Co-reporter:Viswanath Arutla, Joseph Leal, Xiaowei Liu, Sriram Sokalingam, Michael Raleigh, Adejimi Adaralegbe, Li Liu, Paul R. Pentel, Sidney M. Hecht, and Yung Chang
ACS Combinatorial Science May 8, 2017 Volume 19(Issue 5) pp:286-286
Publication Date(Web):April 6, 2017
DOI:10.1021/acscombsci.6b00179
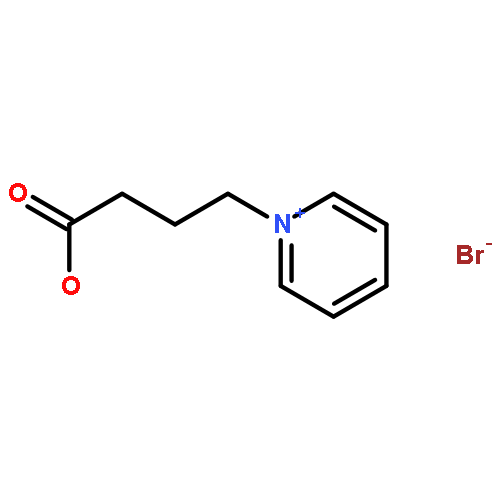
Since the demonstration of nicotine vaccines as a possible therapeutic intervention for the effects of tobacco smoke, extensive effort has been made to enhance nicotine specific immunity. Linker modifications of nicotine haptens have been a focal point for improving the immunogenicity of nicotine, in which the evaluation of these modifications usually relies on in vivo animal models, such as mice, rats or nonhuman primates. Here, we present two in vitro screening strategies to estimate and predict the immunogenic potential of our newly designed nicotine haptens. One utilizes a competition enzyme-linked immunoabsorbent assay (ELISA) to profile the interactions of nicotine haptens or hapten-protein conjugates with nicotine specific antibodies, both polyclonal and monoclonal. Another relies on computational modeling of the interactions between haptens and amino acid residues near the conjugation site of the carrier protein to infer linker-carrier protein conjugation effect on antinicotine antibody response. Using these two in vitro methods, we ranked the haptens with different linkers for their potential as viable vaccine candidates. The ELISA-based hapten ranking was in an agreement with the results obtained by in vivo nicotine pharmacokinetic analysis. A correlation was found between the average binding affinity (IC50) of the haptens to an anti-Nic monoclonal antibody and the average brain nicotine concentration in the immunized mice. The computational modeling of hapten and carrier protein interactions helps exclude conjugates with strong linker-carrier conjugation effects and low in vivo efficacy. The simplicity of these in vitro screening strategies should facilitate the selection and development of more effective nicotine conjugate vaccines. In addition, these data highlight a previously under-appreciated contribution of linkers and hapten-protein conjugations to conjugate vaccine immunogenicity by virtue of their inclusion in the epitope that binds and activates B cells.Keywords: B cells; computational modeling; nicotine hapten linkers; pharmacokinetics; vaccine candidates;
Co-reporter:Basab Roy, Poulami Talukder, Hyun-Jin Kang, Shujian S. Tsuen, Mohammad P. Alam, Laurence H. Hurley, and Sidney M. Hecht
Journal of the American Chemical Society 2016 Volume 138(Issue 34) pp:10950-10962
Publication Date(Web):August 2, 2016
DOI:10.1021/jacs.6b05036
Co-reporter:Arnaud Chevalier, Yanmin Zhang, Omar M. Khdour, and Sidney M. Hecht
Organic Letters 2016 Volume 18(Issue 10) pp:2395-2398
Publication Date(Web):May 11, 2016
DOI:10.1021/acs.orglett.6b00882
Acylation of 3-(N-formylamino)salicylic acids resulted in transacylation with loss of the formyl moiety. The reaction proceeds through a bis-N-acylated intermediate, which undergoes facile deformylation. This transacylation reaction has been employed for the site-specific functionalization of the mitochondrial poison antimycin A, affording several novel derivatives. The selective cytotoxicity of some of these derivatives toward cultured A549 human lung epithelial adenocarcinoma cells, in comparison with WI-38 normal human lung fibroblasts, illustrates one application of this transacylation reaction.
Co-reporter:Sandipan Roy Chowdhury, Pradeep S. Chauhan, Larisa M. Dedkova, Xiaoguang Bai, Shengxi Chen, Poulami Talukder, and Sidney M. Hecht
Biochemistry 2016 Volume 55(Issue 17) pp:2427-2440
Publication Date(Web):April 6, 2016
DOI:10.1021/acs.biochem.6b00102
Described herein are the synthesis and photophysical characterization of a library of aryl-substituted oxazole- and thiazole-based dipeptidomimetic analogues, and their incorporation into position 66 of green fluorescent protein (GFP) in lieu of the natural fluorophore. These fluorescent analogues resemble the fluorophore formed naturally by GFP. As anticipated, the photophysical properties of the analogues varied as a function of the substituents at the para position of the phenyl ring. The fluorescence emission wavelength maxima of compounds in the library varied from ∼365 nm (near-UV region) to ∼490 nm (visible region). The compounds also exhibited a large range of quantum yields (0.01–0.92). The analogues were used to activate a suppressor tRNACUA and were incorporated into position 66 of GFP using an in vitro protein biosynthesizing system that employed engineered ribosomes selected for their ability to incorporate dipeptides. Four analogues with interesting photophysical properties and reasonable suppression yields were chosen, and the fluorescent proteins (FPs) containing these fluorophores were prepared on a larger scale for more detailed study. When the FPs were compared with the respective aminoacyl-tRNAs and the actual dipeptide analogues, the FPs exhibited significantly enhanced fluorescence intensities at the same concentrations. Part of this was shown to be due to the presence of the fluorophores as an intrinsic element of the protein backbone. There were also characteristic shifts in the emission maxima, indicating the environmental sensitivity of these probes. Acridon-2-ylalanine and oxazole 1a were incorporated into positions 39 and 66 of GFP, respectively, and were shown to form an efficient Förster resonance energy transfer (FRET) pair, demonstrating that the analogues can be used as FRET probes.
Co-reporter:Arnaud Chevalier, Mohammad Parvez Alam, Omar M. Khdour, Margaret Schmierer, Pablo M. Arce, Cameron D. Cripe, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 21) pp:5206-5220
Publication Date(Web):1 November 2016
DOI:10.1016/j.bmc.2016.08.039
Previously we described a novel series of pyrimidinol antioxidants and their structural optimization as potential therapeutic agents for neurodegenerative and mitochondrial disorders. Our initial lead compound was a potent antioxidant in vitro, but was subsequently found to exhibit poor stability to oxidative metabolism. The current study focused on balancing potency with metabolic stability through structural modification, and involved modifications at positions 2 and 4 of the pyrimidinol redox core, likely sites of oxidative metabolism. Eight new analogues have been prepared and their ability to suppress lipid peroxidation and reactive oxygen species (ROS), and to preserve mitochondrial membrane potential (Δψm) and support ATP production, has been investigated. The metabolic stability of the prepared compounds was also assessed in vitro using bovine liver microsomes to obtain preliminary insight on this class of compounds. This study revealed the complexity of balancing reasonable metabolic stability with efficient antioxidant properties. While a few analogues appear promising, especially in terms of metabolic stability, a 4-isopropoxy derivative conserved the favorable biological activity and exhibited good metabolic stability. The favorable metabolic stability conferred by the combination of the azetidine and isopropoxy moieties in analogue 6 makes this compound an excellent candidate for further evaluation.
Co-reporter:Poulami Talukder, Larisa M. Dedkova, Andrew D. Ellington, Petro Yakovchuk, Jaebum Lim, Eric V. Anslyn, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 18) pp:4177-4187
Publication Date(Web):15 September 2016
DOI:10.1016/j.bmc.2016.07.008
Proteins which bind to nucleic acids and regulate their structure and functions are numerous and exceptionally important. Such proteins employ a variety of strategies for recognition of the relevant structural elements in their nucleic acid substrates, some of which have been shown to involve rather subtle interactions which might have been difficult to design from first principles. In the present study, we have explored the preparation of proteins containing unnatural amino acids having nucleobase side chains. In principle, the introduction of multiple nucleobase amino acids into the nucleic acid binding domain of a protein should enable these modified proteins to interact with their nucleic acid substrates using Watson-Crick and other base pairing interactions. We describe the synthesis of five alanyl nucleobase amino acids protected in a fashion which enabled their attachment to a suppressor tRNA, and their incorporation into each of two proteins with acceptable efficiencies. The nucleobases studied included cytosine, uracil, thymine, adenine and guanine, i.e. the major nucleobase constituents of DNA and RNA. Dihydrofolate reductase was chosen as one model protein to enable direct comparison of the facility of incorporation of the nucleobase amino acids with numerous other unnatural amino acids studied previously. The Klenow fragment of DNA polymerase I was chosen as a representative DNA binding protein whose mode of action has been studied in detail.
Co-reporter:Diego Mastroeni, Omar M. Khdour, Pablo M. Arce, Sidney M. Hecht, and Paul D. Coleman
ACS Chemical Neuroscience 2015 Volume 6(Issue 4) pp:588
Publication Date(Web):February 10, 2015
DOI:10.1021/cn500323q
Alzheimer’s disease is associated with metabolic deficits and reduced mitochondrial function, with the latter due to the effects of oligomeric amyloid beta peptide (AβO) on the respiratory chain. Recent evidence has demonstrated reduction of epigenetic markers, such as DNA methylation, in Alzheimer’s disease. Here we demonstrate a link between metabolic and epigenetic deficits via reduction of mitochondrial function which alters the expression of mediators of epigenetic modifications. AβO-induced loss of mitochondrial function in differentiated neuronal cells was reversed using two novel antioxidants (1 and 2); both have been shown to mitigate the effects of reactive oxygen species (ROS), and compound 1 also restores adenosine triphosphate (ATP) levels. While both compounds were effective in reducing ROS, restoration of ATP levels was associated with a more robust response to AβO treatment. Our in vitro system recapitulates key aspects of data from Alzheimer’s brain samples, the expression of epigenetic genes in which are also shown to be normalized by the novel analogues.Keywords: Alzheimer’s disease; epigenetics; Mitochondria; multifunctional radical quenchers
Co-reporter:Poulami Talukder, Shengxi Chen, Basab Roy, Petro Yakovchuk, Michelle M. Spiering, Mohammad P. Alam, Manikandadas M. Madathil, Chandrabali Bhattacharya, Stephen J. Benkovic, and Sidney M. Hecht
Biochemistry 2015 Volume 54(Issue 51) pp:7457-7469
Publication Date(Web):November 30, 2015
DOI:10.1021/acs.biochem.5b01085
Described herein are the syntheses and photophysical characterization of three novel cyanotryptophans, and their efficient incorporation into proteins as fluorescent probes. Photophysical characteristics indicated that each was significantly brighter and red-shifted in fluorescence emission relative to tryptophan. Each analogue was used to activate a suppressor tRNA transcript and was incorporated with good efficiency into two different positions (Trp22 and Trp74) of Escherichia coli dihydrofolate reductase (ecDHFR). The Trp analogues could be monitored selectively in the presence of multiple native Trp residues in DHFR. 6-CNTrp (A) formed an efficient Förster resonance energy transfer (FRET) pair with l-(7-hydroxycoumarin-4-yl)ethylglycine (HCO, D) at position 17. Further, 6-CNTrp (A) was incorporated into two DNA binding proteins, including the Klenow fragment of DNA polymerase I and an RNA recognition motif (RRM2) of heterogeneous nuclear ribonucleoprotein L-like (hnRNP LL). Using these proteins, we demonstrated the use of FRET involving A as a fluorescence donor and benzo[g]quinazoline-2,4-(1H,3H)-dione 2′-deoxyriboside (Tf) or 4-aminobenzo[g]quinazoline-2-one 2′-deoxyriboside (Cf) as fluorescent acceptors to study the binding interaction of the Klenow fragment with duplex DNA oligomers (labeled with Tf), or the domain-specific association between hnRNP LL and the BCL2 i-motif DNA (labeled with Cf). Thus, the non-natural amino acid could be used as a FRET partner for studying protein–nucleic acid interactions. Together, these findings demonstrate the potential utility of 6-CNTrp (A) as a fluorescence donor for the study of protein conformational events.
Co-reporter:Poulami Talukder, Shengxi Chen, Pablo M. Arce, and Sidney M. Hecht
Organic Letters 2014 Volume 16(Issue 2) pp:556-559
Publication Date(Web):January 6, 2014
DOI:10.1021/ol403429e
Two new fluorescent probes of protein structure and dynamics have been prepared by concise asymmetric syntheses using the Schöllkopf chiral auxiliary. The site-specific incorporation of one probe into dihydrofolate reductase is reported. The utility of these tryptophan derivatives lies in their absorption and emission maxima which differ from those of tryptophan, as well as in their large Stokes shifts and high molar absorptivities.
Co-reporter:Mohammad Parvez Alam, Omar M. Khdour, Pablo M. Arce, Yana Chen, Basab Roy, Walter G. Johnson, Sriloy Dey, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 17) pp:4935-4947
Publication Date(Web):1 September 2014
DOI:10.1016/j.bmc.2014.06.040
As part of our ongoing efforts to identify compounds having potential utility in treating neurodegenerative and mitochondrial disorders, a series of pyridinol analogues have been prepared. The synthetic route employed for the preparation of the new analogues is different, and considerably more efficient, than that used in previously reported studies. The new route yields a pair of pyridinol regioisomers that can be readily separated and evaluated. Their ability to quench lipid peroxidation and reactive oxygen species (ROS), and to preserve mitochondrial membrane potential (Δψm) and support ATP synthesis is reported. The optimal side chain length was found to be 16 carbon atoms. The metabolic stability of those compounds having optimal biological activities was evaluated in vitro using bovine liver microsomes. The omission of any side chain hydroxyl group and introduction of an azetidine moiety at position 6 of the pyridinol redox core (8 and 9) increased their microsomal stability as compared to the exocyclic dimethylamino group. The favorable metabolic stability conferred by the azetidine moiety in compounds 8 and 9 makes these compounds excellent candidates for further evaluation.
Co-reporter:Poulami Talukder, Shengxi Chen, C. Tony Liu, Edwin A. Baldwin, Stephen J. Benkovic, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2014 Volume 22(Issue 21) pp:5924-5934
Publication Date(Web):1 November 2014
DOI:10.1016/j.bmc.2014.09.015
With the continuing interest in deciphering the interplay between protein function and conformational changes, small fluorescence probes will be especially useful for tracking changes in the crowded protein interior space. Presently, we describe the potential utility of six unnatural amino acid fluorescence donors structurally related to tryptophan and show how they can be efficiently incorporated into a protein as fluorescence probes. We also examine the various photophysical properties of the new Trp analogues, which are significantly redshifted in their fluorescence spectra relative to tryptophan. In general, the Trp analogues were well tolerated when inserted into Escherichia coli DHFR, and did not perturb enzyme activity, although substitution for Trp22 did result in a diminution in DHFR activity. Further, it was demonstrated that D and E at position 37 formed efficient FRET pairs with acridon-2-ylalanine (Acd) at position 17. The same was also true for a DHFR construct containing E at position 79 and Acd at position 17. Together, these findings demonstrate that these tryptophan analogues can be introduced into DHFR with minimal disruption of function, and that they can be employed for the selective study of targeted conformational changes in proteins, even in the presence of unmodified tryptophans.
Co-reporter:Rumit Maini, Dan T. Nguyen, Shengxi Chen, Larisa M. Dedkova, Sandipan Roy Chowdhury, Rafael Alcala-Torano, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2013 Volume 21(Issue 5) pp:1088-1096
Publication Date(Web):1 March 2013
DOI:10.1016/j.bmc.2013.01.002
Ribosomes containing modifications in three regions of 23S rRNA, all of which are in proximity to the ribosomal peptidyltransferase center (PTC), were utilized previously as a source of S-30 preparations for in vitro protein biosynthesis experiments. When utilized in the presence of mRNAs containing UAG codons at predetermined positions + β-alanyl–tRNACUA, the modified ribosomes produced enhanced levels of full length proteins via UAG codon suppression. In the present study, these earlier results have been extended by the use of substituted β-amino acids, and direct evidence for β-amino acid incorporation is provided. Presently, five of the clones having modified ribosomes are used in experiments employing four substituted β-amino acids, including α-methyl-β-alanine, β,β-dimethyl-β-alanine, β-phenylalanine, and β-(p-bromophenyl)alanine. The β-amino acids were incorporated into three different positions (10, 18 and 49) of Escherichia coli dihydrofolate reductase (DHFR) and their efficiencies of suppression of the UAG codons were compared with those of β-alanine and representative α-l-amino acids. The isolated proteins containing the modified β-amino acids were subjected to proteolytic digestion, and the derived fragments were characterized by mass spectrometry, establishing that the β-amino acids had been incorporated into DHFR, and that they were present exclusively in the anticipated peptide fragments. DHFR contains glutamic acid in position 17, and it has been shown previously that Glu-C endoproteinase can hydrolyze DHFR between amino acids residues 17 and 18. The incorporation of β,β-dimethyl-β-alanine into position 18 of DHFR prevented this cleavage, providing further evidence for the position of incorporation of the β-amino acid.
Co-reporter:David M. Fash, Omar M. Khdour, Sunil J. Sahdeo, Ruth Goldschmidt, Jennifer Jaruvangsanti, Sriloy Dey, Pablo M. Arce, Valérie C. Collin, Gino A. Cortopassi, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2013 21(8) pp: 2346-2354
Publication Date(Web):
DOI:10.1016/j.bmc.2013.01.075
Co-reporter:Ruth Goldschmidt, Pablo M. Arce, Omar M. Khdour, Valérie C. Collin, Sriloy Dey, Jennifer Jaruvangsanti, David M. Fash, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2013 21(4) pp: 969-978
Publication Date(Web):
DOI:10.1016/j.bmc.2012.11.051
Co-reporter:Pablo M. Arce, Ruth Goldschmidt, Omar M. Khdour, Manikandadas M. Madathil, Jennifer Jaruvangsanti, Sriloy Dey, David M. Fash, Jeffrey S. Armstrong, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 17) pp:5188-5201
Publication Date(Web):1 September 2012
DOI:10.1016/j.bmc.2012.07.005
Selected pyridinol analogues of the experimental neuroprotective drug idebenone have been synthesized and evaluated as antioxidants capable of preserving mitochondrial function. The compounds, having a different redox core but the same side chain as idebenone, exhibited a range of potencies, reflecting differences in their structures. The results obtained provide guidance in the design of such analogues with improved properties. Analogues were identified that have significantly improved antioxidant activity compared with idebenone in cultured lymphocytes, and which exhibit lesser inhibition of the electron transport chain.
Co-reporter:Shengxi Chen, Nour Eddine Fahmi, Ryan C. Nangreave, Youcef Mehellou, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 8) pp:2679-2689
Publication Date(Web):15 April 2012
DOI:10.1016/j.bmc.2012.02.024
N,S-diprotected l-thiothreonine and l-allo-thiothreonine derivatives were synthesized using a novel chemical strategy, and used for esterification of the dinucleotide pdCpA. The aminoacylated dinucleotides were then employed for the preparation of activated suppressor tRNACUA transcripts. Thiothreonine and allo-thiothreonine were incorporated into a predetermined position of a catalytically competent dihydrofolate reductase (DHFR) analogue lacking cysteine, and the elaborated proteins were derivatized site-specifically at the thiothreonine residue with a fluorophore.
Co-reporter:Xiaoqing Cai, Omar M. Khdour, Jennifer Jaruvangsanti, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 11) pp:3584-3595
Publication Date(Web):1 June 2012
DOI:10.1016/j.bmc.2012.03.075
Bicyclic pyridinol antioxidants have been reported to suppress the autoxidation of methyl linoleate more effectively than α-tocopherol in benzene solution. A few novel lipophilic analogues have recently been synthesized by conjugating a pyridinol core with the phytyl side chain of α-tocopherol; these have been shown to possess potent antioxidant activity. However, the complexity of the synthetic routes has hampered their further development. Herein, we describe a facile approach, involving only five synthetic steps, to simplified analogues (1a–1c) and their acetate ester precursors (2a–2c). Simple alkyl chains of different lengths were attached to the 6-methyl group of the antioxidant core via regioselective metalation. These analogues were found to retain biological activity and exhibit protective behaviour under conditions of induced oxidative stress, which could lead to the development of more readily accessible analogues as potential antioxidants capable of preserving mitochondrial function.
Co-reporter:Xiaoqing Cai, Paul A. Zaleski, Ali Cagir, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 12) pp:3831-3844
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmc.2011.04.047
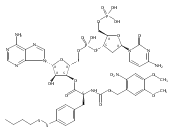
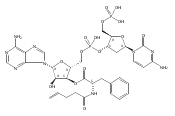
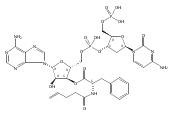
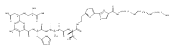
Previous studies have indicated that the methylvalerate subunit of bleomycin (BLM) plays an important role in facilitating DNA cleavage by BLM and deglycoBLM. Eleven methylvalerate analogues have been synthesized and incorporated into deglycoBLM congeners by the use of solid-phase synthesis. The effect of the valerate moiety in the deglycoBLM analogues has been studied by comparison with the parent deglycoBLM A5 using supercoiled DNA relaxation and sequence-selective DNA cleavage assays. All of the deglycoBLM analogues were found to effect the relaxation of the plasmid DNA. Those analogues having aromatic C4-substituents exhibited cleavage efficiency comparable to that of deglycoBLM A5. Some, but not all, of the deglycoBLM analogues were also capable of mediating sequence-selective DNA cleavage.
Co-reporter:Jun Lu, Xiaoqing Cai, and Sidney M. Hecht
Organic Letters 2010 Volume 12(Issue 22) pp:5189-5191
Publication Date(Web):October 12, 2010
DOI:10.1021/ol102217c
The recently reported bicyclic pyridinols 1 and 2 are highly effective antioxidants exhibiting 88- and 28-fold greater potency, respectively, than α-tocopherol when assayed for their ability to suppress the autoxidation of methyl linoleate. Described herein is a short, economical, and scalable strategy for the synthesis of this novel group of antioxidants, as well as analogues 3−6. Key reactions involved the cyclocondensation reaction of lactam acetals with enaminone 7 and selective functionalizaton of the heterocyclic systems.
Co-reporter:Damien Y. Duveau, Pablo M. Arce, Robert A. Schoenfeld, Nidhi Raghav, Gino A. Cortopassi, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 17) pp:6429-6441
Publication Date(Web):1 September 2010
DOI:10.1016/j.bmc.2010.06.104
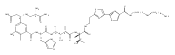
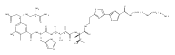
Analogues of mitoQ and idebenone were synthesized to define the structural elements that support oxygen consumption in the mitochondrial respiratory chain. Eight analogues were prepared and fully characterized, then evaluated for their ability to support oxygen consumption in the mitochondrial respiratory chain. While oxygen consumption was strongly inhibited by mitoQ analogues 2–4 in a chain length-dependent manner, modification of idebenone by replacement of the quinone methoxy groups by methyl groups (analogues 6–8) reduced, but did not eliminate, oxygen consumption. Idebenone analogues 6–8 also displayed significant cytoprotective properties toward cultured mammalian cells in which glutathione had been depleted by treatment with diethyl maleate.
Co-reporter:Jun Lu, Omar M. Khdour, Jeffrey S. Armstrong, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 21) pp:7628-7638
Publication Date(Web):1 November 2010
DOI:10.1016/j.bmc.2010.08.030
An efficient synthesis has provided access to a novel α-tocopherol analogue (2), as well as its trifluoroacetate salt and acetate ester. An annulation reaction was used to establish the pyridinol core structure and a Stille coupling reaction was employed for conjugation with the tocopherol side chain. This analogue was shown to suppress the levels of reactive oxygen species in cultured cells, and to quench peroxidation of mitochondrial membranes.
Co-reporter:Simon J. Leiris, Omar M. Khdour, Zachary J. Segerman, Krystal S. Tsosie, Jean-Charles Chapuis, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 10) pp:3481-3493
Publication Date(Web):15 May 2010
DOI:10.1016/j.bmc.2010.03.070
Verticipyrone has recently been isolated from the culture broth of Verticillium sp. and shown to inhibit NADH fumarate reductase, as well as NADH oxidoreductase (complex I) of the mitochondrial electron transport chain. In order to assess the structural elements in verticipyrone essential for complex I inhibitor, 15 structural analogues were prepared and analyzed for their effects on mitochondrial NADH oxidoreductase and NADH oxidase activities. Also measured were the abilities of several of the analogues to inhibit respiration as judged by a shift to glycolysis, and to inhibit the growth of several mammalian cell lines. The nature of the pyrone ring was shown to be important to potency of inhibition, as was the length and nature of substituents in the side chain of the analogues.
Co-reporter:Jean-Charles Chapuis, Omar Khdour, Xiaoqing Cai, Jun Lu, Sidney M. Hecht
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 6) pp:2204-2209
Publication Date(Web):15 March 2009
DOI:10.1016/j.bmc.2008.10.071
Four Δlac-acetogenins have been prepared and characterized as inhibitors of the mitochondrial respiratory chain, to define the effects of unsaturation within the alkyl substituents. In keeping with earlier reports, the presence of acetylenic functionalities within the alkyl substituents slightly diminished their potency of inhibition of NADH oxidase activity, which measures the overall transfer of electrons from NADH to oxygen through mitochondrial complexes I, III, and IV. In contrast, both of the acetylenic Δlac-acetogenins were far more active in a NADH–ubiquinone Q1 oxidoreductase assay that measures complex I function per se.
Co-reporter:Arnaud Chevalier, Omar M. Khdour, Margaret Schmierer, Indrajit Bandyopadhyay, Sidney M. Hecht
Bioorganic & Medicinal Chemistry (1 March 2017) Volume 25(Issue 5) pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.bmc.2017.01.030
Recently, we described the optimization of novel pyrimidinol-based antioxidants as potential therapeutic molecules for targeting mitochondrial diseases. That study focused on improving the potency and metabolic stability of pyrimidinol antioxidants. This led us to consider the possibility of altering the positions of the exocyclic alkoxy and alkylamino substituents on the pyrimidinol scaffold. Twelve new analogues were prepared and their biological activities were investigated. The metabolic stability of the prepared regioisomers was also assessed in vitro using bovine liver microsomes. Unexpectedly, the 2-alkoxy-4-alkylamino substituted pyrimidinol antioxidants were found to have properties in protecting mitochondrial function superior to the isomeric 4-alkoxy-2-alkylamino substituted pyrimidinols evaluated in all earlier studies. This observation suggests a possible mode of action involving the intermediacy of an ortho-iminoquinone, a species not previously associated with mitochondrial respiratory chain function.
.jpg)
![1H-INDOLE-7-CARBONITRILE, 3-FORMYL-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/863500/863407-38-9.png)
![1H-INDOLE-7-CARBONITRILE, 3-FORMYL-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/863500/863407-38-9_b.png)

![1H-INDOLE-6-CARBONITRILE, 3-FORMYL-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/468800/468718-26-5.png)
![1H-INDOLE-6-CARBONITRILE, 3-FORMYL-1-[(4-METHYLPHENYL)SULFONYL]-](http://img.cochemist.com/ccimg/468800/468718-26-5_b.png)
![GLYCINE, N,N-BIS[(1,1-DIMETHYLETHOXY)CARBONYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/153400/153393-01-2.png)
![GLYCINE, N,N-BIS[(1,1-DIMETHYLETHOXY)CARBONYL]-, METHYL ESTER](http://img.cochemist.com/ccimg/153400/153393-01-2_b.png)
![D-Lysine, N6-[(1,1-dimethylethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/63400/63328-49-4.png)
![D-Lysine, N6-[(1,1-dimethylethoxy)carbonyl]-, methyl ester](http://img.cochemist.com/ccimg/63400/63328-49-4_b.png)













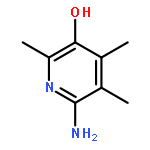
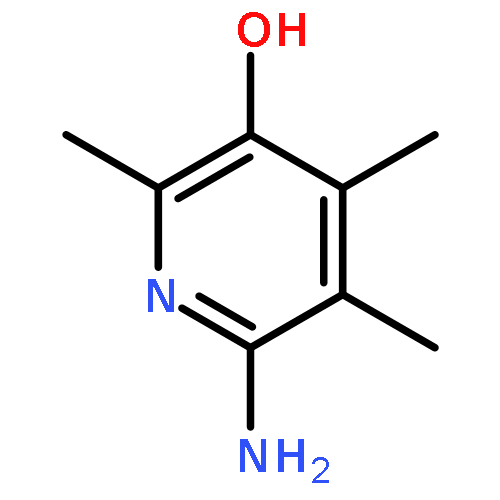
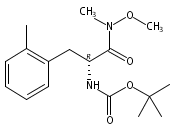
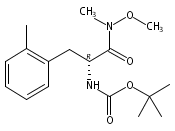
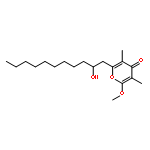
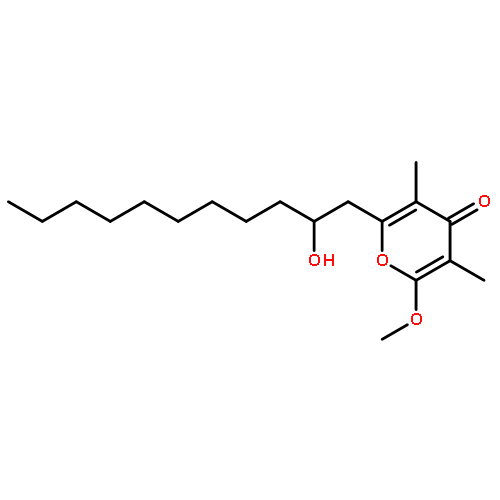


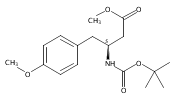
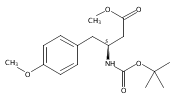
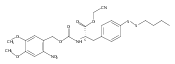
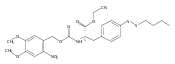
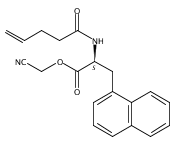

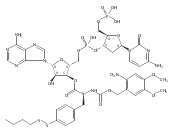





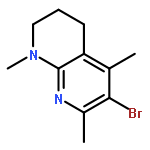
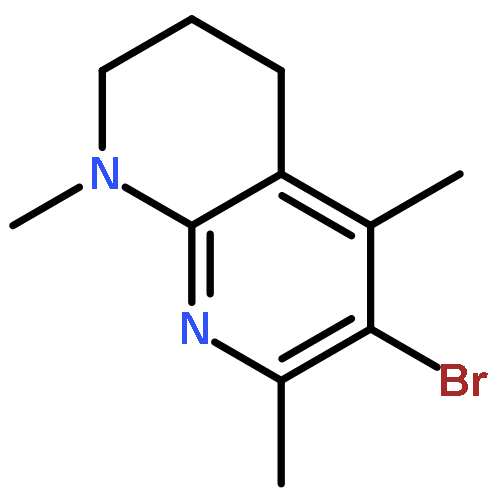
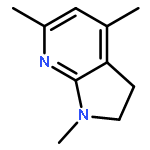
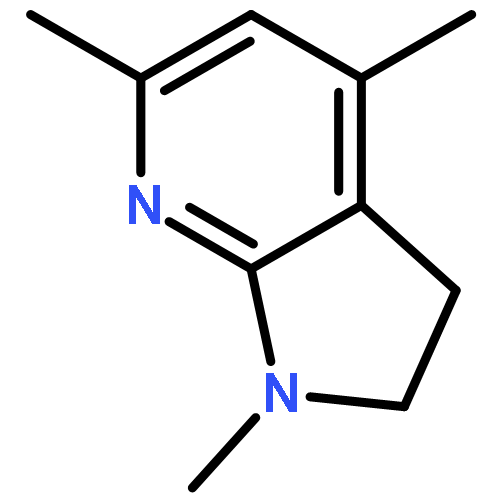
![2,3-dihydro-1,4,6-trimethyl-1H-Pyrrolo[2,3-b]pyridin-5-ol](http://img.cochemist.com/ccimg/627100/627098-04-8.png)
![2,3-dihydro-1,4,6-trimethyl-1H-Pyrrolo[2,3-b]pyridin-5-ol](http://img.cochemist.com/ccimg/627100/627098-04-8_b.png)
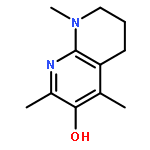
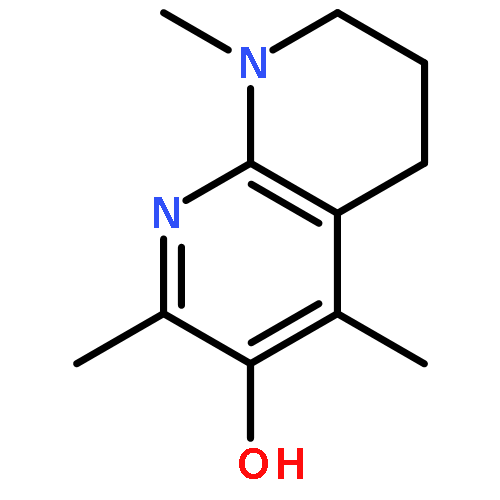



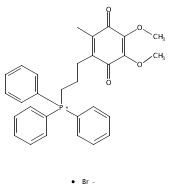
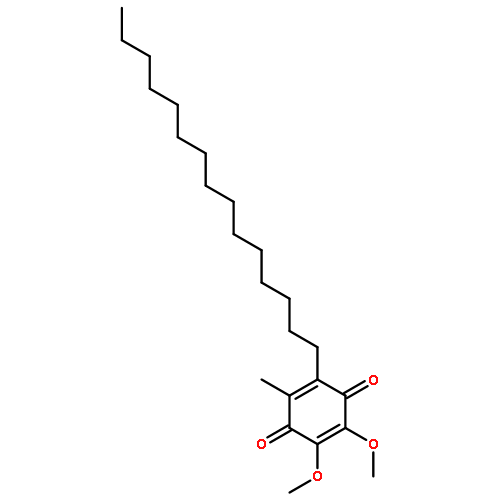
![Phosphonium,[10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadien-1-yl)decyl]triphenyl-,bromide (1:1)](http://img.cochemist.com/ccimg/336200/336184-91-9.png)
![Phosphonium,[10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadien-1-yl)decyl]triphenyl-,bromide (1:1)](http://img.cochemist.com/ccimg/336200/336184-91-9_b.png)







![Carbamic acid, [(1R)-2-cyclohexyl-1-formylethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/160900/160820-17-7.png)
![Carbamic acid, [(1R)-2-cyclohexyl-1-formylethyl]-, 1,1-dimethylethylester](http://img.cochemist.com/ccimg/160900/160820-17-7_b.png)










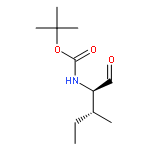
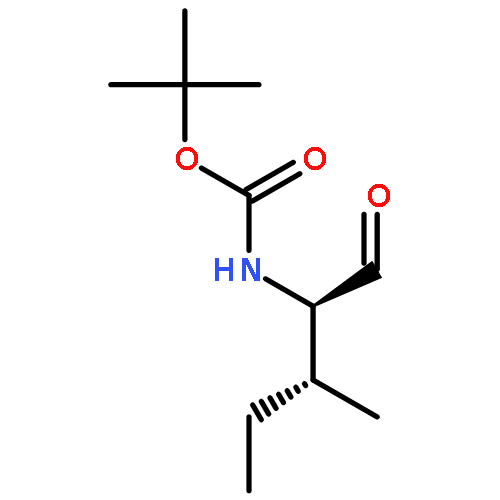


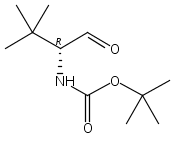


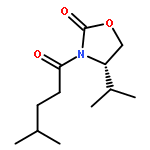
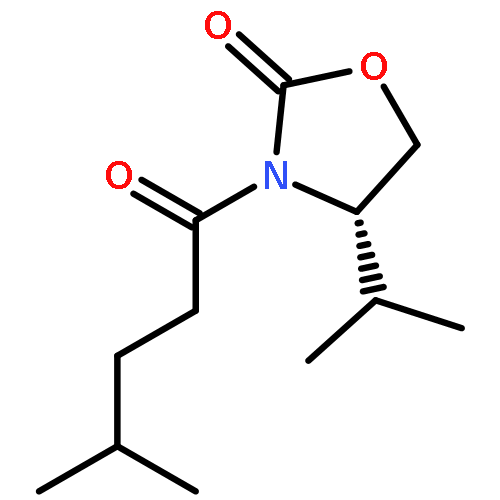
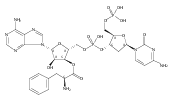
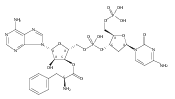


![Carbamic acid, [(1R)-1-formyl-3-methylbutyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/116700/116662-96-5.png)
![Carbamic acid, [(1R)-1-formyl-3-methylbutyl]-, 1,1-dimethylethyl ester](http://img.cochemist.com/ccimg/116700/116662-96-5_b.png)




![Bleomycinamide,N1-[4-[(aminoiminomethyl)amino]butyl]-41-O-de[2-O-[3-O-(aminocarbonyl)-a-D-mannopyranosyl]-a-L-gulopyranosyl]- (9CI)](http://img.cochemist.com/ccimg/78400/78314-57-5.png)
![Bleomycinamide,N1-[4-[(aminoiminomethyl)amino]butyl]-41-O-de[2-O-[3-O-(aminocarbonyl)-a-D-mannopyranosyl]-a-L-gulopyranosyl]- (9CI)](http://img.cochemist.com/ccimg/78400/78314-57-5_b.png)

![4-Pyrimidinecarboxylicacid,6-amino-2-[(1S)-3-amino-1-[[(2S)-3-amino-2-[[(1,1-dimethylethoxy)carbonyl]amino]-3-oxopropyl]amino]-3-oxopropyl]-5-methyl-](http://img.cochemist.com/ccimg/75500/75452-30-1.png)
![4-Pyrimidinecarboxylicacid,6-amino-2-[(1S)-3-amino-1-[[(2S)-3-amino-2-[[(1,1-dimethylethoxy)carbonyl]amino]-3-oxopropyl]amino]-3-oxopropyl]-5-methyl-](http://img.cochemist.com/ccimg/75500/75452-30-1_b.png)
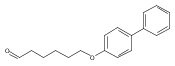
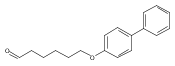
![1-HEXANOL, 6-([1,1'-BIPHENYL]-4-YLOXY)-](http://img.cochemist.com/ccimg/73600/73586-33-1.png)
![1-HEXANOL, 6-([1,1'-BIPHENYL]-4-YLOXY)-](http://img.cochemist.com/ccimg/73600/73586-33-1_b.png)




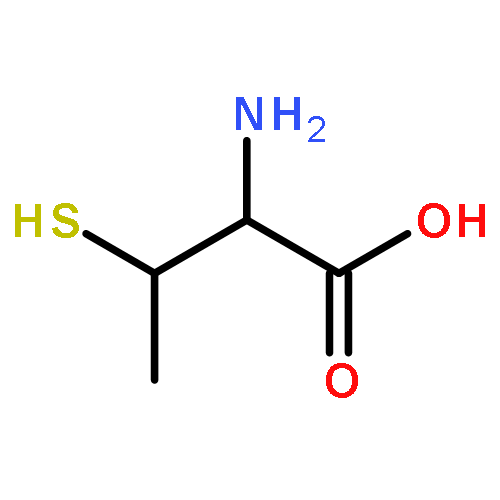

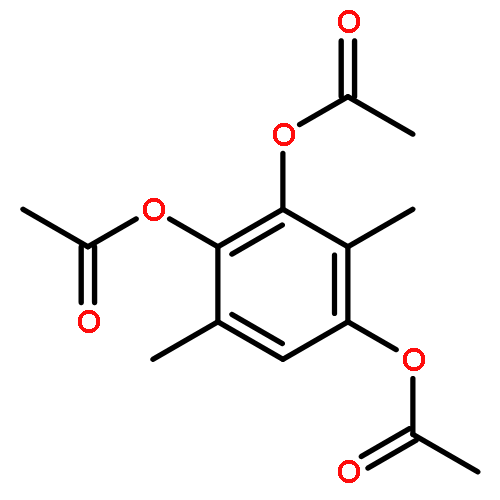
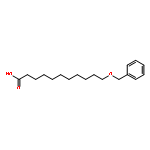
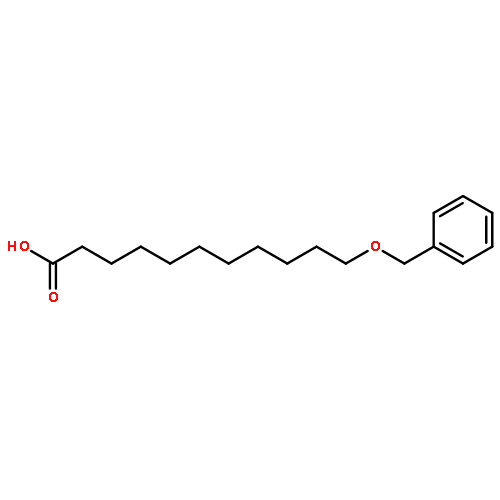
![5'-O-[hydroxy(1H-imidazol-1-yl)phosphoryl]adenosine](http://img.cochemist.com/ccimg/20900/20816-58-4.png)
![5'-O-[hydroxy(1H-imidazol-1-yl)phosphoryl]adenosine](http://img.cochemist.com/ccimg/20900/20816-58-4_b.png)

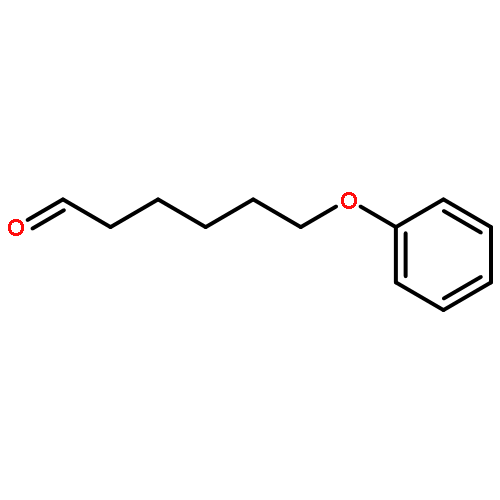
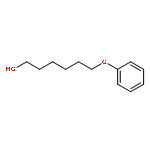








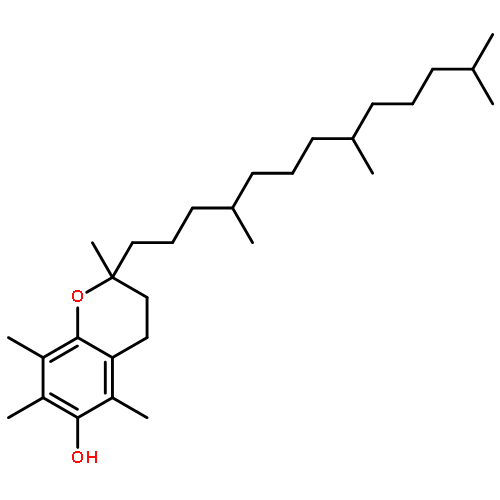
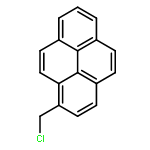
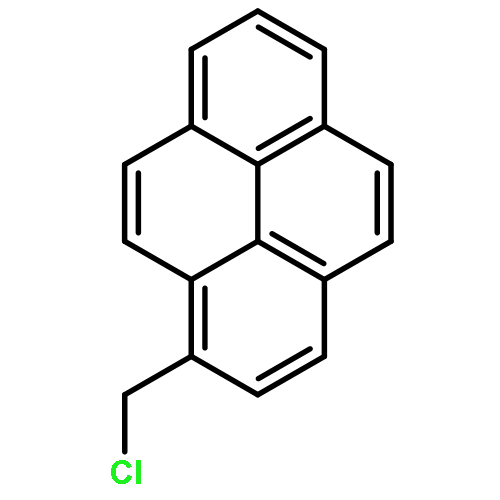
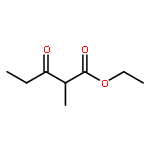












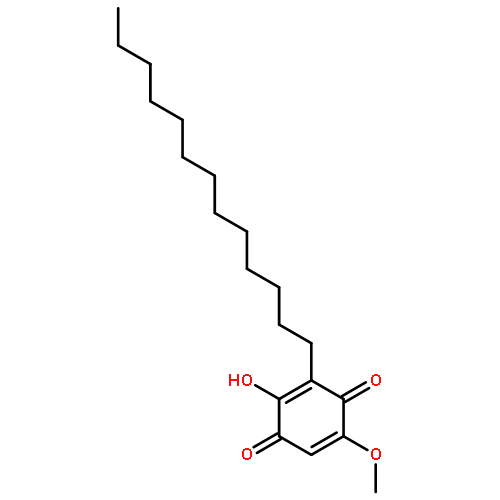
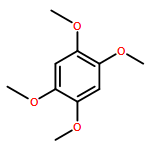
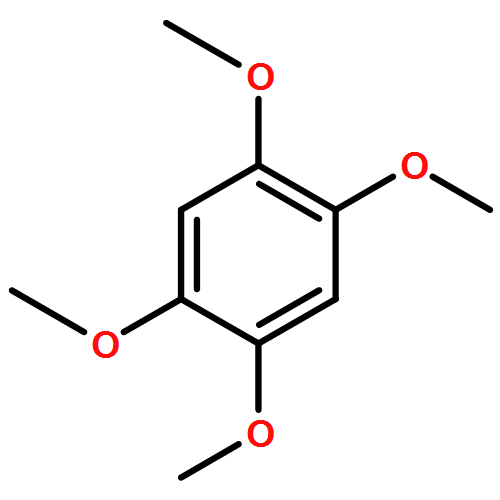




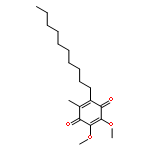



![N1-[3-[(4-aminobutyl)amino]propyl]-](http://img.cochemist.com/ccimg/11200/11116-32-8.png)
![N1-[3-[(4-aminobutyl)amino]propyl]-](http://img.cochemist.com/ccimg/11200/11116-32-8_b.png)