Co-reporter:Nicholas B. Tito, Scott T. Milner and Jane E. G. Lipson
Soft Matter 2015 vol. 11(Issue 39) pp:7792-7801
Publication Date(Web):27 Aug 2015
DOI:10.1039/C5SM01701G
The diffusion of mobility in bulk and thin film fluids near their glass transition is examined with a kinetic lattice model, and compared to recent experiments on bulk liquids and vapor-deposited thin film glasses. The “limited mobility” (LM) lattice model exhibits dynamic heterogeneity of mobility when the fluid is near its kinetic arrest transition; a finite-parameter second-order critical point in the LM model bearing strong resemblance to the glass transition in real fluids. The spatial heterogeneity of mobility near kinetic arrest leads to dynamics that violate the Stokes–Einstein relation. To make connections with experiment, LM model simulations of self-diffusion constants in fluids near kinetic arrest are compared to those in two organic glass-formers. In addition, simulations of mobility in films that have been temperature-jumped above kinetic arrest (starting from an arrested state) are carried out. The films develop a “front” of mobility at their free surface that progresses into the film interior at a constant rate, thereby mobilising the entire film to fluidity. The velocity of the front scales with the self-diffusion constant for analogous bulk systems—an observation consistent with experiments on vapor-deposited molecular thin films.
Co-reporter:Jeffrey DeFelice and Jane E. G. Lipson
Macromolecules 2014 Volume 47(Issue 16) pp:5643-5654
Publication Date(Web):August 8, 2014
DOI:10.1021/ma501199n
In this article, we connect the experimental miscibility of several polymer/supercritical carbon dioxide (scCO2) mixtures with their pure component properties, such as free volume and interaction energy. We directly address the experimental observations that suggest free volume-rich polymers and those with weak polymer segment–segment interaction energies mix more favorably with scCO2. By applying our simple locally correlated lattice (LCL) theory to model the pressure–volume–temperature (PVT) behavior of the pure polymers and supercritical solvent, we obtain characteristic molecular parameters which are then used to predict the key physical properties of interest. We probe the underlying thermodynamic contributions (entropic and enthalpic) to the free energies of mixing and show that our LCL theory can explain the experimental miscibility ranking based solely on our characterization of the pure components.
Co-reporter:Nicholas B. Tito, Jane E. G. Lipson and Scott T. Milner
Soft Matter 2013 vol. 9(Issue 11) pp:3173-3180
Publication Date(Web):07 Feb 2013
DOI:10.1039/C3SM25679K
We present a new model of glass-forming liquids in which free volume fluctuations drive dynamic heterogeneity and diffusion of mobility. Our kinetic model is developed from a microscopic perspective where free volume creation requires existing free volume nearby as a source of mobility; free volume diffusion and destruction is spontaneous by virtue of its own mobility. Simulations indicate “molten” and kinetically arrested “glassy” regions in parameter space, and a critical point where dynamic length and intermittency time scales grow as power laws. Systems near the critical point contain colonies of free volume that propagate around bubbles of glassified material.
Co-reporter:Nicholas B. Tito, Jane E. G. Lipson and Scott T. Milner
Soft Matter 2013 vol. 9(Issue 39) pp:9403-9413
Publication Date(Web):15 Aug 2013
DOI:10.1039/C3SM51287H
A simple kinetic lattice model of free volume and mobility transport in fluids is applied to study the enhancement of mobility at a free surface in thin fluid films, as well as proximity effects in fluid bilayers consisting of materials with different local mobility. Consistent with experimental observations on fluid and polymeric thin films, our model predicts the presence of a mobile layer of material near the free surface of a kinetically arrested (glassy) film. The mobile layer extends deeper into the film, in front-like fashion, as the sample approaches the transition to complete fluidity. The extent of enhanced mobility is independent of film thickness at a given temperature, thus we find that thinner films have more suppressed sample-average glass transition temperatures compared to bulk material. This theme repeats itself in our simulations of fluid bilayers; slabs of material with suppressed or enhanced mobility respectively cause premature or delayed glassification of the whole system.
Co-reporter:E.A. Clark, J.E.G. Lipson
Polymer 2012 Volume 53(Issue 2) pp:536-545
Publication Date(Web):24 January 2012
DOI:10.1016/j.polymer.2011.11.045
We introduce a model for polymer solutions and blends that display both an upper and a lower critical solution temperature (UCST, LCST). Using our simple analytic lattice theory along with a fixed parameter set independent of both composition and temperature, we study solutions and blends exhibiting complicated miscibility patterns, including cases in which a UCST is below an LCST, or above. With respect to the former, we examine the conditions under which a so-called hourglass phase diagram may evolve. Where possible we compare directly with experiment, however, we also explore the behaviour of a number of hypothetical solutions and blends. One experimental solution of particular interest comprises star polystyrene (PS) and cyclohexane; in this case we supply what we believe are the only measured pressure-volume-temperature data for a high molecular weight (5.2 × 105 g/mol) star PS.
Co-reporter:Nicholas B. Tito, Scott T. Milner, and Jane E. G. Lipson
Macromolecules 2012 Volume 45(Issue 18) pp:7607-7620
Publication Date(Web):September 14, 2012
DOI:10.1021/ma3011558
The conformation of a linear gradient copolymer chain in a homopolymer melt is investigated using theory and numerical solutions of self-consistent field equations. In the limit of large comonomer immiscibility and chain length, it is found that the copolymer collapses into a globule with monomers self-assembled into a “ball-of-yarn” conformation. The spatial heterogeneity of monomers within the globule is in striking contrast to the “tadpole” conformation of a collapsed symmetric diblock copolymer and the disordered globular state of a collapsed homopolymer or random copolymer. By simple free energy calculations, we find that the same thermodynamic factors which drive a melt of linear gradient copolymers to self-assemble into lamellar microphases in the strong-segregation regime act to drive a single copolymer to self-assemble its own monomers into the yarn ball conformation when in a homopolymer melt with which it is immiscible. Moreover, by considering self-assembly of monomers within the globule of a collapsed copolymer, we find that the thermodynamic stability of a linear gradient is less than a symmetric diblock—a conclusion that is not possible to obtain by assuming that the copolymers pack their monomers randomly upon collapse.
Co-reporter:Ronald P. White and Jane E. G. Lipson and Julia S. Higgins
Macromolecules 2012 Volume 45(Issue 21) pp:8861-8871
Publication Date(Web):2017-2-22
DOI:10.1021/ma3018124
Fundamental insight regarding what drives polymer blend immiscibility/miscibility requires understanding the enthalpic and entropic contributions to the free energy of mixing. In this modeling investigation we show that two quantities, connected to the molecular characterization parameters, serve as separate “controls” on these thermodynamic mixing functions. The g parameter is defined as g = εij/(εiiεjj)1/2, with ε representing a segment–segment interaction energy, which may be obtained using minimal data on the mixture (for example, a phase separation temperature), appears to be correlated with the enthalpy of mixing. Characterization of the pure components, for example by fitting equation of state data, yields εii values. In this work we present evidence that |εii – εjj| controls the entropy of mixing. Furthermore, by analyzing the separate ideal and excess contributions to the entropy of mixing, we demonstrate that it is the excess contribution in particular that is strongly influenced by the value of |εii – εjj|, becoming increasingly unfavorable as |εii – εjj| increases. This sheds further light on a correlation noted in recent work of ours [White; Macromolecules 2012, 45, 1076−1084], that for LCST-type blends an increasingly favorable εij (meaning an increasing value of g) is needed to offset a greater mismatch in εii and εjj values (meaning an increasing |εii – εjj| difference) in order to maintain even partial miscibility. Given the importance of the excess entropy of mixing in driving miscibility, especially for LCST-type blends, we conclude that knowledge of |εii – εjj| can lead to a degree of a priori insight in assessing the mixture thermodynamics in the absence of any mixture data.
Co-reporter:Jane E. G. Lipson and Scott T. Milner
Macromolecules 2010 Volume 43(Issue 23) pp:9874-9880
Publication Date(Web):November 15, 2010
DOI:10.1021/ma101099n
In the companion paper (DOI 10.1021/ma101098d), we presented a quantitative theory for the suppression of the glass transition in a thin polymer film. Our delayed glassification (DG) model follows a proposal by de Gennes that free volume can be transmitted from surface to film interior via kinks transported along molecular strands or loops. In this paper, we use the DG model to predict the effects of molecular weight and film thickness on the film-averaged glass transition for a polystyrene sample. Our predictions for both freestanding and supported films of polystyrene illustrate that the DG model is able to account for some, but not all, of the experimental trends. This leads us to confront a number of issues, including how to average local glass transitions to yield a sample value as well as how to rationalize the nature of the molecular weight dependence for transitions in the thinnest freestanding films.
Co-reporter:Nicholas B. Tito, Scott T. Milner, and Jane E. G. Lipson
Macromolecules 2010 Volume 43(Issue 24) pp:10612-10620
Publication Date(Web):December 3, 2010
DOI:10.1021/ma102296r
We investigate lamellar microphases in linear gradient copolymer melts by a combination of techniques, including numerical solutions of self-consistent field equations, scaling theory, and analysis of the strong-segregation limit. In particular, we construct a Flory theory to predict the scaling of the equilibrium lamellar width Leq as a function of comonomer incompatibility measured by χN. The Flory theory balances chain stretching with increased monomer repulsive interactions resulting from conformational fluctuations about a state of uniformly stretched chains, that obtains in the strong-segregation limit. We find Leq/Rg ∼ (χN)1/6, which agrees well with numerical results. Remarkably, this is the same result as for symmetric diblock copolymers, although for quite different physical reasons.
Co-reporter:Antonis Gitsas and George Floudas, Ronald P. White and Jane E. G. Lipson
Macromolecules 2009 Volume 42(Issue 15) pp:5709-5716
Publication Date(Web):July 2, 2009
DOI:10.1021/ma900831h
The effect of pressure on the segmental dynamics in two symmetric blends of PMPS and PS is studied for pressures up to 250 MPa. In these blends, there is interplay between spinodal decomposition and glass transition, resulting in the enrichment of the high Tg component by the more mobile component. The distinctly different pressure sensitivities of PS and PMPS are used as fingerprints of the phase state, allowing for identification of the origin of the two dynamic processes arising from the PMPS segmental dynamics in PMPS-rich and PS-rich domains. Model calculations using a lattice-based equation of state lead to prediction of the phase diagram, as well as the effect of pressure on the critical temperature for the same PS/PMPS blend. The weak pressure sensitivity of the critical temperature (dTc/dP), compared to the two segmental relaxations, suggests that a transition to a thermodynamically miscible but dynamically heterogeneous state takes place for pressures above 300 MPa.
Co-reporter:Jeffrey DeFelice, Julia S. Higgins, Jane E.G. Lipson
Polymer (7 April 2017) Volume 114() pp:149-160
Publication Date(Web):7 April 2017
DOI:10.1016/j.polymer.2017.02.089
.jpg)
![Poly[oxycarbonyl-1,3-phenylenecarbonyloxy-1,4-phenylene(1-methylethylidene)-1,4-phenylene]](http://img.cochemist.com/ccimg/25300/25212-77-5.png)
![Poly[oxycarbonyl-1,3-phenylenecarbonyloxy-1,4-phenylene(1-methylethylidene)-1,4-phenylene]](http://img.cochemist.com/ccimg/25300/25212-77-5_b.png)
![POLY[OXYCARBONYL-1,3-PHENYLENECARBONYLOXY(2,6-DIMETHYL-1,4-PHENYLENE)(1-METHYLETHYLIDENE)(3,5-DIMETHYL-1,4-PHENYLENE)]](http://img.cochemist.com/ccimg/62900/62804-43-7.png)
![POLY[OXYCARBONYL-1,3-PHENYLENECARBONYLOXY(2,6-DIMETHYL-1,4-PHENYLENE)(1-METHYLETHYLIDENE)(3,5-DIMETHYL-1,4-PHENYLENE)]](http://img.cochemist.com/ccimg/62900/62804-43-7_b.png)




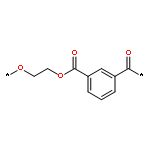
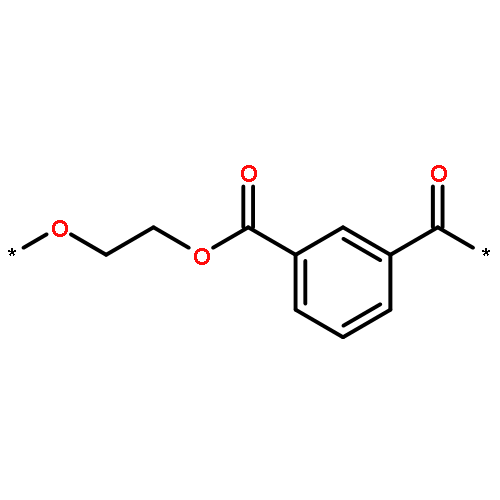
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4.png)
![Poly[oxy(1-oxo-1,6-hexanediyl)]](http://img.cochemist.com/ccimg/25300/25248-42-4_b.png)










![Poly[oxycarbonyloxy(2,6-dimethyl-1,4-phenylene)(1-methylethylidene)(3,
5-dimethyl-1,4-phenylene)]](http://img.cochemist.com/ccimg/38800/38797-88-5.png)
![Poly[oxycarbonyloxy(2,6-dimethyl-1,4-phenylene)(1-methylethylidene)(3,
5-dimethyl-1,4-phenylene)]](http://img.cochemist.com/ccimg/38800/38797-88-5_b.png)