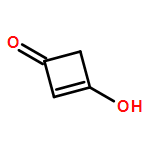
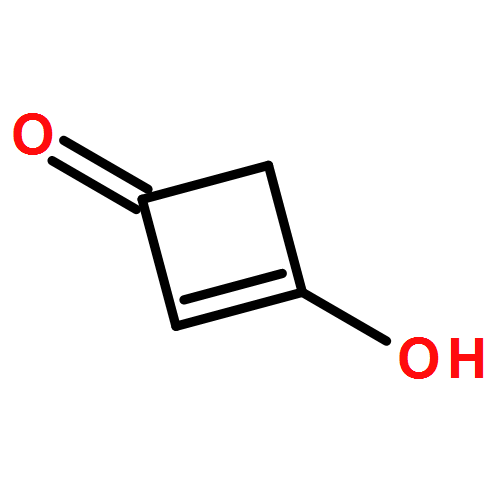
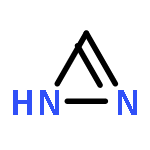
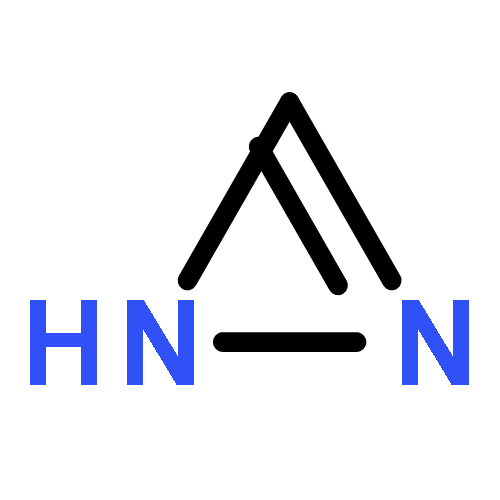
Co-reporter:Jianqi Sun;Xiangjuan Zheng;Xinjie Wu;Dong Li;Guomin Xia;Shuxian Yu;Qiming Yu
Analytical Methods (2009-Present) 2017 vol. 9(Issue 48) pp:6830-6838
Publication Date(Web):2017/12/14
DOI:10.1039/C7AY02218B
A cationic squaraine-based chemosensor SQM was synthesized and its sensing behaviors towards nucleophiles followed by CO2 gas were described in detail by interpreting the results from UV-Vis, ESI-MS, and 1H NMR spectral analysis. The results indicated that there were two different paths for the bleaching of SQM, depending on whether water existed or not when the nucleophilic addition reaction between SQM and a superbase, such as TBD or DBU, occurred. More interestingly, the bleached SQM in MeCN–H2O (v/v = 90 : 10) could be colorimetrically restored with bubbling CO2 gas, which enabled it to act as a highly sensitive “naked-eye” CO2 gas detector in an aqueous medium. However, when treated in the same way, the SQM bleached in MeCN remained unchanged, unexpectedly. Combining theoretical analysis and experimental tests, a plausible sensing mechanism was proposed to illustrate the bleaching of SQM and the response of the bleached SQM to CO2 gas in an aqueous medium.
Co-reporter:Jianqi Sun;Xiangjuan Zheng;Xinjie Wu;Dong Li;Guomin Xia;Shuxian Yu;Qiming Yu
Analytical Methods (2009-Present) 2017 vol. 9(Issue 48) pp:6830-6838
Publication Date(Web):2017/12/14
DOI:10.1039/C7AY02218B
A cationic squaraine-based chemosensor SQM was synthesized and its sensing behaviors towards nucleophiles followed by CO2 gas were described in detail by interpreting the results from UV-Vis, ESI-MS, and 1H NMR spectral analysis. The results indicated that there were two different paths for the bleaching of SQM, depending on whether water existed or not when the nucleophilic addition reaction between SQM and a superbase, such as TBD or DBU, occurred. More interestingly, the bleached SQM in MeCN–H2O (v/v = 90 : 10) could be colorimetrically restored with bubbling CO2 gas, which enabled it to act as a highly sensitive “naked-eye” CO2 gas detector in an aqueous medium. However, when treated in the same way, the SQM bleached in MeCN remained unchanged, unexpectedly. Combining theoretical analysis and experimental tests, a plausible sensing mechanism was proposed to illustrate the bleaching of SQM and the response of the bleached SQM to CO2 gas in an aqueous medium.
Co-reporter:Jianqi Sun, Benfei Ye, Guomin Xia, Hongming Wang
Sensors and Actuators B: Chemical 2017 Volume 249(Volume 249) pp:
Publication Date(Web):1 October 2017
DOI:10.1016/j.snb.2017.03.134
•SQ exhibited a high sensitivity for detecting Al3+, Zn2+ and Cd2+.•SQ can restore Aβ1–42-Al complex to Aβ1–42 efficiently and prevent or revert Aβ aggregation.•DFT/TDDFT calculations showed the chemosensor SQ followed a turn on mechanism with the inhibition of CN isomerization (cis-trans).A novel multi-response squaraine-based fluorescent chemosensor SQ was designed and synthesized, which exhibited a high sensitivity for detecting Al3+, Zn2+ and Cd2+ in ethanol-water buffer solution and selectively responded to them in an order of Al3+ to Zn2+ to Cd2+ due to the differences of stability and binding mode when forming their respective metal-ligand complexes. Furthermore, in solution, SQ can restore Aβ1–42-Al complex involving the potential relationships with the conformational aspects of Alzheimer’s disease (AD) to Aβ1–42 efficiently by binding Al3+ in Aβ1–42-Al complex, suggesting SQ may act as a protective agent by preventing or reverting Aβ aggregation in the extracellular spaces, which can provide a better biological understanding for developing successful therapeutics for the treatment of AD. The binding properties of sensor SQ with metal ions were investigated by UV–vis, fluorescence, 1H NMR and mass spectra. The results of various experiments and DFT/TDDFT calculations showed the chemosensor SQ followed a turn on mechanism with the inhibition of CN isomerization (cis-trans) and the activation of chelation enhanced fluorescence (CHEF).Download high-res image (126KB)Download full-size image
Co-reporter:Guomin Xia, Hongming Wang
Journal of Photochemistry and Photobiology C: Photochemistry Reviews 2017 Volume 31(Volume 31) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.jphotochemrev.2017.03.001
•Summary of frequently-used synthetic strategies to SQ dyes.•SQ-based colorimetric and fluorescence probe for metal ions, anions, pH, thiol-based compounds and biomolecules.•Current challenges and perspectives in the field of synthesis methods and optical applications of SQ dyes.Squaraine dyes, a four-membered ring system with structural rigidity, possess unique photoelectrical properties and are marked by their exceptionally sharp and intense absorption associated with a strong fluorescent emission in solution. These favorable characteristics have prompted their exploitation in a number of state of the art applications including photoconductivity, data storage, light-emitting field-effect transistors, solar cells and fluorescent histological probes. In this review, we first summarize the recently proposed novel methods in the synthesis of these versatile derivatives. Subsequently, their extensive applications in the prevalent optical detection of the surrounding medium such as ions, pH, thiol-based compounds, biomolecules and cell over the past decades are covered and discussed. In addition, different categories for the synthesis and sensing mechanisms for various squaric acid-based chemo-/bio- sensors are illustrated. Finally, the challenges and opportunities in the synthesis and application of these derivatives are also briefly discussed.Download high-res image (180KB)Download full-size image
Co-reporter:Kaikai Xu;Yihu Dai;Benfei Ye
Dalton Transactions 2017 vol. 46(Issue 33) pp:10780-10785
Publication Date(Web):2017/08/22
DOI:10.1039/C7DT02527K
Two dimensional (2D) metalloporphyrin-based covalent organic framework (COF) composites were synthesized and employed to catalyze the coupling of CO2 and epoxides to form cyclic carbonates. With the aid of a co-catalyst, a satisfactory selectivity (∼100%) and activity (99.2%) for the synthesis of 1,2-butylene carbonate were obtained on COF-366-Zn under mild reaction conditions. Their great recyclability and adaptability for various substrates were also demonstrated. The excellent performance may be benefited from their unique 2D COF structure and the facilitation effects on central metalloporphyrin sites from the internal environment of COFs.
Co-reporter:Jianqi Sun, Benfei Ye, Guomin Xia, Xiaohong Zhao, Hongming Wang
Sensors and Actuators B: Chemical 2016 Volume 233() pp:76-82
Publication Date(Web):5 October 2016
DOI:10.1016/j.snb.2016.04.052
•A squaraine-based system was developed to fluorescently and colorimetrically sense CO2 gas with high sensitivity.•A plausible mechanism that the deprotonated SQ-NH2 with fluoride ion will rapidly restore on condition that acidic carbon dioxide gas bubbles is proposed.•Allowing colorimetric and fluorescent visualizing the presence of CO2 gas.A squaraine-based system was developed and demonstrated to fluorescently and colorimetrically sense CO2 gas with high sensitivity in DMSO in the presence of fluoride ion. From results, large hypsochromic shifts were observed both in the absorption spectra (134 nm) and fluorescence spectra (126 nm), and the color change of solution could be clearly observed by the naked eye in response to carbon dioxide gas. Combining 1H NMR titration tests and theoretical calculations, a plausible sensing mechanism that the deprotonated SQ-NH2 with fluoride ion will rapidly restore on condition that acidic carbon dioxide gas bubbles in DMSO is proposed and further revealed. Moreover, theoretical studies agree with experimental data well.
Co-reporter:Guomin Xia, Yang Liu, Benfei Ye, Jianqi Sun and Hongming Wang
Chemical Communications 2015 vol. 51(Issue 72) pp:13802-13805
Publication Date(Web):21 Jul 2015
DOI:10.1039/C5CC04755B
An unsymmetrical squaraine-based chemosensor SH2 has been synthesized, and its sensing behavior towards CO2 gas was described in detail by UV-vis and 1H NMR spectroscopies in DMSO. The results indicated that the extremely sensitive “naked-eye” CO2 gas detection can be operated in the presence of excess [Bu4N]F (TBAF) and the sensor is easy to recycle. These properties enable SH2 to act as a CO2 and F− controlled “OFF–ON–OFF” switch. Combining theoretical analyses, a plausible sensing mechanism was proposed to illustrate how the receptor SH2 works as a CO2 sensitive and selective colorimetric probe in the present system.
Co-reporter:Liujie Wang, Xiaoliang Zhang, Longhua Yang, Chao Wang and Hongming Wang
Catalysis Science & Technology 2015 vol. 5(Issue 10) pp:4800-4805
Publication Date(Web):10 Jul 2015
DOI:10.1039/C5CY00772K
A new method for the reduction of CO2 to CH3OH coupled with selective alcohol oxidation using a photocatalytic method has been developed in the present paper. CO2 as the oxidant was reduced to methanol by photo-induced electrons, and aromatic alcohols were used as the reductants to react with photo-generated holes and were converted to aromatic aldehydes with high selectivity. The maximum conversion of aromatic alcohol to aldehyde was 91.7% with a selectivity of 98%, and the highest yield of methanol was 358.7 μmol g−1. This study could provide a valuable method not only for carbon dioxide conversion into fuel which could remission energy shortages, but also for the selective oxidation of alcohols to produce aldehydes.
Co-reporter:Guomin Xia, Chengyan Ruan and Hongming Wang
Analyst 2015 vol. 140(Issue 15) pp:5099-5104
Publication Date(Web):09 Jun 2015
DOI:10.1039/C5AN00947B
In the present paper, a “light-up” chemsensor with a high specificity for carbon dioxide detection using a pyrimido[1,2-a]benzimidazole derivative (P1H) in liquid media has been developed. The results show that P1H reacts with carbon dioxide activated by a basic ion to form a carboxylic acid compound (P1-COOH). These results also provide a possible method for carboxylation reactions of P1H using carbon dioxide based on a vinyl carbanion. The complete reaction mechanism cycle was also described using DFT calculations.
Co-reporter:Yanli Yuan, Xiping Gong and Hongming Wang
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 17) pp:11375-11381
Publication Date(Web):23 Mar 2015
DOI:10.1039/C5CP00011D
The synergistic effect of graphene and MoS2 was investigated by using density functional theory (DFT) calculations on the enhanced photocatalytic H2 production activity of TiO2/graphene/MoS2 ternary nanoparticles. Our results indicate that it can form a weak covalent bond between the Ti atom of TiO2 nanocluster and the nearest C atom on graphene, which not only makes the original degenerate C(2p) orbital level of the graphene (part of the conduction band energy level) split, resulting in the production of a lower level of C(2p) that makes it easier to accept the excited electron from the Ti(3d) orbital, but also forms a +/− sequence electric field in the interface between them. It is conclusive that the electron moves from the TiO2 cluster to the graphene. In addition, we also find that the band gap of the TiO2 cluster can be doped by the graphene and MoS2, and the conduction band consists predominantly of C(2p), S(3p) and Mo(4d) orbital energy level near the Fermi level. These results illustrate that the excited electron will eventually accumulate in the graphene or MoS2 film, which can effectively enhance the separation between the excited electrons and the holes in the TiO2 clusters, thereby increasing the efficiency of hydrogen evolution. Our results are consistent with the experimental results, and can provide some valuable information for the design of photocatalytic composites.
Co-reporter:Yang Liu, Guoming Xia, Chao Luo, Jianqi Sun, Benfei Ye, Yanli Yuan, Hongming Wang
Tetrahedron Letters 2015 Volume 56(Issue 36) pp:5071-5075
Publication Date(Web):2 September 2015
DOI:10.1016/j.tetlet.2015.07.050
An efficient method for the preparation of pyrimido[1,2-a]benzimidazole derivatives utilizing squaric acid dichloride has been developed at room temperature without catalyst. This method provided a rapid synthesis of pyrimido[1,2-a]benzimidazole system. The result indicates that those expeditious reactions could be carried out only in the presence of alcohols, affording the corresponding alkyl 2-chloropyrimido[1,2-a]benzimidazole-3-carboxylates. Furthermore, the yield of products seems to be closely relevant to the steric hindrance of the added alcohols. On the basis of experimental and theoretical analyses, a plausible mechanism has been proposed and corroborated through DFT calculations for exploring a practical way to efficiently synthesize those highly versatile substituted homologs.
Co-reporter:Chengyan Ruan, Longhua Yang, Yanli Yuan, Yan Ju, Hongming Wang
Computational and Theoretical Chemistry 2015 Volume 1058() pp:34-40
Publication Date(Web):15 April 2015
DOI:10.1016/j.comptc.2015.02.001
•We report a DFT study on the mechanism of CO2 and alkynylaniline derivative.•The reaction catalyzed by silver salt and base is the optimal path with comparison.•Silver salt can reduce the whole energy of the system.•The use of base makes the process of hydrogen transfer more easily.Benzoxazine-2-one derivatives are important heterocyclic molecules because of their various pharmaceutical activities. The reaction mechanisms for synthesizing benzoxazine-2-one derivative, (Z)-4-benzylidene-1-methyl-1H- benzo[d][1,3]oxazin-2(4H)-one, via the coupled reaction of carbon dioxide with o-alkynylaniline derivatives, N-methyl-2-(2-phenylethynyl)benzenamine, have been studied computationally using density functional theory (DFT). Three reaction systems have been studied for comparison, with no catalyst, silver salt catalyst and copper salt catalyst, respectively. The results indicate that the silver salt can efficiently catalyze the reaction in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).We investigated a DFT study on the reaction mechanism of CO2 and alkynylaniline derivative. Three reaction systems have been studied for comparison, with no catalyst, silver salt catalyst and copper salt catalyst, respectively. The results indicate that the reaction catalyzed by silver salt in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) is the optimal path and it involves two parts: the transfer of hydrogen, the insertion of CO2 molecule. In this path, silver salt can reduce the whole energy of the system, and DBU makes the hydrogen transfer more easily. Moreover, all the energy barriers of this path are very low, so that it is more likely for the reaction to proceed at normal temperatures and pressures.
Co-reporter:Longhua Yang, Yanli Yuan, Hongming Wang, Ning Zhang and Sanguo Hong
RSC Advances 2014 vol. 4(Issue 61) pp:32457-32466
Publication Date(Web):24 Jun 2014
DOI:10.1039/C4RA00254G
The reaction mechanisms of copper(I)–NHC-catalyzed C–H carboxylation of terminal alkynes with CO2 were investigated by DFT calculations (NHCN-heterocyclic carbene). Three types of reaction mechanisms were designed, explored and compared. The optimal reaction channels of corresponding pathways were selected. It was investigated that the formation of new C–C bond in the insertion process of activated CO2 by NHC was induced by the formation of Cu–O bond. Also, the functions of NHC were determined. Our calculations investigated that (1) the special difunctional roles of NHC can indeed facilitate the reaction process after the formation of CO2–NHC–Cu cocatalyst, whereas the unexpected low energy of this cocatalyst results in its ultrastability and then hinders the dropping of energy barrier in the whole reaction and (2) the additional interaction of NHC with the same metal atom will promote the insertion process of CO2 through increasing the electrophilicity of the metal center.
Co-reporter:Longhua Yang ;Dr. Hongming Wang
ChemSusChem 2014 Volume 7( Issue 4) pp:962-998
Publication Date(Web):
DOI:10.1002/cssc.201301131
Abstract
In the last two decades, CO2 emission has caused a lot of environmental problems. To mitigate the concentration of CO2 in the atmosphere, various strategies have been implemented, one of which is the use of N-heterocyclic carbenes (NHCs) and related complexes to accomplish the capture, fixation, and activation of CO2 effectively. In this review, we summarize CO2 capture, fixation, and activation by utilizing NHCs and related complexes; homogeneous reactions and their reaction mechanisms are discussed. Free NHCs and NHC salts can capture CO2 in both direct and indirect ways to form imidazolium carboxylates, and they can also catalyze the reaction of aromatic aldehydes with CO2 to form carboxylic acids and derivatives. Moreover, associated with transition metals (TMs), NHCs can form NHC–TM complexes to transform CO2 into industrial acid or esters. Non-metal–NHC complexes can also catalyze the reactions of silicon and boron complexes with CO2. In addition, catalytic cycloaddition of epoxides with CO2 is another effective function of NHC complexes, and NHC ionic liquids perform excellently in this aspect.
Co-reporter:Longhua Yang, Hongming Wang, Ning Zhang and Sanguo Hong
Dalton Transactions 2013 vol. 42(Issue 31) pp:11186-11193
Publication Date(Web):22 May 2013
DOI:10.1039/C3DT50337B
The reaction mechanism of CO2 hydrogenation catalyzed by [FeH(PP3)]BF4 (PP3 = P(CH2CH2PPh2)3) had been investigated by DFT calculations. Our calculations indicated that the reduction of carbon dioxide could be carried out via two spin states, the high-spin (HS) triplet state and the low-spin (LS) singlet state. The minimum energy crossing points (MECPs) on the seam of two intersecting PESs (potential energy surfaces) were searched out. Some interesting phenomena, such as the open-loop phenomenon, and the O-rebound process, were demonstrated to be the important causes of the spin crossover. All these calculations gave us insight into the essence of the related experiment from the macro point of view, and helped to verify which spin states the related complexes pertinent were in. All of these researches would help advance the development of efficient and structurally tailorable CO2 hydrogenation catalysts.
Co-reporter:Guomin Xia, Zhiwei Wu, Yanli Yuan and Hongming Wang
RSC Advances 2013 vol. 3(Issue 39) pp:18055-18061
Publication Date(Web):23 Jul 2013
DOI:10.1039/C3RA43608J
In this paper, a synthetic method to produce 1,2-benzimidazole squaraines with a yield of up to 89% is developed. It is found that both strong organic and inorganic bases have a satisfactory catalytic activity for this reaction. Theoretical studies provide detailed explanations for the 1,2 versus 1,3 condensation regiochemistry of the squaraines. The experimental and theoretical studies agree well with each other, paving a practical way to efficiently synthesize 1,2-squaraines.
Co-reporter:X. Ren;Y. Yuan;Y. Ju;H. Wang
Chemistry of Heterocyclic Compounds 2013 Volume 49( Issue 2) pp:260-272
Publication Date(Web):2013 May
DOI:10.1007/s10593-013-1243-z
In this paper, we report theoretical studies of the addition–cyclization–isomerization reaction of propargyl cyanamides with thiol and methanol by density functional theory (DFT) calculation. The results reveal that this reaction takes place via five steps: 1) nucleophilic attack of S or O atom to C atom in the cyanogen group of propargyl cyanamide to form a cisoid-intermediate; 2) the conversion of the latter to its trans-conformer; 3) nucleophilic attack by N atom at the alkyne group to produce a five-membered thermodynamically unstable zwitterionic 4-ethylidene-4,5-dihydroimidazole intermediate; 4) proton transfer from N to C(4) atom to produce a more stable intermediate; 5) proton transfer from C(5) to ethylidene group to form the final 4-ethyl-1,5-dimethyl-2-methylsulfanyl- or 4-ethyl-2-methoxy-1,5-dimethylimidazole. We find that the autocatalysis by thiol or methanol is able to largely decrease the energy barrier of intramolecular proton transfer in the isomerization step and the proton transfer in the addition step.
Co-reporter:Longhua Yang;Xingye Ren;Ning Zhang
Research on Chemical Intermediates 2012 Volume 38( Issue 1) pp:113-133
Publication Date(Web):2012 January
DOI:10.1007/s11164-011-0330-y
Density functional theory calculations have been carried out to explore the reaction mechanisms for the reactions of “frustrated Lewis pairs” (FLPs) with small molecules. Four reactions were studied in the present investigation. A new N-heterocyclic carbene borane, boron amidinate compound HC(iPrN)2B(C6F5)2 (1), classified as FLPs, was chosen as the common reactant of these reactions. It was used to react with CO2, CO, and two terminal alkynes, methylacetylene and phenylacetylene. The reactions of 1 with CO2 and CO can both be regarded as the concerted addition mechanisms. In these reactions, the formations of the C2–N2 and B–O1 bonds take place simultaneously. For the reactions of 1 and methylacetylene or phenylacetylene, our calculations indicated that a deprotonation pathway and the connection between B atoms and terminal alkyne C atoms occur by a concerted manner simultaneously, together with the connection between N2 and hydrogen atoms. We also investigated the reaction mechanisms according to the frontier molecular orbital (FMO) theory and carried out electric charge analyses, finding that the two results were consistent with each other perfectly. Electric charges transfer from HOMO of 1_OPEN to LUMO of CO2 or CO. In contrast, electric charges transfer from HOMO of methylacetylene or phenylacetylene to LUMO of 1_OPEN.
Co-reporter:Yanli Yuan;Peiyu Chen;Xingye Ren ;Dr. Hongming Wang
ChemPhysChem 2012 Volume 13( Issue 3) pp:741-750
Publication Date(Web):
DOI:10.1002/cphc.201100730
Abstract
The mechanism of the 1,3-dipolar cycloaddition reaction of azidotrimethylsilane (ATS) onto nanographene (NG) is thoroughly investigated at the B3LYP/6-31G(d,p) level. Calculations reveal that the reaction occurs through a two-step reaction mechanism. The first step is the chemical adsorption and the second one is the decomposition of the thereby formed nitride upon thermal activation, giving rise to an N-bridged product ultimately. The latter is the rate-determining step. Two possible pathways are compared to show that the [3+2] channel is favored over the [3+4] channel. The former is a symmetric synchronous process, whereas the latter follows an asymmetric concerted way, which can be rationalized by means of the frontier molecular orbital (FMO) theory. The reactivity of NG is discussed in detail, revealing that it is the electron density at the functionalization site which dominates the reactivity rather than the energetic effect. As a result, the edge area is calculated to be much more reactive than the centre.
Co-reporter:Dr. Xingye Ren;Dr. Yanli Yuan;Dr. Yan Ju ;Dr. Hongming Wang
ChemCatChem 2012 Volume 4( Issue 12) pp:1943-1951
Publication Date(Web):
DOI:10.1002/cctc.201200529
Abstract
The mechanism of the CO2 splitting reaction with N-heterocyclic carbene (NHC) as a catalyst and cinnamic aldehydes as the oxygen acceptor was thoroughly investigated at the B3LYP/6-31G(d,p) level. Owing to its nucleophilicity, NHC can initiate the reaction through two distinct channels (I and II) by nucleophilic attack. The results reveal that without the assistance by H2O, channel II is slightly more favorable than channel I, and the reaction path II-D is the optimum reaction path. However, the reaction is difficult to conduct at room temperature because of its high activation energy barrier. Although the results suggest that most of the high activation energy barriers involved in proton transfer decrease if the reaction is assisted by H2O, all highest energy barriers for all reaction channels are still above 40 kcal mol−1. This indicates that this reaction cannot be performed at room temperature, which is also proven by experiments.
Co-reporter:Han Wu, Yanni Wang, Hongming Wang, Yan Ju
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2012 Volume 94() pp:222-227
Publication Date(Web):August 2012
DOI:10.1016/j.saa.2012.03.063
The photophysical properties of a hydrazone-based switch, which can be induced using pH to yield three stable configurations (QPH-E, QPH-Z-H+ and QPH-Z-H22+) and another unstable configuration (QPH-Z). The three stable configuration have been investigated by means of density functional theory (DFT) and time-dependent density functional theory (TDDFT). A generalized gradient approximation (GGA) functional (PBE) and two hybrid-type functional (B3LYP and BH&HLYP) as well as four popular basis sets (6-31G(d,p), SVP, TZVP and DZP) have been selected to calculate the photophysical properties. The solvent effects were in view with the Conductor-like Polarizable Continuum Model (CPCM). Besides, the charge distribution of this switch had been investigated by the pop at the level of the B3YLP/6-31G(d,p) in gas. Results show that the B3LYP functional is more accurate in all simulations.Graphical abstractHighlights► The results show that the B3LYP function is more accurate in these simulations. ► The solvent polarity would affect the photophysics properties of this switch. ► The basis sets appear to have only a small influence on the TDDFT calculation.
Co-reporter:Xingye Ren, Chongfa Xiao and Hongming Wang
Dalton Transactions 2011 vol. 40(Issue 14) pp:3576-3583
Publication Date(Web):03 Mar 2011
DOI:10.1039/C0DT01021A
The reaction mechanism for the reduction of CO2 gas activated by (tBuArN)3MN was studied by the means of density functional theory (DFT) calculations. The calculations indicated that this reaction has a two step reaction mechanism. From our calculations, we found that (tBuArN)3TaN held the best activity among the three (tBuArN)3MN complexes studied. Our results also indicated that the reaction of (tBuArN)3MN with CO2 occurred under orbital control involving the HOMO-3 orbital of (tBuArN)3MN, which could give higher overlapping with the LUMO of the CO2 molecule. The substitutions on the amino donor ligands studied here took larger effect on the HOMO structure of the (tBuArN)3MN molecules. The electronic structure of the (tBuArN)3MN complexes also showed their ability for activating CO2 molecules, in the order of M = V < Nb < Ta.
Co-reporter:Xingye Ren, Hongming Wang, Yan Ju
Computational and Theoretical Chemistry 2011 Volume 967(Issue 1) pp:129-135
Publication Date(Web):July 2011
DOI:10.1016/j.comptc.2011.04.005
The experiment study indicates that tantalaziridine–hydride ((Ar[tBuCH2]N)2(η2-tBu(H)CNAr)TaH) is able to convert to the isomeric hydride ((Ar[tBuCH2]N)2(κ2-CH2C(Me)2CH2NAr)TaH). And also, the both of tantalaziridine–hydride were found to be able to react with CO2 to give bimetallic methlene diolates. In this paper, the mechanism of the tautomerism had been investigated theoretically by DFT. The results reveal that this reaction is accomplished by two steps via tridentateligand intermediate. This kind of intermediate can be considered as the activating reagent to activate the CH bond. Further calculations showed that the reduction of CO2 with the terminal tantalum hydrides was carried out by two-step reaction mechanisms, via formate intermediate. In these studies, fully geometry optimized reactants, products, transition states, and intermediates were obtained.
Co-reporter:Jiajian Gao, Chunmiao Jia, Liping Zhang, Hongming Wang, Yanhui Yang, Sung-Fu Hung, Ying-Ya Hsu, Bin Liu
Journal of Catalysis (September 2016) Volume 341() pp:82-90
Publication Date(Web):1 September 2016
DOI:10.1016/j.jcat.2016.06.009
•Fe-, Co-, Ni-, and Cu-doped α-MnO2 nanowires were synthesized by a one-step hydrothermal method.•All doped MnO2 nanowires exhibited much enhanced CO oxidation activity.•The Cu-doped MnO2 nanowires had a maximum TOF of 9.1 × 10−3 s−1 at 70 °C.•Cu-doped MnO2 could maintain 50 h without obvious deactivation with 2% water moisture.•Cu doping makes the formation of oxygen vacancies easier in MnO2.Replacing a small fraction of cations in a host metal oxide with a different cation (also known as doping) provides a useful strategy for improving the catalytic activity. Here, we report transition metal (Fe, Co, Ni, and Cu)-doped α-MnO2 nanowires synthesized by a one-step hydrothermal method as CO oxidation catalysts. The as-prepared catalysts displayed morphology, crystal structure, and specific surface area similar to those of the pure MnO2 nanowires. A catalytic activity test showed that all doped MnO2 nanowires exhibited much enhanced CO oxidation activity, with the Cu-doped ones being the most active (TOF of 9.1 × 10−3 s−1 at 70 °C). The Cu-doped MnO2 nanowires showed nearly 100% conversion of CO at 100 °C at an hourly gas space velocity of 36,000 mL g−1 h−1, which could last for 50 h without obvious deactivation even in the presence of 2% water vapor. Density functional theory calculations suggested that Cu doping could make the formation of oxygen vacancies in MnO2, which is the rate-determining step for CO oxidation reaction, easier than for Fe-, Co-, and Ni-doped and pristine MnO2. Our work demonstrates a facile and promising strategy for improving the catalytic activity for oxide-based catalysts, which should be applicable for a variety of different chemical reactions.Download high-res image (138KB)Download full-size image
Co-reporter:Guomin Xia, Benfei Ye, Dong Li, Hongming Wang
Sensors and Actuators B: Chemical (June 2017) Volume 244() pp:252-258
Publication Date(Web):June 2017
DOI:10.1016/j.snb.2016.12.143
Co-reporter:Guomin Xia, Yang Liu, Benfei Ye, Jianqi Sun and Hongming Wang
Chemical Communications 2015 - vol. 51(Issue 72) pp:NaN13805-13805
Publication Date(Web):2015/07/21
DOI:10.1039/C5CC04755B
An unsymmetrical squaraine-based chemosensor SH2 has been synthesized, and its sensing behavior towards CO2 gas was described in detail by UV-vis and 1H NMR spectroscopies in DMSO. The results indicated that the extremely sensitive “naked-eye” CO2 gas detection can be operated in the presence of excess [Bu4N]F (TBAF) and the sensor is easy to recycle. These properties enable SH2 to act as a CO2 and F− controlled “OFF–ON–OFF” switch. Combining theoretical analyses, a plausible sensing mechanism was proposed to illustrate how the receptor SH2 works as a CO2 sensitive and selective colorimetric probe in the present system.
Co-reporter:Chao Luo, Qiming Yu and Hongming Wang
Environmental Science: Nano 2016 - vol. 18(Issue 12) pp:
Publication Date(Web):
DOI:10.1039/C6EM00420B
Co-reporter:Longhua Yang, Hongming Wang, Ning Zhang and Sanguo Hong
Dalton Transactions 2013 - vol. 42(Issue 31) pp:NaN11193-11193
Publication Date(Web):2013/05/22
DOI:10.1039/C3DT50337B
The reaction mechanism of CO2 hydrogenation catalyzed by [FeH(PP3)]BF4 (PP3 = P(CH2CH2PPh2)3) had been investigated by DFT calculations. Our calculations indicated that the reduction of carbon dioxide could be carried out via two spin states, the high-spin (HS) triplet state and the low-spin (LS) singlet state. The minimum energy crossing points (MECPs) on the seam of two intersecting PESs (potential energy surfaces) were searched out. Some interesting phenomena, such as the open-loop phenomenon, and the O-rebound process, were demonstrated to be the important causes of the spin crossover. All these calculations gave us insight into the essence of the related experiment from the macro point of view, and helped to verify which spin states the related complexes pertinent were in. All of these researches would help advance the development of efficient and structurally tailorable CO2 hydrogenation catalysts.
Co-reporter:Xingye Ren, Chongfa Xiao and Hongming Wang
Dalton Transactions 2011 - vol. 40(Issue 14) pp:NaN3583-3583
Publication Date(Web):2011/03/03
DOI:10.1039/C0DT01021A
The reaction mechanism for the reduction of CO2 gas activated by (tBuArN)3MN was studied by the means of density functional theory (DFT) calculations. The calculations indicated that this reaction has a two step reaction mechanism. From our calculations, we found that (tBuArN)3TaN held the best activity among the three (tBuArN)3MN complexes studied. Our results also indicated that the reaction of (tBuArN)3MN with CO2 occurred under orbital control involving the HOMO-3 orbital of (tBuArN)3MN, which could give higher overlapping with the LUMO of the CO2 molecule. The substitutions on the amino donor ligands studied here took larger effect on the HOMO structure of the (tBuArN)3MN molecules. The electronic structure of the (tBuArN)3MN complexes also showed their ability for activating CO2 molecules, in the order of M = V < Nb < Ta.
Co-reporter:Liujie Wang, Xiaoliang Zhang, Longhua Yang, Chao Wang and Hongming Wang
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 10) pp:NaN4805-4805
Publication Date(Web):2015/07/10
DOI:10.1039/C5CY00772K
A new method for the reduction of CO2 to CH3OH coupled with selective alcohol oxidation using a photocatalytic method has been developed in the present paper. CO2 as the oxidant was reduced to methanol by photo-induced electrons, and aromatic alcohols were used as the reductants to react with photo-generated holes and were converted to aromatic aldehydes with high selectivity. The maximum conversion of aromatic alcohol to aldehyde was 91.7% with a selectivity of 98%, and the highest yield of methanol was 358.7 μmol g−1. This study could provide a valuable method not only for carbon dioxide conversion into fuel which could remission energy shortages, but also for the selective oxidation of alcohols to produce aldehydes.
Co-reporter:Yanli Yuan, Xiping Gong and Hongming Wang
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 17) pp:NaN11381-11381
Publication Date(Web):2015/03/23
DOI:10.1039/C5CP00011D
The synergistic effect of graphene and MoS2 was investigated by using density functional theory (DFT) calculations on the enhanced photocatalytic H2 production activity of TiO2/graphene/MoS2 ternary nanoparticles. Our results indicate that it can form a weak covalent bond between the Ti atom of TiO2 nanocluster and the nearest C atom on graphene, which not only makes the original degenerate C(2p) orbital level of the graphene (part of the conduction band energy level) split, resulting in the production of a lower level of C(2p) that makes it easier to accept the excited electron from the Ti(3d) orbital, but also forms a +/− sequence electric field in the interface between them. It is conclusive that the electron moves from the TiO2 cluster to the graphene. In addition, we also find that the band gap of the TiO2 cluster can be doped by the graphene and MoS2, and the conduction band consists predominantly of C(2p), S(3p) and Mo(4d) orbital energy level near the Fermi level. These results illustrate that the excited electron will eventually accumulate in the graphene or MoS2 film, which can effectively enhance the separation between the excited electrons and the holes in the TiO2 clusters, thereby increasing the efficiency of hydrogen evolution. Our results are consistent with the experimental results, and can provide some valuable information for the design of photocatalytic composites.

![3-Cyclobutene-1,2-dione, 3-[4-(dimethylamino)phenyl]-4-hydroxy-](/data/chemimg/353200/107885-39-2.png)
![3-Cyclobutene-1,2-dione, 3-[4-(dimethylamino)phenyl]-4-hydroxy-](/data/chemimg/353200/107885-39-2_b.png)