeff). The method is applied to the test cases of ubiquitin, lipase, dihydrofolate reductase and cold shock proteins with series of εp and εeff. It is found that the optimal values of these dielectric constants lead to very promising results, both for the relative stability and the absolute folding energy. Consideration of the specific values of the optimal dielectric constants leads to an exciting conceptual description of the reorganization effect during the folding process. Although this description should be examined by further microscopic studies, the practical use of the current approach seems to offer a powerful tool for protein design and for studies of the energetics of protein folding.
Co-reporter:Shina Caroline Lynn Kamerlin and Arieh Warshel
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 22) pp:NaN10411-10411
Publication Date(Web):2011/04/27
DOI:10.1039/C0CP02823A
Recent years have witnessed a tremendous explosion in computational power, which in turn has resulted in great progress in the complexity of the biological and chemical problems that can be addressed by means of all-atom simulations. Despite this, however, our computational time is not infinite, and in fact many of the key problems of the field were resolved long before the existence of the current levels of computational power. This review will start by presenting a brief historical overview of the use of multiscale simulations in biology, and then present some key developments in the field, highlighting several cases where the use of a physically sound simplification is clearly superior to a brute-force approach. Finally, some potential future directions will be discussed.
.jpg)
![3-[[(4r,5s,6s,7r)-4,7-dibenzyl-5,6-dihydroxy-2-oxo-3-[[3-(1,3-thiazol-2-ylcarbamoyl)phenyl]methyl]-1,3-diazepan-1-yl]methyl]-n-(1,3-thiazol-2-yl)benzamide](http://img.cochemist.com/ccimg/183900/183854-11-7.png)
![3-[[(4r,5s,6s,7r)-4,7-dibenzyl-5,6-dihydroxy-2-oxo-3-[[3-(1,3-thiazol-2-ylcarbamoyl)phenyl]methyl]-1,3-diazepan-1-yl]methyl]-n-(1,3-thiazol-2-yl)benzamide](http://img.cochemist.com/ccimg/183900/183854-11-7_b.png)
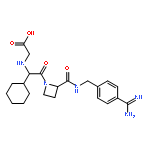
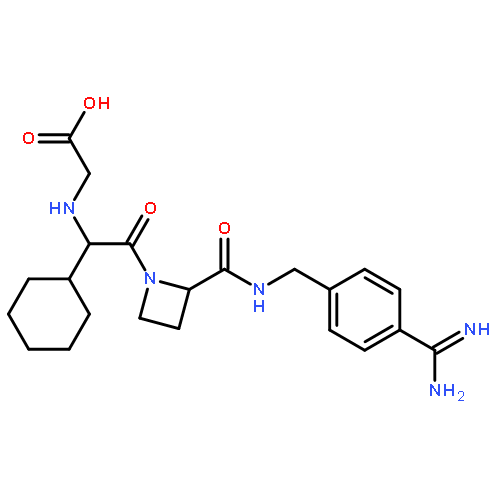
![Imidazo[4,5,1-jk][1,4]benzodiazepine-2(1H)-thione,9-chloro-4,5,6,7-tetrahydro-5-methyl-6-(3-methyl-2-butenyl)-, (R)- (9CI)](http://img.cochemist.com/ccimg/131700/131645-71-1.png)
![Imidazo[4,5,1-jk][1,4]benzodiazepine-2(1H)-thione,9-chloro-4,5,6,7-tetrahydro-5-methyl-6-(3-methyl-2-butenyl)-, (R)- (9CI)](http://img.cochemist.com/ccimg/131700/131645-71-1_b.png)




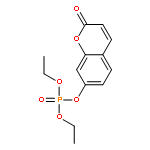
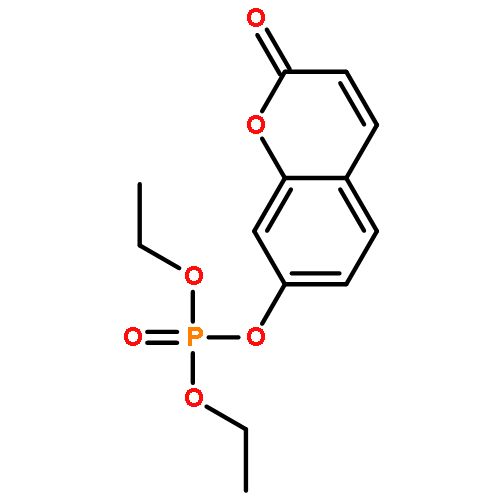


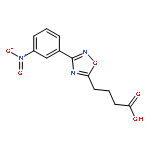
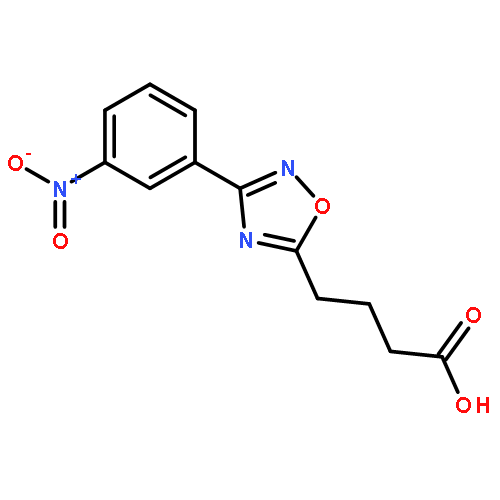


![2-Oxabicyclo[3.3.1]non-6-ene-3,5-dicarboxylic acid, 8-hydroxy-, (1R,3S,5S,8R)-](/data/chemimg/3753600/139012-48-9.png)
![2-Oxabicyclo[3.3.1]non-6-ene-3,5-dicarboxylic acid, 8-hydroxy-, (1R,3S,5S,8R)-](/data/chemimg/3753600/139012-48-9_b.png)
![Imidazo[4,5,1-jk][1,4]benzodiazepin-2(1H)-one, 4,5,6,7-tetrahydro-5-methyl-6-(3-methyl-2-buten-1-yl)-, (5R)-](/data/chemimg/459900/131613-18-8.png)
![Imidazo[4,5,1-jk][1,4]benzodiazepin-2(1H)-one, 4,5,6,7-tetrahydro-5-methyl-6-(3-methyl-2-buten-1-yl)-, (5R)-](/data/chemimg/459900/131613-18-8_b.png)





![1H-Thieno[3,4-d]imidazole-6-pentanoicacid, 2-amino-3a,4,6,6a-tetrahydro-, (3aR,6S,6aS)-](http://img.cochemist.com/ccimg/13400/13395-35-2.png)
![1H-Thieno[3,4-d]imidazole-6-pentanoicacid, 2-amino-3a,4,6,6a-tetrahydro-, (3aR,6S,6aS)-](http://img.cochemist.com/ccimg/13400/13395-35-2_b.png)











![2H-1,3-Diazepin-2-one,hexahydro-5,6-dihydroxy-1,3-bis[[4-(hydroxymethyl)phenyl]methyl]-4,7-bis(phenylmethyl)-,(4R,5S,6S,7R)-](http://img.cochemist.com/ccimg/151900/151867-81-1.png)
![2H-1,3-Diazepin-2-one,hexahydro-5,6-dihydroxy-1,3-bis[[4-(hydroxymethyl)phenyl]methyl]-4,7-bis(phenylmethyl)-,(4R,5S,6S,7R)-](http://img.cochemist.com/ccimg/151900/151867-81-1_b.png)

