Co-reporter:Kejing Lao, Jie Sun, Chong Wang, Weiting Lyu, Boshen Zhou, Ruheng Zhao, Qian Xu, Qidong You, Hua Xiang
Steroids 2017 Volume 124(Volume 124) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.steroids.2017.05.011
•A series of novel steroidal androst-3,5-diene-3-carboxylic acids have been designed and synthesized.•Most of the synthesized compounds displayed excellent 5α-reductase inhibitory potency.•16a was found to be the most potential inhibitor against type 1 and 2 isozymes.•16a showed good prostate weight reduction effect and pharmacokinetic properties.5α-Reductase is a key enzyme responsible for dihydrotestosterone biosynthesis and has been recognized as an important target for discovering new drugs against benign prostatic hyperplasia (BPH). In this study, a series of novel steroidal androst-3,5-diene-3-carboxylic acids have been designed and synthesized. Biological evaluations were performed on their 5α-reductase inhibitory activities by both in vitro enzyme inhibition assay and in vivo by prostate weighing method. Results showed that most of them displayed excellent 5α-reductase inhibitory potency. Detailed evaluation indicated that most of the compounds displayed slightly higher inhibition potency towards type 2 isozyme. Among all the compounds, 16a was found to be the most potential inhibitor with the IC50 of 0.25 μM and 0.13 μM against type 1 and 2 isozymes respectively. In vivo 5a-reductase inhibitory evaluation of 16a also showed a more significant reduction effect (p < 0.001) in rat prostate weight than epristeride. Furthermore, the results of in silico ADME study indicated that compound 16a exhibited good pharmacokinetic properties. Thus, 16a could serve as promising lead candidates for further study.Download high-res image (115KB)Download full-size image
Co-reporter:Guoshun Luo, Xinyu Li, Guoqing Zhang, Chengzhe Wu, Zhengpu Tang, Linyi Liu, Qidong You, Hua Xiang
European Journal of Medicinal Chemistry 2017 Volume 140(Volume 140) pp:
Publication Date(Web):10 November 2017
DOI:10.1016/j.ejmech.2017.09.015
•A series of coumarin-based analogues were rationally designed and synthesized.•42d inhibited cancer cell proliferation and migration as well as angiogenesis.•Synergetic effect of 42d on ERα and VEGFR-2/Raf-1/MAPK/ERK pathway.•42d turned out to be a promising dual targeting candidate for breast cancer.There is considerable interest in developing new SERMs as multifunctional agents in women's health. Development of dual selective estrogen receptor modulators/VEGFR-2 inhibitors (SERMs/V-2I) has been an attractive strategy for the discovery of new breast cancer therapeutic agents. Our previous efforts led to the preparation of a series of 3-aryl-4-anilino-2H-chromen-2-ones endowed with potent estrogen receptor binding affinity and anti-proliferative efficacy. In this study, various structurally related 3-aryl-4-anilino/aryloxy-2H-chromen-2-one analogues were rationally designed, synthesized and evaluated as a new chemo-type of dual ERα and VEGFR-2 inhibitors. Most of the derivatives exhibited potent activities in both enzymatic and cellular assays. SAR investigation revealed that introducing of bioisosteric O atom at the C-4 position of coumarin scaffold is beneficial to improve the inhibitory potency, especially in ERα binding affinity assay. Furthermore, most of the piperidyl substituted compounds showed better inhibitory activity against MCF-7 and Ishikawa cells than lead compounds BL-18d, tamoxifen and Vandetanib. Optimization of the hit compound, identified in an ERα binding affinity assay, led to compound 42d, exhibiting an IC50 for ERα binding affinity of 2.19 μM while retaining an excellent inhibition on VGFR-2 as well as a potent suppression on the growth of angiogenesis-related cells. In RT-PCR assay, 42d exerted significantly antiestrogenic property via suppressing the expression of progesterone receptor (PgR) mRNA in MCF-7 cells, which was consistent with the ERα antagonistic property of a selective estrogen receptor modulator. Further mechanism investigation demonstrated that compound 42d could inhibit the activation of VEGFR-2 and subsequent signaling transduction of Raf-1/MAPK/ERK pathway in MCF-7 cells. All these results together with molecular modeling studies open a new avenue for the development of multifunctional agents targeting ERα and VEGFR-2 in the therapy of some breast cancers.Download high-res image (222KB)Download full-size image
Co-reporter:Kejing Lao, Yejun Wang, Mingqi Chen, Jingjing Zhang, Qidong You, Hua Xiang
European Journal of Medicinal Chemistry 2017 Volume 139(Volume 139) pp:
Publication Date(Web):20 October 2017
DOI:10.1016/j.ejmech.2017.08.016
•A series of 11α-substituted 2-methoxyestradiol analogs have been designed and synthesized.•Most of the synthesized compounds displayed excellent good anti-proliferative activities.•24c and 30c could induce G2/M cell cycle arrest as well as significant anti-estrogenic activity.•24c and 30c presented significantly anti-angiogenesis activity comparable with 2-ME2.2-methoxyestradiol is a novel agent showing both anti-angiogenic and vascular disrupting properties. In this study, a series of 11α-substituted 2-methoxyestradiol analogs have been designed and synthesized targeting dual ERα and microtubulin. Biological evaluation was performed on their anti-proliferative activities against 5 different cell lines. The results indicated that most compounds exhibited good activities, in which compound 24c and 30c showed the best activity with low micromolar IC50 (2.73 μM −7.75 μM) in all cell lines. The investigation of ER affinity showed that the majority of the compounds displayed good activity at the concentration of 50 μM. In further mechanism study, it was observed that 24c and 30c could induce G2/M cell cycle arrest as well as significant anti-estrogenic activity. In CAM assay, compound 24c and 30c presented significantly anti-angiogenesis activity comparable with 2-methoxyestradiol. Overall, based on biological activities data, 24c and 30c can be identified as a potential lead molecule which might be of therapeutic importance for cancer treatment.Download high-res image (106KB)Download full-size image
Co-reporter:Kejing Lao, Jie Sun, Chong Wang, Ying Wang, Qidong You, Hong Xiao, Hua Xiang
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 17(Issue 17) pp:
Publication Date(Web):1 September 2017
DOI:10.1016/j.bmcl.2017.05.078
Prostate cancer (PCa) is the second leading cause of death in men. Recently, some researches have showed that 5α-reductase inhibitors were beneficial in PCa treatment as well. In this study, a series of novel 3-oxo-4-oxa-5α-androst-17β-amide derivatives have been designed and synthesized in a more simple and convenient method. Most of the synthesized compounds displayed good 5α-reductase inhibitory activities and androgen receptor binding affinities. Their anti-proliferation activities in PC-3 and LNCaP cell lines were also evaluated and the results indicated that most of the synthesized compounds exhibited potent anti-proliferative activities. It is obvious that the androgen-dependent cell line LNCaP was much more sensitive than the androgen-independent cell line PC-3. Among all the synthesized compounds, 11d and 11k displayed the best inhibition activity with 4-fold more sensitive toward LNCaP than PC-3, which was consistent with their high affinities observed in AR binding assay. Molecular modeling studies suggested that 11k could bind to AR in a manner similar to the binding of dihydrotestosterone to AR. Compared to the finasteride, 11k showed a longer plasma half-life (4 h) and a better bioavailability. Overall, based on biological activities data, compound 11d and 11k can be identified as potential dual 5α-reductase inhibitors and AR antagonists which might be of therapeutic importance for prostate cancer treatment.Download high-res image (108KB)Download full-size image
Co-reporter:Guoshun Luo, Mingqi Chen, Weiting Lyu, Ruheng Zhao, Qian Xu, Qidong You, Hua Xiang
Bioorganic & Medicinal Chemistry Letters 2017 Volume 27, Issue 12(Issue 12) pp:
Publication Date(Web):15 June 2017
DOI:10.1016/j.bmcl.2017.04.029
The estrogen receptor (ER) has played an important role in breast cancer development and progression and is a central target for anticancer drug discovery. In order to develop novel selective ERα modulators (SERMs), we designed and synthesized 18 novel 3-aryl-4-anilino-2H-chromen-2-one derivatives based on previously reported lead compounds. The biological results indicated that most of the compounds presented potent ERα binding affinity and possessed better anti-proliferative activities against MCF-7 and Ishikawa cell lines than the positive control tamoxifen. The piperidyl substituted compounds such as 16d and 18d demonstrated strong ERα binding affinities and excellent anti-proliferative activities respectively. Compound 18d displayed the most potent ERα binding affinity with RBA value of 2.83%, while 16d exhibited the best anti-proliferative activity against MCF-7 cells with IC50 value of 4.52 ± 2.47 μM. Further molecular docking studies were also carried out to investigate binding pattern of the newly synthesized compounds with ERα. All these results together with the structure–activity relationships (SARs) indicated that these 3-aryl-4-anilino-2H-chromen-2-one derivatives with basic side chain could serve as promising leads for further optimization as novel SERMs.Download high-res image (155KB)Download full-size image
Co-reporter:Zhichao Tang, Chengzhe Wu, Tianlin Wang, Kejing Lao, Yejun Wang, Linyi Liu, Moses Muyaba, Pei Xu, Conghui He, Guoshun Luo, Zhouyang Qian, Shaoxiong Niu, Lijun Wang, Ying Wang, Hong Xiao, Qidong You, Hua Xiang
European Journal of Medicinal Chemistry 2016 Volume 118() pp:328-339
Publication Date(Web):8 August 2016
DOI:10.1016/j.ejmech.2016.04.029
•Designed multiple ligands may exert improved efficacy with lower incidence of side effects.•A series of 6-aryl-indenoisoquinolone derivatives were described as dual ERα and VEGFR-2 inhibitors.•Compound 21c turned out to be a promising dual targeting candidate for breast cancer.The estrogen receptors have played important roles in breast cancer development and progression. Selective estrogen receptor modulators, such as Tamoxifen, have showed great benefits in the treatment and prevention of breast cancer. But the disadvantages of induction of endometrial cancer and drug resistance have limited their use. Multiple ligand which act at multiple biomolecular targets may exert favorable advantages of improved efficacy with lower incidence of side effects. In this work, we described the synthesis and evaluation of a series of 6-aryl-indenoisoquinolone derivatives as dual ERα and VEGFR-2 inhibitors. These compounds presented good ERα binding affinity and ERα antagonistic activity, as well as potent VEGFR-2 inhibitory potency. They also possessed excellent anti-proliferative activities against MCF-7, MDA-MB-231, Ishikawa and HUVEC cell lines. Further investigation of selective compound 21c showed that it was able to inhibit the activation of VEGFR-2 and the signaling transduction of Raf-1/MAPK/ERK pathway in MCF-7 cells.
Co-reporter:Xinge Zhao, Wei Huang, Yazhou Wang, Minhang Xin, Qiu Jin, Jianfeng Cai, Feng Tang, Yong Zhao, Hua Xiang
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 4) pp:891-901
Publication Date(Web):15 February 2015
DOI:10.1016/j.bmc.2014.10.043
A series of novel reversible BTK inhibitors was designed based on the structure of the recently reported preclinical drug RN486. Knowledge of the binding mode of RN486 led to the design of new inhibitors that utilized pyrrolo[2,3-d]pyrimidine to conformationally restrain key pharmacophoric groups within the molecule. Comprehensive SAR was disclosed and the most promising compound 4x displayed superior activity both in BTK enzyme (IC50 = 4.8 nM) and cellular inhibition (IC50 = 17 nM) assays to that of RN486.Graphical abstract
Co-reporter:Xinge Zhao, Minhang Xin, Wei Huang, Yanliang Ren, Qiu Jin, Feng Tang, Hailong Jiang, Yazhou Wang, Jie Yang, Shifu Mo, Hua Xiang
Bioorganic & Medicinal Chemistry 2015 Volume 23(Issue 2) pp:348-364
Publication Date(Web):15 January 2015
DOI:10.1016/j.bmc.2014.11.006
•Novel 5-phenylpyridin-2(1H)-one derivatives were designed, synthesized and evaluated for Btk inhibitors.•Some compounds exhibited excellent Btk inhibition in vitro assay.•Compound 16b possessed better PK properties and favorable anti-RA activity in vivo.A series of novel reversible Btk inhibitors has been designed based on the structure of the recently reported preclinical drug RN486. The synthesis and SAR of these compounds are described. Among these derivatives, compound 16b was identified to be a potent and orally available reversible agent with satisfactory Btk enzymatic and cellular inhibition in vitro, as well as favorable PK properties and inhibition of arthritis in vivo.
Co-reporter:Xinge Zhao, Wei Huang, Yazhou Wang, Minhang Xin, Qiu Jin, Jianfeng Cai, Feng Tang, Yong Zhao, Hua Xiang
Bioorganic & Medicinal Chemistry 2015 23(15) pp: 4344-4353
Publication Date(Web):
DOI:10.1016/j.bmc.2015.06.023
Co-reporter:Zhichao Tang, Shaoxiong Niu, Fei Liu, Kejing Lao, Jingshan Miao, Jinzi Ji, Xiang Wang, Ming Yan, Luyong Zhang, Qidong You, Hong Xiao, Hua Xiang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 9) pp:2129-2133
Publication Date(Web):1 May 2014
DOI:10.1016/j.bmcl.2014.03.042
The estrogen receptor α is recognized as important pharmaceutical target for breast cancer therapy, and vascular endothelial growth factor receptors (VEGFRs) play important roles in tumor angiogenesis including breast cancer. A series of 2,3-diaryl isoquinolinone derivatives were designed and synthesized targeting both estrogen receptor α (ERα) and VEGFR-2. Bioactivity evaluation showed that compounds 7c, 7d and 7f exhibited significant anti-proliferative and anti-angiogenesis activities via ERα and VEGFR-2 dependent mechanisms.
Co-reporter:Guoshun Luo, Moses Muyaba, Weiting Lyu, Zhichao Tang, Ruheng Zhao, Qian Xu, Qidong You, Hua Xiang
Bioorganic & Medicinal Chemistry Letters (15 February 2017) Volume 27(Issue 4) pp:
Publication Date(Web):15 February 2017
DOI:10.1016/j.bmcl.2017.01.013
Various 3-substituted 4-anilino-coumarin derivatives have been designed, synthesized and their anti-proliferative properties have been studied. The in vitro cytotoxicity screening was performed against MCF-7, HepG2, HCT116 and Panc-1 cancer cell lines by MTT assay. Most of the synthesized compounds exhibited comparable anti-proliferative activity to the positive control 5-Fluorouracil against these four tested cancer cell lines. Among the different substituents at C-3 position of coumarin scaffold, 3-trifluoroacetyl group showed the most promising results. Especially, compounds 33d (IC50 = 16.57, 5.45, 4.42 and 5.16 μM) and 33e (IC50 = 20.14, 6.71, 4.62 and 5.62 μM) showed excellent anti-proliferative activities on MCF-7, HepG2, HCT116 and Panc-1 cell lines respectively. In addition, cell cycle analysis and apoptosis activation revealed that 33d induced G2/M phase arrest and apoptosis in MCF-7 cells in a dose-dependent manner. Low toxicity of compounds 33d and 33e was observed against human umbilical vein endothelial cells (HUVECs), suggesting their acceptable safety profiles in normal cells. Furthermore, the results of in silico ADME studies indicated that both 33d and 33e exhibited good pharmacokinetic properties.

![4H-1-BENZOPYRAN-4-ONE, 7-HYDROXY-3-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/876300/876294-38-1.png)
![4H-1-BENZOPYRAN-4-ONE, 7-HYDROXY-3-[(4-METHOXYPHENYL)METHYL]-](http://img.cochemist.com/ccimg/876300/876294-38-1_b.png)

![Benzo[b]thiophene, 4-methoxy-3-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/246300/246233-44-3.png)
![Benzo[b]thiophene, 4-methoxy-3-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/246300/246233-44-3_b.png)
![Benzo[b]thiophene, 6-methoxy-3-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/225300/225222-75-3.png)
![Benzo[b]thiophene, 6-methoxy-3-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/225300/225222-75-3_b.png)
![Estra-1,3,5(10)-trien-17-one,3-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-](http://img.cochemist.com/ccimg/180100/180003-17-2.png)
![Estra-1,3,5(10)-trien-17-one,3-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-](http://img.cochemist.com/ccimg/180100/180003-17-2_b.png)






![Benzo[b]thiophene, 4-methoxy-2-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/90500/90433-54-8.png)
![Benzo[b]thiophene, 4-methoxy-2-(4-methoxyphenyl)-](http://img.cochemist.com/ccimg/90500/90433-54-8_b.png)

![5,10-Methanocyclodeca[b]furan, 6,7,8,9-tetrahydro-6,6-dimethyl-](http://img.cochemist.com/ccimg/69300/69219-40-5.png)
![5,10-Methanocyclodeca[b]furan, 6,7,8,9-tetrahydro-6,6-dimethyl-](http://img.cochemist.com/ccimg/69300/69219-40-5_b.png)
![Ethanone, 1-[2-hydroxy-4-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-08-6.png)
![Ethanone, 1-[2-hydroxy-4-(methoxymethoxy)phenyl]-](http://img.cochemist.com/ccimg/65500/65490-08-6_b.png)







![(2E)-3-[4-METHOXY-3-(TRIFLUOROMETHYL)PHENYL]ACRYLIC ACID](http://img.cochemist.com/ccimg/39700/39604-64-3.png)
![(2E)-3-[4-METHOXY-3-(TRIFLUOROMETHYL)PHENYL]ACRYLIC ACID](http://img.cochemist.com/ccimg/39700/39604-64-3_b.png)



























![(1R,3aR,5aR,5bR,7aR,11aR,11bR,13aR,13bR)-3a,5a,5b,8,8,11a-Hexamethyl-1-(prop-1-en-2-yl)icosahydro-1H-cyclopenta[a]chrysen-9-yl acetate](http://img.cochemist.com/ccimg/1700/1617-68-1.png)
![(1R,3aR,5aR,5bR,7aR,11aR,11bR,13aR,13bR)-3a,5a,5b,8,8,11a-Hexamethyl-1-(prop-1-en-2-yl)icosahydro-1H-cyclopenta[a]chrysen-9-yl acetate](http://img.cochemist.com/ccimg/1700/1617-68-1_b.png)










![4-Bromo-2-methyl-1H-pyrrolo[2,3-b]pyridine](http://img.cochemist.com/ccimg/1014700/1014613-64-9.png)
![4-Bromo-2-methyl-1H-pyrrolo[2,3-b]pyridine](http://img.cochemist.com/ccimg/1014700/1014613-64-9_b.png)
![1-Piperazineacetic acid, 4-[5-chloro-2-[(4-methoxyphenyl)thio]phenyl]-2-methyl-, (2R)-](http://img.cochemist.com/ccimg/1014700/1014613-05-8.png)
![1-Piperazineacetic acid, 4-[5-chloro-2-[(4-methoxyphenyl)thio]phenyl]-2-methyl-, (2R)-](http://img.cochemist.com/ccimg/1014700/1014613-05-8_b.png)
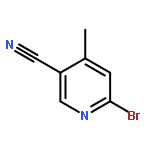
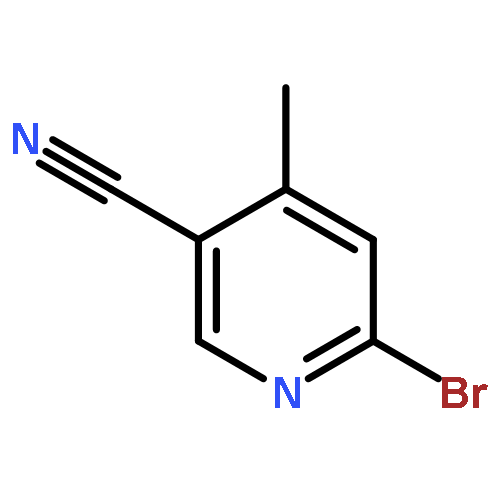
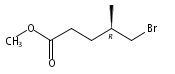
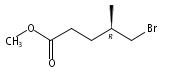
![[1-(tert-Butoxycarbonyl)-1,2,3,6-tetrahydropyridine-4-yl]boronic acid](http://img.cochemist.com/ccimg/844600/844501-00-4.png)
![[1-(tert-Butoxycarbonyl)-1,2,3,6-tetrahydropyridine-4-yl]boronic acid](http://img.cochemist.com/ccimg/844600/844501-00-4_b.png)
![Thieno[3,2-c]pyridin-4-amine, 7-(2-furanyl)-3-(4-phenoxyphenyl)-](http://img.cochemist.com/ccimg/832700/832697-35-5.png)
![Thieno[3,2-c]pyridin-4-amine, 7-(2-furanyl)-3-(4-phenoxyphenyl)-](http://img.cochemist.com/ccimg/832700/832697-35-5_b.png)
![Thieno[3,2-c]pyridin-4-amine, 3-(4-phenoxyphenyl)-7-(4-pyridinyl)-](http://img.cochemist.com/ccimg/832700/832694-02-7.png)
![Thieno[3,2-c]pyridin-4-amine, 3-(4-phenoxyphenyl)-7-(4-pyridinyl)-](http://img.cochemist.com/ccimg/832700/832694-02-7_b.png)
![6-Bromo-4-chloro-7-(phenylsulfonyl)-7H-pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/784200/784150-40-9.png)
![6-Bromo-4-chloro-7-(phenylsulfonyl)-7H-pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/784200/784150-40-9_b.png)






![7H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-6-phenyl-7-(phenylsulfonyl)-](http://img.cochemist.com/ccimg/252800/252723-15-2.png)
![7H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-6-phenyl-7-(phenylsulfonyl)-](http://img.cochemist.com/ccimg/252800/252723-15-2_b.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-cyclopentyl-5-(4-phenoxyphenyl)-](http://img.cochemist.com/ccimg/213800/213743-31-8.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-cyclopentyl-5-(4-phenoxyphenyl)-](http://img.cochemist.com/ccimg/213800/213743-31-8_b.png)
![7-(benzenesulfonyl)-4-chloropyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/186600/186519-89-1.png)
![7-(benzenesulfonyl)-4-chloropyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/186600/186519-89-1_b.png)
![1H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-6-phenyl-](http://img.cochemist.com/ccimg/173500/173459-05-7.png)
![1H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-6-phenyl-](http://img.cochemist.com/ccimg/173500/173459-05-7_b.png)
![4-Chloro-6-(4-methoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/173500/173459-03-5.png)
![4-Chloro-6-(4-methoxyphenyl)-7H-pyrrolo[2,3-d]pyrimidine](http://img.cochemist.com/ccimg/173500/173459-03-5_b.png)




![TERT-BUTYL 3-[2-[2-[2-(4-BROMOBUT-2-ENOXY)ETHOXY]ETHOXY]ETHOXY]PROPANOATE](http://img.cochemist.com/ccimg/143000/142950-86-5.png)
![TERT-BUTYL 3-[2-[2-[2-(4-BROMOBUT-2-ENOXY)ETHOXY]ETHOXY]ETHOXY]PROPANOATE](http://img.cochemist.com/ccimg/143000/142950-86-5_b.png)












![(4-CHLOROPHENYL)-(7-METHYL-3-AZA-7-AZONIABICYCLO[3.3.1]NONAN-3-YL)METHANONE](http://img.cochemist.com/ccimg/41700/41643-81-6.png)
![(4-CHLOROPHENYL)-(7-METHYL-3-AZA-7-AZONIABICYCLO[3.3.1]NONAN-3-YL)METHANONE](http://img.cochemist.com/ccimg/41700/41643-81-6_b.png)



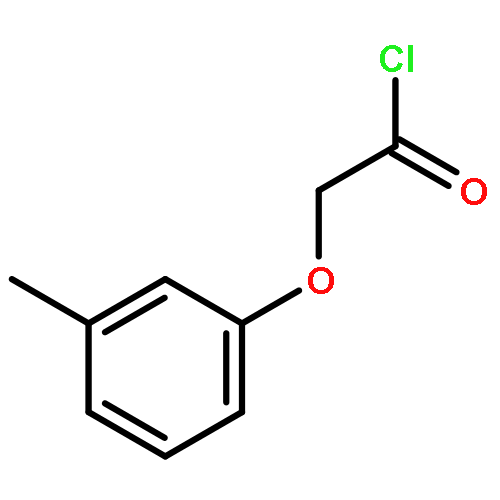



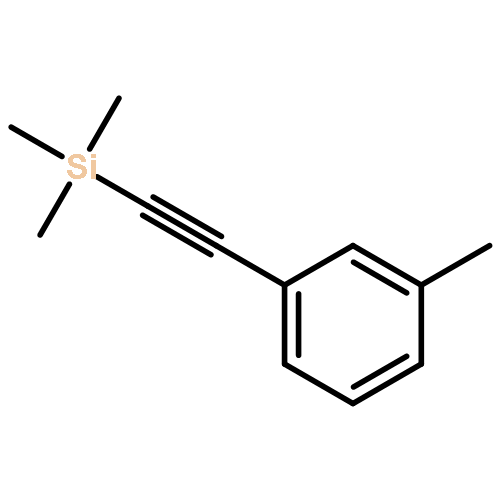






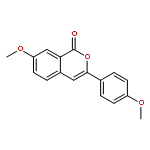
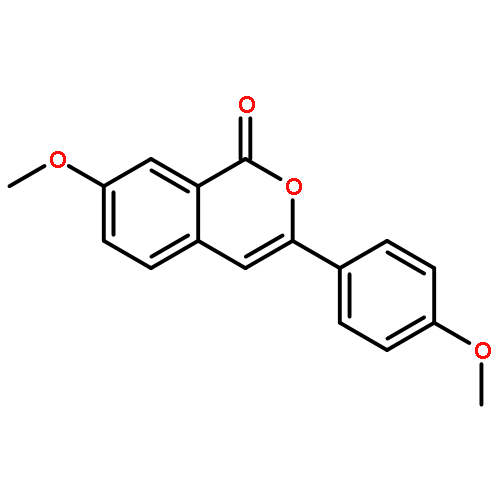




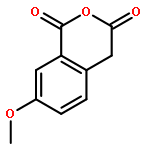
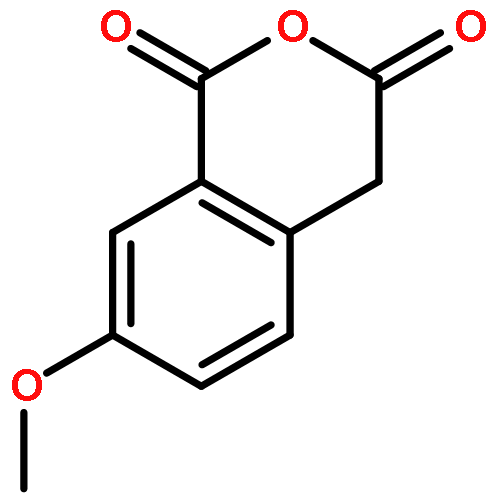
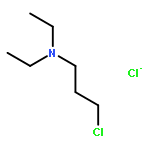
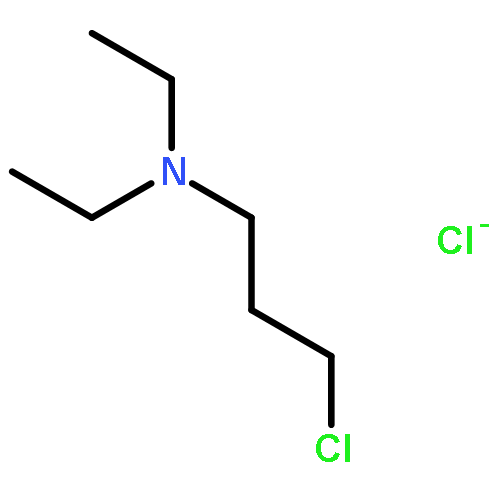



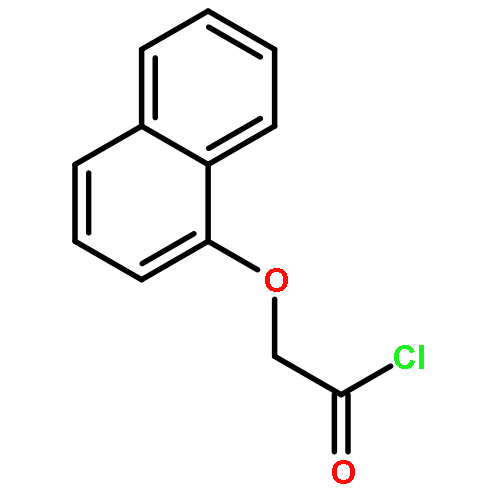

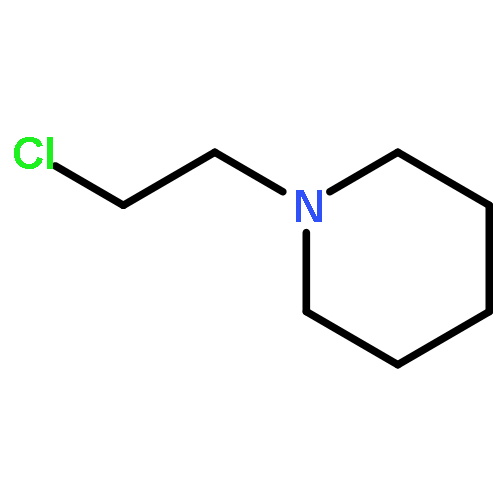
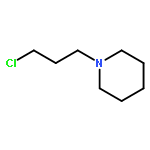
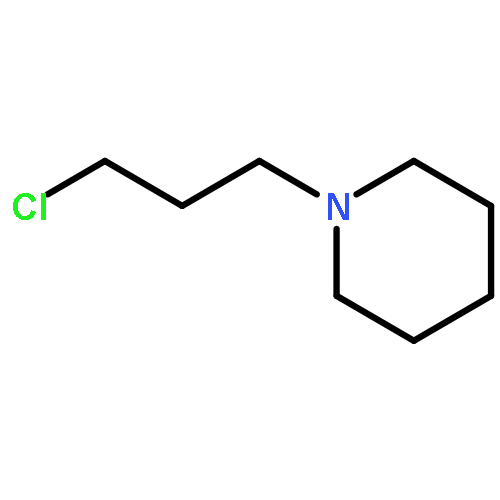



![THIENO[3,2-C]PYRIDIN-4-AMINE, 7-IODO-3-(4-PHENOXYPHENYL)-](http://img.cochemist.com/ccimg/832700/832694-04-9.png)
![THIENO[3,2-C]PYRIDIN-4-AMINE, 7-IODO-3-(4-PHENOXYPHENYL)-](http://img.cochemist.com/ccimg/832700/832694-04-9_b.png)
![Thieno[3,2-c]pyridin-4-amine, 3-(4-phenoxyphenyl)-](http://img.cochemist.com/ccimg/832700/832694-03-8.png)
![Thieno[3,2-c]pyridin-4-amine, 3-(4-phenoxyphenyl)-](http://img.cochemist.com/ccimg/832700/832694-03-8_b.png)


![1(2H)-Isoquinolinone, 6-cyclopropyl-2-[3-[1,6-dihydro-1-methyl-5-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]-6-oxo-3-pyridinyl]-2-](/data/chemimg/409200/1242156-23-5.png)
![1(2H)-Isoquinolinone, 6-cyclopropyl-2-[3-[1,6-dihydro-1-methyl-5-[[5-(4-methyl-1-piperazinyl)-2-pyridinyl]amino]-6-oxo-3-pyridinyl]-2-](/data/chemimg/409200/1242156-23-5_b.png)

