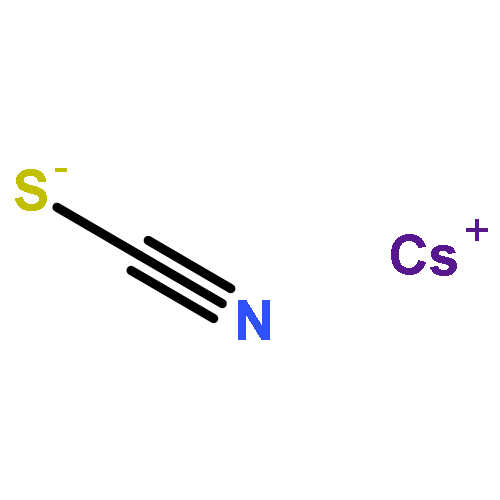
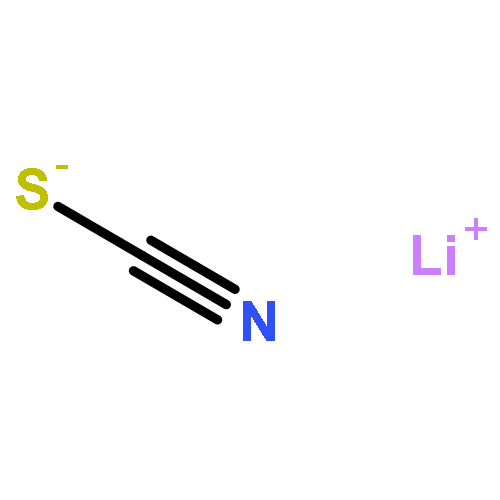
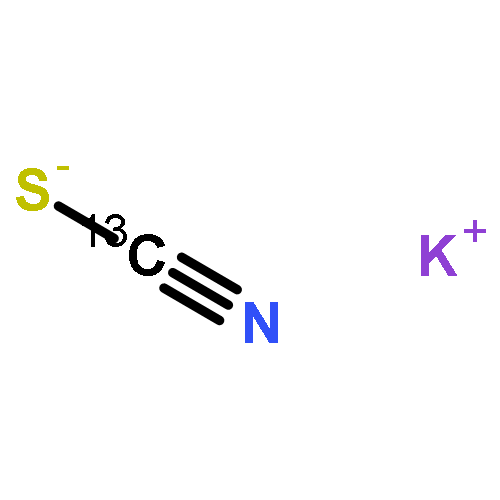
Co-reporter:Chuanqi Ge, Yuneng Shen, Gang-Hua Deng, Yuhuan Tian, Dongqi Yu, Xueming Yang, Kaijun Yuan, and Junrong Zheng
The Journal of Physical Chemistry B 2016 Volume 120(Issue 12) pp:3187-3195
Publication Date(Web):March 11, 2016
DOI:10.1021/acs.jpcb.5b12652
Isotopic effects on the formation and dissociation kinetics of hydrogen bonds are studied in real time with ultrafast chemical exchange spectroscopy. The dissociation time of hydrogen bond between phenol-OH and p-xylene (or mesitylene) is found to be identical to that between phenol-OD and p-xylene (or mesitylene) in the same solvents. The experimental results demonstrate that the isotope substitution (D for H) has negligible effects on the hydrogen bond kinetics. DFT calculations show that the isotope substitution does not significantly change the frequencies of vibrational modes that may be along the hydrogen bond formation and dissociation coordinate. The zero point energy differences of these modes between hydrogen bonds with OH and OD are too small to affect the activation energy of the hydrogen bond dissociation in a detectible way at room temperature.
Co-reporter:Dr. Yupeng Pan;Dr. Cheng-Ling Pan;Yufan Zhang;Dr. Huaifeng Li;Dr. Shixiong Min;Dr. Xunmun Guo;Dr. Bin Zheng;Dr. Hailong Chen;Addison Anders; Zhiping Lai; Junrong Zheng; Kuo-Wei Huang
Chemistry – An Asian Journal 2016 Volume 11( Issue 9) pp:1357-1360
Publication Date(Web):
DOI:10.1002/asia.201600169
Abstract
An unsymmetrically protonated PN3-pincer complex in which ruthenium is coordinated by one nitrogen and two phosphorus atoms was employed for the selective generation of hydrogen from formic acid. Mechanistic studies suggest that the imine arm participates in the formic acid activation/deprotonation step. A long life time of 150 h with a turnover number over 1 million was achieved.
Co-reporter:Dr. Yupeng Pan;Dr. Cheng-Ling Pan;Yufan Zhang;Dr. Huaifeng Li;Dr. Shixiong Min;Dr. Xunmun Guo;Dr. Bin Zheng;Dr. Hailong Chen;Addison Anders; Zhiping Lai; Junrong Zheng; Kuo-Wei Huang
Chemistry – An Asian Journal 2016 Volume 11( Issue 9) pp:
Publication Date(Web):
DOI:10.1002/asia.201600536
Co-reporter:Yuneng Shen, Tianmin Wu, Bo Jiang, Ganghua Deng, Jiebo Li, Hailong Chen, Xunmin Guo, Chuanqi Ge, Yajing Chen, Jieya Hong, Xueming Yang, Kaijun Yuan, Wei Zhuang, and Junrong Zheng
The Journal of Physical Chemistry B 2015 Volume 119(Issue 30) pp:9893-9904
Publication Date(Web):July 2, 2015
DOI:10.1021/acs.jpcb.5b04530
In this work, MD simulations with two different force fields, vibrational energy relaxation and resonant energy transfer experiments, and neutron scattering data are used to investigate ion pairing and clustering in a series of GdmSCN aqueous solutions. The MD simulations reproduce the major features of neutron scattering experimental data very well. Although no information about ion pairing or clustering can be obtained from the neutron scattering data, MD calculations clearly demonstrate that substantial amounts of ion pairs and small ion clusters (subnanometers to a few nanometers) do exist in the solutions of concentrations 0.5 M*, 3 M*, and 5 M* (M* denotes mole of GdmSCN per 55.55 mole of water). Vibrational relaxation experiments suggest that significant amounts of ion pairs form in the solutions. Experiments measuring the resonant energy transfers among the thiocyanate anions in the solutions suggest that the ions form clusters and in the clusters the average anion distance is 5.6 Å (5.4 Å) in the 3 M* (5 M*) Gdm–DSCN/D2O solution.
Co-reporter:Hailong Chen, Qiang Zhang, Xunmin Guo, Xiewen Wen, Jiebo Li, Wei Zhuang, and Junrong Zheng
The Journal of Physical Chemistry A 2015 Volume 119(Issue 4) pp:669-680
Publication Date(Web):December 30, 2014
DOI:10.1021/jp511651t
Energy-gap-dependent vibrational-energy transfers among the nitrile stretches of KSCN/KS13CN/KS13C15N in D2O, DMF, and formamide liquid solutions at room temperature were measured by the vibrational-energy-exchange method. The energy transfers are slower with a larger energy donor/acceptor gap, independent of the calculated instantaneous normal mode (“phonons” in liquids) densities or the terahertz absorption spectra. The energy-gap dependences of the nonresonant energy transfers cannot be described by phonon compensation mechanisms with the assumption that phonons are the instantaneous normal modes of the liquids. Instead, the experimental energy-gap dependences can be quantitatively reproduced by the dephasing mechanism. A simple theoretical derivation shows that the fast molecular motions in liquids randomize the modulations on the energy donor and acceptor by phonons and diminish the phonon compensation efficiency on energy transfer. Estimations based on the theoretical derivations suggest that, for most nonresonant intermolecular vibrational-energy transfers in liquids with energy gaps smaller than the thermal energy, the dephasing mechanism dominates the energy-transfer process.
Co-reporter:Hailong Chen, Hongtao Bian, Jiebo Li, Xiewen Wen, Qiang Zhang, Wei Zhuang, and Junrong Zheng
The Journal of Physical Chemistry B 2015 Volume 119(Issue 12) pp:4333-4349
Publication Date(Web):February 13, 2015
DOI:10.1021/jp512320a
The methodology and principle using vibrational energy transfer to measure molecular distances in liquids are introduced. The application of the method to the studies of ion pairing and clustering in strong electrolyte aqueous solutions is demonstrated with MSCN aqueous solutions where M = Li, Na, K, Cs, and NH4. Experiments suggest that ions in the concentrated aqueous solutions can form substantial quantities of ion clusters in which both cations and anions are involved. More and larger clusters form in solutions that are relatively more concentrated and which include a larger cation. In KSCN solutions, the shortest anionic distance in the ion clusters is the same as that in the KSCN crystal. The rotational time of the anion and the nonresonant vibrational energy transfer time with a gap of 75 cm–1 in the KSCN saturated solution are very similar to those in the KSCN crystal. However, the KSCN ion clusters are closer in structure to the melt. The clusters form an interconnected network with random ionic orientations. Because of ion clustering, the anion and water dynamics behave distinctly in the same solutions. At high concentrations, the anion rotation significantly slows down because of the increase in the size of the ion clusters, but the slowdown amplitude of water rotation is very modest because many of the water molecules still remain in the “bulk” state due to ion clustering. The rotational dynamics of both water and anions are slower in a solution with a smaller cation, primarily because a smaller cation has a stronger cation/anion interaction and a cation/water interaction that leads to more water molecules confined in the ion clusters. Adding ions or molecules into the KSCN solutions can perturb the ion clusters. Weakly hydrated anions can participate in clustering and form mixed ion clusters with KSCN, and strongly hydrated anions can reduce the effective water molecules solvating KSCN and facilitate the formation of more and larger KSCN ion clusters. Similarly, molecules which can strongly bind to SCN– prefer to participate in the KSCN ion clusters. Molecules which are strongly hydrated prefer to remain hydrated and facilitate the ion clustering of KSCN.
Co-reporter:Hailong Chen, Xiewen Wen, Xunmin Guo and Junrong Zheng
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 27) pp:13995-14014
Publication Date(Web):20 May 2014
DOI:10.1039/C4CP01300J
Resonant and nonresonant intermolecular vibrational energy transfers in KSCN/KSC13N/KS13C15N aqueous and DMF solutions and crystals are studied. Both energy-gap and temperature dependent measurements reveal some surprising results, e.g. inverted energy-gap dependent energy transfer rates and opposite temperature dependences of resonant and nonresonant energy transfer rates. Two competing mechanisms are proposed to be responsible for the experimental observations. The first one is the dephasing mechanism in which the measured energy transfer rate originates from the dephasing of the energy donor–acceptor coherence, and the second one is the phonon-compensation mechanism derived from the second order perturbation. It is found that both the nonresonant energy transfers in the liquids and resonant energy transfers in both liquids and solids can be well described by the first mechanism. The second mechanism explains the nonresonant energy transfers in one series of the solid samples very well.
Co-reporter:Kaijun Yuan, Hongtao Bian, Yuneng Shen, Bo Jiang, Jiebo Li, Yufan Zhang, Hailong Chen, and Junrong Zheng
The Journal of Physical Chemistry B 2014 Volume 118(Issue 13) pp:3689-3695
Publication Date(Web):March 14, 2014
DOI:10.1021/jp500877u
The coordination number of Li+ in acetonitrile solutions was determined by directly measuring the rotational times of solvent molecules bound and unbound to it. The CN stretch of the Li+ bound and unbound acetonitrile molecules in the same solution has distinct vibrational frequencies (2276 cm–1 vs 2254 cm–1). The frequency difference allows the rotation of each type of acetonitrile molecule to be determined by monitoring the anisotropy decay of each CN stretch vibrational excitation signal. Regardless of the nature of anions and concentrations, the Li+ coordination number was found to be 4–6 in the LiBF4 (0.2–2 M) and LiPF6 (1–2 M) acetonitrile solutions. However, the dissociation constants of the salt are dependent on the nature of anions. In 1 M LiBF4 solution, 53% of the salt was found to dissociate into Li+, which is bound by 4–6 solvent molecules. In 1 M LiPF6 solution, 72% of the salt dissociates. 2D IR experiments show that the binding between Li+ and acetonitrile is very strong. The lifetime of the complex is much longer than 19 ps.
Co-reporter:Hailong Chen, Xiewen Wen, Jiebo Li, and Junrong Zheng
The Journal of Physical Chemistry A 2014 Volume 118(Issue 13) pp:2463-2469
Publication Date(Web):March 18, 2014
DOI:10.1021/jp500586h
In general, intermolecular distances in condensed phases at the angstrom scale are difficult to measure. We were able to do so by using the vibrational energy transfer method, an ultrafast vibrational analogue of Förster resonance energy transfer. The distances among SCN– anions in KSCN crystals and ion clusters of KSCN aqueous solutions were determined with the method. In the crystalline samples, the closest anion distance was determined to be 3.9 ± 0.3 Å, consistent with the XRD result. In the 1.8 and 1 M KSCN aqueous solutions, the anion distances in the ion clusters were determined to be 4.4 ± 0.4 Å. The clustered anion distances in aqueous solutions are very similar to the closest anion distance in the KSCN crystal but significantly shorter than the average anion distance (0.94–1.17 nm) in the aqueous solutions if ion clustering did not occur. The result suggests that ions in the strong electrolyte aqueous solutions can form clusters inside of which they have direct contact with each other.
Co-reporter:Hailong Chen, Hongtao Bian, Jiebo Li, Xiewen Wen, and Junrong Zheng
The Journal of Physical Chemistry A 2013 Volume 117(Issue 29) pp:6052-6065
Publication Date(Web):February 25, 2013
DOI:10.1021/jp312604v
In this work, through investigating a series of liquid, glassy, and crystalline samples with ultrafast multiple-mode 2D IR and IR transient absorption methods, we demonstrated that the signal anisotropy of vibrational relaxation-induced heat effects is determined by both relative molecular orientations and molecular rotations. If the relative molecular orientations are randomized or molecular rotations are fast compared to heat transfer, the signal anisotropy of heat effects is zero. If the relative molecular orientations are anisotropic and the molecular rotations are slow, the signal anisotropy of heat effects can be nonzero, which is determined by the relative orientations of the energy source mode and the heat sensor mode within the same molecule and in different molecules. We also demonstrated that the correlation between the anisotropy value of heat signal and the relative molecular orientations can be quantitatively calculated.
Co-reporter:Jiebo Li, Hongtao Bian, Hailong Chen, Xiewen Wen, Bryan T. Hoang, and Junrong Zheng
The Journal of Physical Chemistry B 2013 Volume 117(Issue 16) pp:4274-4283
Publication Date(Web):August 28, 2012
DOI:10.1021/jp3053373
KSCN and NH4SCN aqueous solutions were investigated with intermolecular vibrational energy transfer methods. In a KSCN/H2O (1/10 molar ratio) solution, 90% of the initial excitation of the CN stretch (∼2066 cm–1) of the SCN– anion is transferred to the HOH bending mode (∼1636 cm–1) of water molecules with an energy transfer time constant 3.1 ps. In a NH4SCN/H2O (1/10 molar ratio) solution, only 49% of the CN excitation flows to the water HOH bending mode with a time constant 6.3 ps. Most of the remaining CN excitation goes to the NH bending mode (∼1460 cm–1) of the NH+ cation with a time constant of 7.0 ps. The results indicate that about 50% of the energy transfer channel from the CN stretch to the HOH bending observed in the KSCN solution is overpowered by the NH4+ cations in the NH4SCN/H2O solution. Ion concentration dependent measurements support this argument. According to the dipole/dipole approximation, the CN/OH energy transfer occurs most efficiently between SCN– anions and the water molecules closest to them. The experimental results therefore suggest that more than 50% of the water molecules closest to the SCN– anions are replaced by the NH4+ cations in the NH4SCN/H2O (1/10 molar ratio) solution. The percentage is much larger than the NH4+/water ratio of 10%, indicating that the ion association between NH4+ and SCN– is caused by the chemical nature of the solution rather than the statistical “forced contact” because of the high ion concentration.
Co-reporter:Hailong Chen, Hongtao Bian, Jiebo Li, Xunmin Guo, Xiewen Wen, and Junrong Zheng
The Journal of Physical Chemistry B 2013 Volume 117(Issue 49) pp:15614-15624
Publication Date(Web):August 27, 2013
DOI:10.1021/jp406232k
The molecular conformations of crystalline l-cysteine prepared in its orthorhombic form were determined by the vibrational cross angle measurements. Its major dihedral angles of chemical bonds determined by this method are consistent with the results from diffraction experiments. In addition, the relative orientations of the chemical bonds associated with the hydrogen atoms of the NH3+ group and the thiol group are also determined. The results demonstrate that the vibrational cross angle method based on the multiple-mode approach can potentially become a structural tool for determining molecular conformations. The major challenges for the method to become a general molecular structural tool are discussed, and some approaches to address them are proposed.
Co-reporter:Hongtao Bian, Hailong Chen, Qiang Zhang, Jiebo Li, Xiewen Wen, Wei Zhuang, and Junrong Zheng
The Journal of Physical Chemistry B 2013 Volume 117(Issue 26) pp:7972-7984
Publication Date(Web):June 13, 2013
DOI:10.1021/jp4016646
Waiting time dependent rotational anisotropies of SCN– anions and water molecules in alkali thiocyanate (XSCN, X = Li, Na, K, Cs) aqueous solutions at various concentrations were measured with ultrafast infrared spectroscopy. It was found that cations can significantly affect the reorientational motions of both water molecules and SCN– anions. The dynamics are slower in a solution with a smaller cation. The reorientational time constants follow the order of Li+ > Na+ > K+ ≃ Cs+. The changes of rotational time constants of SCN– at various concentrations scale almost linearly with the changes of solution viscosity, but those of water molecules do not. In addition, the concentration-dependent amplitudes of dynamical changes are much more significant in the Li+ and Na+ solutions than those in the K+ and Cs+ solutions. Further investigations on the systems with the ultrafast vibrational energy exchange method and molecular dynamics simulations provide an explanation for the observations: the observed rotational dynamics are the balanced results of ion clustering and cation/anion/water direct interactions. In all the solutions at high concentrations (>5 M), substantial amounts of ions form clusters. The structural inhomogeneity in the solutions leads to distinct rotational dynamics of water and anions. The strong interactions of Li+ and Na+ because of their relatively large charge densities with water molecules and SCN– anions, in addition to the likely geometric confinements because of ion clustering, substantially slow down the rotations of SCN– anions and water molecules inside the ion clusters. The interactions of K+ and Cs+ with water or SCN– are much weaker. The rotations of water molecules inside ion clusters of K+ and Cs+ solutions are not significantly different from those of other water species so that the experimentally observed rotational relaxation dynamics are only slightly affected by the ion concentrations.
Co-reporter:Qiang Zhang, Wenjun Xie, HongTao Bian, Yi Qin Gao, Junrong Zheng, and Wei Zhuang
The Journal of Physical Chemistry B 2013 Volume 117(Issue 10) pp:2992-3004
Publication Date(Web):February 17, 2013
DOI:10.1021/jp400441e
Molecular dynamics simulations were carried out to investigate the microscopic origin of the deviation from Stokes–Einstein behavior observed in the dynamics of KSCN aqueous solutions. When the solution becomes more concentrated, the rotational mobilities of SCN– and water bifurcate significantly as also observed in the experimental ultrafast infrared measurements. The translational mobilities of different components, on the other hand, have similar concentration dependences. Furthermore, when concentrating the solution, the mobilities increase slightly first and then reduce afterward. Our simulations revealed that these phenomena observed in the dynamics originate from the ion assembling in the solution. The RDF and pair residence time analysis further suggest the ion pairing effect has significant contribution to the ion assembling. Results herein thus provide a microscopic insight on the origin of the ion assembling phenomenon and its connection with various experimentally observable dynamical phenomena in the ionic solutions.
Co-reporter:Hailong Chen, Yufan Zhang, Jiebo Li, Hongjun Liu, De-En Jiang, and Junrong Zheng
The Journal of Physical Chemistry A 2013 Volume 117(Issue 35) pp:8407-8415
Publication Date(Web):August 12, 2013
DOI:10.1021/jp406304c
The fluctuations of three-dimensional molecular conformations of a molecule in different environments play critical roles in many important chemical and biological processes. X-ray diffraction (XRD) techniques and nuclear magnetic resonance (NMR) methods are routinely applied to monitor the molecular conformations in condensed phases. However, some special requirements of the methods have prevented them from exploring many molecular phenomena at the current stage. Here, we introduce another method to resolve molecular conformations based on an ultrafast MIR/T-Hz multiple-dimensional vibrational spectroscopic technique. The model molecule (4′-methyl-2′-nitroacetanilide, MNA) is prepared in two of its crystalline forms and liquid samples. Two polarized ultrafast infrared pulses are then used to determine the cross-angles of vibrational transition moment directions by exciting one vibrational band and detecting the induced response on another vibrational band of the molecule. The vibrational cross-angles are then converted into molecular conformations with the aid of calculations. The molecular conformations determined by the method are supported by X-ray diffraction and molecular dynamics simulation results. The experimental results suggest that thermodynamic interactions with solvent molecules are not altering the molecular conformations of MNA in the solutions to control their ultimate conformations in the crystals.
Co-reporter:Jiebo Li, Hongtao Bian, Xiewen Wen, Hailong Chen, Kaijun Yuan, and Junrong Zheng
The Journal of Physical Chemistry B 2012 Volume 116(Issue 40) pp:12284-12294
Publication Date(Web):September 17, 2012
DOI:10.1021/jp306369w
Interactions between model molecules representing building blocks of proteins and the thiocyanate anion, a strong protein denaturant agent, were investigated in aqueous solutions with intermolecular vibrational energy exchange methods. It was found that thiocyanate anions are able to bind to the charged ammonium groups of amino acids in aqueous solutions. The interactions between thiocyanate anions and the amide groups were also observed. The binding affinity between the thiocyanate anion and the charged amino acid residues is about 20 times larger than that between water molecules and the amino acids and about 5–10 times larger than that between the thiocyanate anion and the neutral backbone amide groups. The series of experiments also demonstrates that the chemical nature, rather than the macroscopic dielectric constant, of the ions and molecules plays a critical role in ion/molecule interactions in aqueous solutions.
Co-reporter:Hongtao Bian, Jiebo Li, Qiang Zhang, Hailong Chen, Wei Zhuang, Yi Qin Gao, and Junrong Zheng
The Journal of Physical Chemistry B 2012 Volume 116(Issue 49) pp:14426-14432
Publication Date(Web):November 26, 2012
DOI:10.1021/jp310153n
Microscopic structures and dynamics of aqueous salt solutions were investigated with the ultrafast vibrational energy exchange method and anisotropy measurements. In KSCN aqueous solutions of various concentrations, the rotational time constants of SCN– anions are proportional to the viscosities of the solutions. However, the reorientation dynamics of the water molecules are only slightly affected by the solution viscosity. With the addition of strongly hydrated F– anions, the rotations of both SCN– anions and water molecules slow down. With the addition of weakly hydrated I– anions, only the rotation of SCN– anions slows down with that of water molecules unaffected. Vibrational energy exchange measurements show that the separation among SCN– anions decreases with the addition of F– and increases with the addition of I–. The series of experiments clearly demonstrate that both structures and dynamics of ion and water are segregated in the strong electrolyte aqueous solutions.
Co-reporter:Hongtao Bian, Jiebo Li, Hailong Chen, Kaijun Yuan, Xiewen Wen, Yaqin Li, Zhigang Sun, and Junrong Zheng
The Journal of Physical Chemistry C 2012 Volume 116(Issue 14) pp:7913-7924
Publication Date(Web):March 16, 2012
DOI:10.1021/jp300970p
Knowledge about molecular conformations and nuclear and electronic motions on surfaces of metal nanomaterials is critical for many applications but extremely difficult to obtain. We demonstrate that valuable information of this sort can be determined using multiple-mode multiple-dimensional vibrational spectroscopy. A model compound, 4-mercaptophenol, on the surface of 3.5 nm gold nanoparticles demonstrates the method. Its 3D molecular conformations and vibrational dynamics on the particle surfaces were determined with the method. The experimental results imply that on the particle surfaces, the ligand molecules cannot form energy-optimized hydrogen bonds because of the surface geometry constraint. The conclusion is supported with experiments on the ligand molecules in the crystalline phase and in a dilute solution. Our experiments also showed that the effect of the particle surface nonadiabatic electron/vibration coupling does not play a significant role in the vibrational relaxation of high-frequency modes (>1000 cm–1) about 3 Å away from the surface. Simple theoretical calculations support this observation. The method holds promise as a general tool for the studies of molecular structures and dynamics on the surfaces of nanomaterials. The capability of resolving 3D molecular conformations on nanomaterials surfaces is expected to be helpful for understanding specific intermolecular interactions and conformation-selective reactions (e.g., chirality selectivity) on the surfaces of these materials.
Co-reporter:Hongtao Bian, Hailong Chen, Jiebo Li, Xiewen Wen, and Junrong Zheng
The Journal of Physical Chemistry A 2011 Volume 115(Issue 42) pp:11657-11664
Publication Date(Web):September 14, 2011
DOI:10.1021/jp206937u
The donor/acceptor energy mismatch and vibrational coupling strength dependences of interionic vibrational energy transfer kinetics in electrolyte aqueous solutions were investigated with ultrafast multiple-dimensional vibrational spectroscopy. An analytical equation derived from the Fermi’s Golden rule that correlates molecular structural parameters and vibrational energy transfer kinetics was found to be able to describe the intermolecular mode specific vibrational energy transfer. Under the assumption of the dipole–dipole approximation, the distance between anions in the aqueous solutions was obtained from the vibrational energy transfer measurements, confirmed with measurements on the corresponding crystalline samples. The result demonstrates that the mode-specific vibrational energy transfer method holds promise as an angstrom molecular ruler.
Co-reporter:Hongtao Bian;Xiewen Wen;Jiebo Li;Hailong Chen;Jian Song;Wei Zhuang;Suzee Han;Xiuquan Sun
PNAS 2011 Volume 108 (Issue 12 ) pp:4737-4742
Publication Date(Web):2011-03-22
DOI:10.1073/pnas.1019565108
Despite prolonged scientific efforts to unravel the hydration structures of ions in water, many open questions remain, in
particular concerning the existences and structures of ion clusters in 1∶1 strong electrolyte aqueous solutions. A combined
ultrafast 2D IR and pump/probe study through vibrational energy transfers directly observes ion clustering in aqueous solutions
of LiSCN, NaSCN, KSCN and CsSCN. In a near saturated KSCN aqueous solution (water/KSCN molar ratio = 2.4/1), 95% of the anions
form ion clusters. Diluting the solution results in fewer, smaller, and tighter clusters. Cations have significant effects
on cluster formation. A small cation results in smaller and fewer clusters. The vibrational energy transfer method holds promise
for studying a wide variety of other fast short-range molecular interactions.
Co-reporter:Hongtao Bian, Jiebo Li, Xiewen Wen, Zhigang Sun, Jian Song, Wei Zhuang, and Junrong Zheng
The Journal of Physical Chemistry A 2011 Volume 115(Issue 15) pp:3357-3365
Publication Date(Web):March 25, 2011
DOI:10.1021/jp200516p
The multiple-mode two-dimensional infrared (2D-IR) spectrum in a broad frequency range from 1000 to 3200 cm−1 of a 1-cyanovinyl acetate solution in CCl4 is reported. By analyzing its relative orientations of the transition dipole moments of normal modes that cover vibrations of all chemical bonds, the three-dimensional molecular conformations and their population distributions of 1-cyanovinyl acetate are obtained, with the aid of quantum chemistry calculations that translate the experimental transition dipole moment cross angles into the cross angles among chemical bonds.
Co-reporter:Hailong Chen, Xiewen Wen, Xunmin Guo and Junrong Zheng
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 27) pp:NaN14014-14014
Publication Date(Web):2014/05/20
DOI:10.1039/C4CP01300J
Resonant and nonresonant intermolecular vibrational energy transfers in KSCN/KSC13N/KS13C15N aqueous and DMF solutions and crystals are studied. Both energy-gap and temperature dependent measurements reveal some surprising results, e.g. inverted energy-gap dependent energy transfer rates and opposite temperature dependences of resonant and nonresonant energy transfer rates. Two competing mechanisms are proposed to be responsible for the experimental observations. The first one is the dephasing mechanism in which the measured energy transfer rate originates from the dephasing of the energy donor–acceptor coherence, and the second one is the phonon-compensation mechanism derived from the second order perturbation. It is found that both the nonresonant energy transfers in the liquids and resonant energy transfers in both liquids and solids can be well described by the first mechanism. The second mechanism explains the nonresonant energy transfers in one series of the solid samples very well.
.jpg)