Co-reporter:Joel Tellinghuisen
Analytical Biochemistry 2016 Volume 513() pp:43-46
Publication Date(Web):15 November 2016
DOI:10.1016/j.ab.2016.08.024
Abstract
Isothermal titration calorimetry data for very low c (≡K[M]0) must normally be analyzed with the stoichiometry parameter n fixed — at its known value or at any reasonable value if the system is not well characterized. In the latter case, ΔH° (and hence n) can be estimated from the T-dependence of the binding constant K, using the van't Hoff (vH) relation. An alternative is global or simultaneous fitting of data at multiple temperatures. In this Note, global analysis of low-c data at two temperatures is shown to estimate ΔH° and n with double the precision of the vH method.
Co-reporter:Joel Tellinghuisen
Analytical Biochemistry 2016 Volume 496() pp:1-3
Publication Date(Web):1 March 2016
DOI:10.1016/j.ab.2015.10.016
Abstract
Relative expression ratios are commonly estimated in real-time qPCR studies by comparing the quantification cycle for the target gene with that for a reference gene in the treatment samples, normalized to the same quantities determined for a control sample. For the “standard curve” design, where data are obtained for all four of these at several dilutions, nonlinear least squares can be used to assess the amplification efficiencies (AE) and the adjusted ΔΔCq and its uncertainty, with automatic inclusion of the effect of uncertainty in the AEs. An algorithm is illustrated for the KaleidaGraph program.
Co-reporter:Joel Tellinghuisen
Journal of Chemical Education 2016 Volume 93(Issue 6) pp:1061-1067
Publication Date(Web):April 20, 2016
DOI:10.1021/acs.jchemed.6b00027
The method of least squares (LS) yields exact solutions for the adjustable parameters when the number of data values n equals the number of parameters p. This holds also when the fit model consists of m different equations and m = p, which means that LS algorithms can be used to obtain solutions to systems of equations. In particular, nonlinear LS solves systems of nonlinear equations. An important example in chemistry is the case of reagents whose concentrations are coupled through multiple equilibrium relations. The capability of nonlinear LS in this application is examined for three programming environments, Excel Solver, FORTRAN, and KaleidaGraph, on a number of equilibrium problems having up to 10 unknown concentrations. FORTRAN and KaleidaGraph perform well in all the examples, but Solver presents difficulties that render it inadequate in several cases unless the problem is reformulated in terms of a smaller number of adjustable concentrations. When the input quantities (equilibrium constants, prepared concentrations) have uncertainty, the calculations can also be used to propagate these uncertainties into the derived quantities.
Co-reporter:Joel Tellinghuisen
Analytical Chemistry 2016 Volume 88(Issue 24) pp:
Publication Date(Web):November 15, 2016
DOI:10.1021/acs.analchem.6b03139
The role of partition volume variability, or polydispersity, in digital polymerase chain reaction methods is examined through formal considerations and Monte Carlo simulations. Contrary to intuition, polydispersity causes little precision loss for low average copy number per partition μ and can actually improve precision when μ exceeds ∼4. It does this by negatively biasing the estimates of μ, thus increasing the number of negative (null) partitions N0. In keeping with binomial statistics, this increases the relative precision of N0 and hence of the biased estimate m of μ. Below μ = 1, the precision loss and the bias are both small enough to be negligible for many applications. For higher μ the bias becomes more important than the imprecision, making accuracy dependent on knowledge of the partition volume distribution function. This information can be gained with optical microscopy or through calibration with reference materials.
Co-reporter:Joel Tellinghuisen , Andrej-Nikolai Spiess
Analytical Chemistry 2015 Volume 87(Issue 17) pp:8925
Publication Date(Web):August 3, 2015
DOI:10.1021/acs.analchem.5b02057
Monte Carlo simulations are used to examine the bias and loss of precision that result from experimental error and analysis procedures in real-time quantitative polymerase chain reaction (PCR). In the limit of small copy numbers (N0), Poisson statistics govern the dispersion in estimates of the quantification cycle (Cq) for replicate experiments, permitting the estimation of N0 from the Cq variance, which is inversely proportional to N0. We derive corrections to expressions given previously for this determination. With increasing N0, the Poisson contribution decreases and other effects, like pipet volume uncertainty (typically >3%), dominate. Cycle-to-cycle variability in the amplification efficiency E produces scale dispersion similar to that for variability in the sensitivity of fluorescence detection. When this E variability is proportional to just the amplification (E – 1), there is insignificant effect on Cq if scale-independent definitions are used for this marker. Single-reaction analysis methods based on the exponential growth equation are inherently low-biased in E and high-biased in N0, and these biases can amount to factor-of-4 or greater error in N0. For estimating Cq, their greatest limitation is use of a constant absolute threshold, making them inefficient for data that exhibit scale variability.
Co-reporter:Joel Tellinghuisen , Andrej-Nikolai Spiess
Analytical Chemistry 2015 Volume 87(Issue 3) pp:1889
Publication Date(Web):January 11, 2015
DOI:10.1021/acs.analchem.5b00077
The quantification cycle (Cq) is widely used for calibration in real-time quantitative polymerase chain reaction (qPCR), to estimate the initial amount, or copy number (N0), of the target DNA. Cq may be defined several ways, including the cycle where the detected fluorescence achieves a prescribed threshold level. For all methods of defining Cq, the standard deviation from replicate experiments is typically much greater than the estimated standard errors from the least-squares fits used to obtain Cq. For moderate-to-large copy number (N0 > 102), pipet volume uncertainty and variability in the amplification efficiency (E) likely account for most of the excess variance in Cq. For small N0, the dispersion of Cq is determined by the Poisson statistics of N0, which means that N0 can be estimated directly from the variance of Cq. The estimation precision is determined by the statistical properties of χ2, giving a relative standard deviation of ∼(2/n)1/2, where n is the number of replicates, for example, a 20% standard deviation in N0 from 50 replicates.
Co-reporter:Joel Tellinghuisen
Journal of Chemical Education 2015 Volume 92(Issue 5) pp:864-870
Publication Date(Web):February 12, 2015
DOI:10.1021/ed500888r
The method of least-squares (LS) has a built-in procedure for estimating the standard errors (SEs) of the adjustable parameters in the fit model: They are the square roots of the diagonal elements of the covariance matrix. This means that one can use least-squares to obtain numerical values of propagated errors by defining the target quantities as adjustable parameters in an appropriate LS fit model. Often this will be an exact, weighted, nonlinear fit, requiring special precautions to circumvent program idiosyncrasies and extract the desired a priori SEs. These procedures are reviewed for several commercial programs and illustrated specifically for the KaleidaGraph program. Examples include the estimation of ΔH°, ΔS°, ΔG°, and K°(T) and their SEs from K° (equilibrium constant) values at two temperatures, with and without uncertainty in T, which is included using the effective variance method, a general-purpose LS procedure for including uncertainty in independent variables. In some cases, the target quantities can be obtained from the original data analysis, by redefining the fit model to include the quantity of interest as an adjustable parameter, automatically handling correlation problems. Examples include the uncertainty in the fit function itself, line areas from spectral line profile data, and the analysis of spectrophotometric data for complex formation.
Co-reporter:Joel Tellinghuisen, Andrej-Nikolai Spiess
Analytical Biochemistry 2014 Volume 449() pp:76-82
Publication Date(Web):15 March 2014
DOI:10.1016/j.ab.2013.12.020
Abstract
New methods are used to compare seven qPCR analysis methods for their performance in estimating the quantification cycle (Cq) and amplification efficiency (E) for a large test data set (94 samples for each of 4 dilutions) from a recent study. Precision and linearity are assessed using chi-square (χ2), which is the minimized quantity in least-squares (LS) fitting, equivalent to the variance in unweighted LS, and commonly used to define statistical efficiency. All methods yield Cqs that vary strongly in precision with the starting concentration N0, requiring weighted LS for proper calibration fitting of Cq vs log(N0). Then χ2 for cubic calibration fits compares the inherent precision of the Cqs, while increases in χ2 for quadratic and linear fits show the significance of nonlinearity. Nonlinearity is further manifested in unphysical estimates of E from the same Cq data, results which also challenge a tenet of all qPCR analysis methods — that E is constant throughout the baseline region. Constant-threshold (Ct) methods underperform the other methods when the data vary considerably in scale, as these data do.
Co-reporter:Joel Tellinghuisen, Andrej-Nikolai Spiess
Analytical Biochemistry 2014 Volume 464() pp:94-102
Publication Date(Web):1 November 2014
DOI:10.1016/j.ab.2014.06.015
Abstract
Most methods for analyzing real-time quantitative polymerase chain reaction (qPCR) data for single experiments estimate the hypothetical cycle 0 signal y0 by first estimating the quantification cycle (Cq) and amplification efficiency (E) from least-squares fits of fluorescence intensity data for cycles near the onset of the growth phase. The resulting y0 values are statistically equivalent to the corresponding Cq if and only if E is taken to be error free. But uncertainty in E usually dominates the total uncertainty in y0, making the latter much degraded in precision compared with Cq. Bias in E can be an even greater source of error in y0. So-called mechanistic models achieve higher precision in estimating y0 by tacitly assuming E = 2 in the baseline region and so are subject to this bias error. When used in calibration, the mechanistic y0 is statistically comparable to Cq from the other methods. When a signal threshold yq is used to define Cq, best estimation precision is obtained by setting yq near the maximum signal in the range of fitted cycles, in conflict with common practice in the y0 estimation algorithms.
Co-reporter:Joel Tellinghuisen
Analytical Biochemistry 2012 Volume 424(Issue 2) pp:211-220
Publication Date(Web):15 May 2012
DOI:10.1016/j.ab.2011.12.035
Literature recommendations for designing isothermal titration calorimetry (ITC) experiments to study 1:1 binding, M + X ⇄ MX, are not consistent and have persisted through time with little quantitative justification. In particular, the “standard protocol” employed by most workers involves 20 to 30 injections of titrant to a final titrant/titrand mole ratio (Rm) of ∼ 2—a scheme that can be far from optimal and can needlessly limit applicability of the ITC technique. These deficiencies are discussed here along with other misconceptions. Whether a specific binding process can be studied by ITC is determined less by c (the product of binding constant K and titrand concentration [M]0) than by the total detectable heat qtot and the extent to which M can be converted to MX. As guidelines, with 90% conversion to MX, K can be estimated within 5% over the range 10 to 108 M−1 when qtot/σq ≈ 700, where σq is the standard deviation for estimation of q. This ratio drops to ∼ 150 when the stoichiometry parameter n is treated as known. A computer application for modeling 1:1 binding yields realistic estimates of parameter standard errors for use in protocol design and feasibility assessment.
Co-reporter:Joel Tellinghuisen, John D. Chodera
Analytical Biochemistry 2011 Volume 414(Issue 2) pp:297-299
Publication Date(Web):15 July 2011
DOI:10.1016/j.ab.2011.03.024
In the study of 1:1 binding by isothermal titration calorimetry, reagent concentration errors are fully absorbed in the data analysis, giving incorrect values for the key parameters—K, ΔH, and n—with no effect on the least-squares statistics. Reanalysis of results from an interlaboratory study of a selected biochemical process demonstrates that concentration errors are likely responsible for most of the overall statistical error in these parameters. The concentration errors are approximately 10%, greatly exceeding expected levels. Furthermore, examination of selected data sets reveals a surprising sensitivity to the baseline, suggesting a need for great care in treating dilution heats.
Co-reporter:Joel Tellinghuisen, Carl H. Bolster
Chemometrics and Intelligent Laboratory Systems 2011 Volume 105(Issue 2) pp:220-222
Publication Date(Web):15 February 2011
DOI:10.1016/j.chemolab.2011.01.004
R2 can be used correctly to select from among competing least-squares fit models when the data are fitted in common form and with common weighting. However, when models are compared by fitting data that have been mathematically transformed in different ways, R2 is a flawed statistic, even when the data are properly weighted in accord with the transformations. The reason is that in its most commonly used form, R2can be expressed in terms of the excess variance (s2) and the total variance in y (sy2) — the first of which is either invariant or approximately so with proper weighting, but the second of which can vary substantially in data transformations. When given data are analyzed “as is” with different models and fixed weights, sy2 remains constant and R2 is a valid statistic. However, then s2, and χ2 in weighted fitting, are arguably better metrics for such comparisons.
Co-reporter:Joel Tellinghuisen, Carl H. Bolster
Environmental Science & Technology 2010 Volume 44(Issue 13) pp:5029-5034
Publication Date(Web):June 14, 2010
DOI:10.1021/es100535b
Phosphorus soil sorption data are typically fitted to simple isotherms for the purpose of compactly summarizing experimental results and extrapolating beyond the range of measurements. Here, the question of which of the commonly preferred models—Langmuir and Freundlich—is better, is addressed using weighted least-squares, with weights obtained by variance function analysis of replicate data. Proper weighting in this case requires attention to a special problem—that the dependent variable S is not measured, rather is calculated from the measured equilibrium concentration C. The latter is commonly taken as the independent variable but is subject to experimental error, violating a fundamental least-squares assumption. This problem is handled through an effective variance treatment. When the data are fitted to the Langmuir, Freundlich, and Temkin isotherms, only the Freundlich model yields a statistically adequate χ2 value, and then only when S is taken to include labile residual P (S0) estimated from isotope-exchange experiments. The Freundlich model also yields good estimates of S0 when this is treated as an adjustable parameter rather than a known quantity—of relevance to studies in which S0 is not measured. By contrast, neglect of weights and labile P can lead to a mistaken preference for the Langmuir model.
Co-reporter:Qiaoling Charlene Zeng, Elizabeth Zhang, Hong Dong, Joel Tellinghuisen
Journal of Chromatography A 2008 Volume 1206(Issue 2) pp:147-152
Publication Date(Web):10 October 2008
DOI:10.1016/j.chroma.2008.08.036
For minimum-variance estimation of parameters by the method of least squares, heteroscedastic data should be weighted inversely as their variance, wi∝1/σi2. Here the instrumental data variance for a commercial high-performance liquid chromatography (HPLC) instrument is estimated from 5 to 11 replicate measurements on more than 20 samples for each of four different analytes. The samples span a range of over four orders of magnitude in concentration and HPLC peak area, over which the sampling variance estimates s2 are well represented as a sum of a constant term and a term proportional to the square of the peak area. The latter contribution is dominant over most of the range used in routine HPLC analysis and represents approximately 0.2% of peak area for all four analytes studied here. It includes a contribution from uncertainty in the syringe injection volume, which is found to be ±0.008 μL. The dominance of proportional error justifies the use of 1/x2 or 1/y2 weighting in routine calibration with such data; however, the constant variance term means that these weighting formulas are not correct in the low-signal limit relevant for analysis at trace levels. Least-squares methods for both direct and logarithmic fitting of variance sampling estimates are described. Since such estimates themselves have proportional uncertainty, direct fitting requires iterative adjustment of the weights, while logarithmic fitting does not.
Co-reporter:Joel Tellinghuisen
Analytical Biochemistry 2008 Volume 373(Issue 2) pp:395-397
Publication Date(Web):15 February 2008
DOI:10.1016/j.ab.2007.08.039
In the study of 1:1 binding, M + X ⇄ MX, isothermal titration calorimetry (ITC) can be used successfully at values of c = K[M]0 well below the value 1.0 that is often considered its lower limit. However, analysis of low-c ITC data may require freezing the stoichiometry parameter n, and that is thought to be prohibitive for biological systems, where n can be poorly known. Here it is noted that the least-squares estimates of the binding constant K are virtually independent of errors in n at low c, permitting reliable determination of K and, from its temperature dependence, ΔH° and n, down to c = 10−4 or lower, ligand solubility permitting.
Co-reporter:Joel Tellinghuisen
The Journal of Physical Chemistry A 2008 Volume 112(Issue 26) pp:5902-5907
Publication Date(Web):June 10, 2008
DOI:10.1021/jp8020358
The equilibrium constant for the dimerization reaction, 2Br2(g) ⇄ Br4(g), is estimated using the classic spectrophotometric method with precise data and a multiwavelength fitting approach. The analysis is very sensitive to small errors in the data, requiring that parameters for the baseline absorption be included at each wavelength. To that end spectra for 18 Br2 pressures in the range 6−119 Torr are augmented by six baseline scans to facilitate estimation of three baseline constants and two molar absorptivities at each wavelength, yielding Kc = 2.5 ± 0.4 L/mol at 22 °C. This value is more than double the only previous estimate, which was based on analysis of PVT data. With adoption of a literature estimate of ΔH° = −9.5 kJ/mol, the new K implies ΔS° = −51 J mol−1 K−1 (ideal gas, 1 bar reference). The spectra for monomer absorption (peak 227 nm) and dimer absorption (205 nm) are obtained with unprecedented precision.
Co-reporter:Joel Tellinghuisen
Analytical Biochemistry 2007 Volume 360(Issue 1) pp:47-55
Publication Date(Web):1 January 2007
DOI:10.1016/j.ab.2006.10.015
An isothermal titration calorimeter of the perfusion type (MicroCal model VP-ITC) is calibrated using the heat of dilution of NaCl in water. The relative apparent molar enthalpy function (Lϕ) for NaCl(aq) varies strongly and nonlinearly with concentration in the low-concentration region (<0.2 M) that is sampled easily and extensively in a single program of injections of NaCl solution into water. This nonlinearity makes it possible to calibrate with respect to two quantities: the measured heat and the active cell volume. The heat factor is determined with typical standard error 0.003; its value in the current case is 0.987. The cell volume factor is 0.93 but is quite sensitive to possible systematic errors in the temperature and in the literature values for Lϕ. Both correction factors are closely tied to the delivered volume from the injection syringe, which required a correction factor of 0.973, attributed to an instrumental gear ratio error. Temperature calibration of the instrument showed a small offset of 0.12 K at the temperature 25 °C of the experiments, but the error increased to more than 1 K at 46 °C. The experiments were not able to distinguish clearly between mixing algorithms that assume instantaneous mixing on injection and those that assume instantaneous injection followed by mixing; however, examination of these algorithms has revealed an error in a program widely used to analyze isothermal titration calorimetry data.
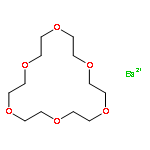
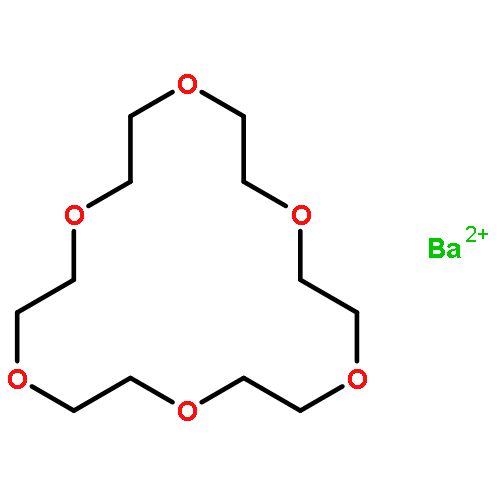
Co-reporter:Laura S Mizoue, Joel Tellinghuisen
Biophysical Chemistry 2004 Volume 110(1–2) pp:15-24
Publication Date(Web):1 July 2004
DOI:10.1016/j.bpc.2003.12.011
The 1:1 complexation reaction between Ba2+ and 18-crown-6 ether is re-examined using isothermal titration calorimetry (ITC), with the goal of clarifying previously reported discrepancies between reaction enthalpies estimated directly (calorimetric) and indirectly, from the temperature dependence of the reaction equilibrium constant K (van't Hoff). The ITC thermograms are analyzed using three different non-linear fit models based on different assumptions about the data error: constant, proportional to the heat and proportional but correlated. The statistics of the fitting indicate a preference for the proportional error model, in agreement with expectations for the conditions of the experiment, where uncertainties in the delivered titrant volume should dominate. With attention to proper procedures for propagating statistical error in the van't Hoff analysis, the differences between ΔHcal and ΔHvH are deemed statistically significant. In addition, statistically significant differences are observed for the ΔHcal estimates obtained for two different sources of Ba2+, BaCl2 and Ba(NO3)2. The effects are tentatively attributed to deficiencies in the standard procedure in ITC of subtracting a blank obtained for pure titrant from the thermogram obtained for the sample.
Co-reporter:Laura S. Mizoue, Joel Tellinghuisen
Analytical Biochemistry 2004 Volume 326(Issue 1) pp:125-127
Publication Date(Web):1 March 2004
DOI:10.1016/j.ab.2003.10.048
Co-reporter:Joel Tellinghuisen
Analytical Biochemistry 2003 Volume 321(Issue 1) pp:79-88
Publication Date(Web):1 October 2003
DOI:10.1016/S0003-2697(03)00406-8
In isothermal titration calorimetry (ITC), the two main sources of random (statistical) error are associated with the extraction of the heat q from the measured temperature changes and with the delivery of metered volumes of titrant. The former leads to uncertainty that is approximately constant and the latter to uncertainty that is proportional to q. The role of these errors in the analysis of ITC data by nonlinear least squares is examined for the case of 1:1 binding, M + X ⇄ MX. The standard errors in the key parameters—the equilibrium constant Ko and the enthalpy ΔHo—are assessed from the variance–covariance matrix computed for exactly fitting data. Monte Carlo calculations confirm that these “exact” estimates will normally suffice and show further that neglect of weights in the nonlinear fitting can result in significant loss of efficiency. The effects of the titrant volume error are strongly dependent on assumptions about the nature of this error: If it is random in the integral volume instead of the differential volume, correlated least-squares is required for proper analysis, and the parameter standard errors decrease with increasing number of titration steps rather than increase.
Co-reporter:Joel Tellinghuisen
Biochimica et Biophysica Acta (BBA) - General Subjects (May 2016) Volume 1860(Issue 5) pp:861-867
Publication Date(Web):May 2016
DOI:10.1016/j.bbagen.2015.10.011
.jpg)