Co-reporter:Nicholas R. Rightmire and Timothy P. Hanusa
Dalton Transactions 2016 vol. 45(Issue 6) pp:2352-2362
Publication Date(Web):05 Jan 2016
DOI:10.1039/C5DT03866A
Solvent-based syntheses have long been normative in all areas of chemistry, although mechanochemical methods (specifically grinding and milling) have been used to good effect for decades in organic, and to a lesser but growing extent, inorganic coordination chemistry. Organometallic synthesis, in contrast, represents a relatively underdeveloped area for mechanochemical research, and the potential benefits are considerable. From access to new classes of unsolvated complexes, to control over stoichiometries that have not been observed in solution routes, mechanochemical (or ‘M-chem’) approaches have much to offer the synthetic chemist. It has already become clear that removing the solvent from an organometallic reaction can change reaction pathways considerably, so that prediction of the outcome is not always straightforward. This Perspective reviews recent developments in the field, and describes equipment that can be used in organometallic synthesis. Synthetic chemists are encouraged to add mechanochemical methods to their repertoire in the search for new and highly reactive metal complexes and novel types of organometallic transformations.
Co-reporter:Nicholas R. Rightmire, David L. Bruns, Timothy P. Hanusa, and William W. Brennessel
Organometallics 2016 Volume 35(Issue 11) pp:1698-1706
Publication Date(Web):May 5, 2016
DOI:10.1021/acs.organomet.6b00151
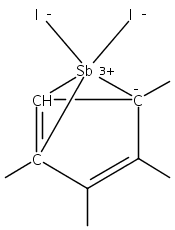
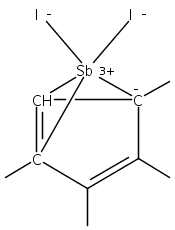
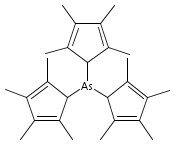
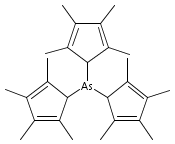
The stereochemical outcomes of reactions conducted in solution and those under mechanochemical conditions need not be the same; this is a well-established observation in organic synthesis, but few examples are known in organometallic systems. Halide metathesis is now shown to be a type of mechanochemical reaction that can produce different ratios of stereoisomers depending on whether the reagents are dissolved or ball-milled. Trihalides of As (X = I), Sb (X = Cl), and Bi (X = Cl) react with K[A′] (A′ = 1,3-(SiMe3)2C3H3) (As, Sb) or [AlA′3] (Bi) to generate the tris(allyl) complexes [EA′3]. All three complexes are found in two diastereomeric forms of C1 (R,S,S) and C3 (R,R,R) symmetry, and mechanochemical synthesis increases the C1:C3 ratio relative to that produced in hexanes solution (up to 3.3× in the case of [AsA′3]). The stereoselectivity of the metathesis in the solid state can be correlated with the asymmetric environment found in the group 15 trihalides; mechanochemical induction provides a new tool for influencing this important class of synthetic reactions.
Co-reporter:Nicholas C. Boyde, Stephen C. Chmely, Timothy P. Hanusa, Arnold L. Rheingold, and William W. Brennessel
Inorganic Chemistry 2014 Volume 53(Issue 18) pp:9703-9714
Publication Date(Web):August 29, 2014
DOI:10.1021/ic501232z
The tris(bistrimethylsilylamido) species P[N(SiMe3)2]3 (1) and As[N(SiMe3)2]3 (2) have been prepared through halide metathesis in high yield. Their single crystal X-ray structures, along with that of Sb[N(SiMe3)2]3 (3), complete the series of structurally authenticated group 15 M[N(SiMe3)2]3 complexes (the bismuth analogue (4) has been previously reported). All four complexes possess the expected pyramidal geometries, with progressively longer M–N bond distances from P to Bi but closely similar N–M–N angles (107–104°). The structures of 1–4 also display distortions that are similar to those in f-element M[N(SiMe3)2]3 and M[CH(SiMe3)2]3 complexes, in which M···(β-Si–C) interactions have been identified. Such structural features include distorted M–(N,CH)–Si and (N,CH)–Si–C angles and close M···C and M···Si contacts. DFT calculations confirm that there are no M···(β-Si–C) interactions in 1–4; the bond distortions appear to result from the particular steric crowding that arises in pyramidal M[(N,CH)(SiMe3)2]3 complexes. This is likely the source of the most of the distortions in the structures of the f-element analogues as well, even though the latter possess attractive M···Si–C interactions.
Co-reporter:Nicholas R. Rightmire, Timothy P. Hanusa, and Arnold L. Rheingold
Organometallics 2014 Volume 33(Issue 21) pp:5952-5955
Publication Date(Web):October 8, 2014
DOI:10.1021/om5009204
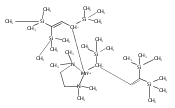
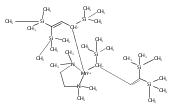
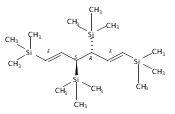
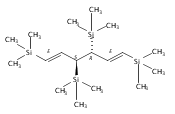
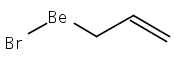
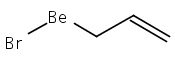
Unsolvated tris(allyl)aluminum, (C3H5)3Al, has never been isolated, although adducts with THF, OPPh3, and pyridine are known. Attempts to make a base-free derivative with the bulky 1,3-bis(trimethylsilyl)allyl anion (A′) from the reaction of aluminum halides and K[A′] in ethers or by deprotonation of HA′ with R3Al (R = Me, Et) were not successful. However, grinding AlX3 and K[A′] in the solid state, followed by extraction of the mixture with hexanes, produces A′3Al in high yield. A single-crystal X-ray structure demonstrated that the monomeric molecule contains σ-bonded allyl ligands (average Al–C 1.964(3) Å) and, like the known gallium analogue, is fluxional in solution. A DFT study of the parent (C3H5)3Al indicates that the ligands would be of mixed hapticity, i.e., (η1-C3H5)2(η3-C3H5)Al, although the trimethylsilylated ligands evidently prevent that conformation in A′3Al. A′3Al reacts immediately with benzophenone in hexanes to produce Al(OCPh2A′)3 but negligibly with the bulky alcohol HOCPh3. Grinding A′3Al with ScCl3 yields no observable product, although the same reaction with K[A′] produces the hexane-soluble A′3Sc, whose X-ray crystal structure reveals three π-bound A′ ligands.
Co-reporter:Nicholas R. Rightmire, Keith T. Quisenberry, and Timothy P. Hanusa
Organometallics 2014 Volume 33(Issue 20) pp:5678-5685
Publication Date(Web):June 10, 2014
DOI:10.1021/om5004573
Passage of CO at atmospheric pressure through solutions of A′2M (M = Fe, Co, Ni; A′ = [1,3-(SiMe3)2C3H3]−) in hexanes produces the corresponding allyl complexes A′2Fe(CO)2, A′Co(CO)3, and A′2Ni(CO), respectively. Although the iron and nickel species can be isolated as pure liquids, the cobalt complex is accompanied by the coupling product 1,3,4,6-tetrakis(trimethylsilyl)-1,5-hexadiene. A′Co(CO)3 was independently prepared from the reaction of Co2(CO)8, A′Br, and PhCH2N(C2H5)3+Cl– in aqueous base. The IR stretching frequencies of A′2Fe(CO)2 and A′Co(CO)3 are lower than those in the unsubstituted analogues, indicating that the trimethylsilated allyl ligand is a better electron donor than the parent version. Density functional theory calculations were performed on various conformations of the complexes, which reproduced the frequency-lowering effect of the trimethylsilyl groups. They also indicate that the thermodynamics of the formation of A′2Ni(CO) and the unknown (C3H5)2Ni(CO) are similar, suggesting that the thermal stability of the former is of kinetic origin. Oxidative coupling of the allyl ligands in A′2Fe and A′2Co is induced with I2; this is different from the case with A′2Ni, which has previously been shown to produce the mixed allyl halide complex [A′Ni(μ-I)2]2.
Co-reporter:Crispin Lichtenberg ; Thomas P. Spaniol ; Ilja Peckermann ; Timothy P. Hanusa ;Jun Okuda
Journal of the American Chemical Society 2012 Volume 135(Issue 2) pp:811-821
Publication Date(Web):December 14, 2012
DOI:10.1021/ja310112e
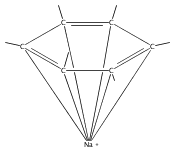
Starting from bis(allyl)magnesium [Mg(C3H5)2], a set of cationic, neutral, anionic, and dianionic allyl magnesium compounds has been isolated and characterized, including [Mg(C3H5)(THF)5][B(C6F5)4] (3), [Mg(C3H5)2(1,4-dioxane)(THF)] (2), [KMg(C3H5)3(THF)] (6), and [MMg(C3H5)4] (8: M = K2; 9: M = Ca). In solution, the allyl ligands of the compounds display fluxional behavior, even at low temperatures. Single crystal X-ray analysis reveals unusual μ2-η1:η3- and unprecedented μ3-η1:η3:η3-coordination modes in the heterobimetallic compounds 6 and [8·(THF)2]. Density functional theory calculations confirm that these metal–allyl conformations are energetically stable. The magnesium compounds have been investigated as initiators for butadiene polymerization and ethylene oligomerization. The heterobimetallic compounds display initiation properties, including higher reaction rates, that are distinctively different from those of the monometallic species. Reactivity trends depend on the formal charge of the magnesium compounds (dianionic, higher-order magnesiate > monoanionic, lower-order magnesiate) and on the nature of the counterion (K+ > Ca2+).
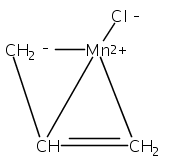
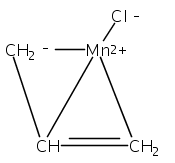
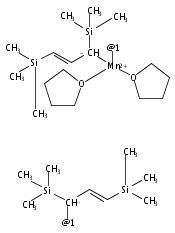
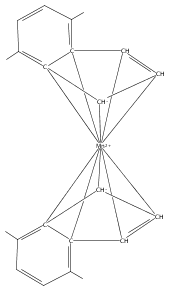
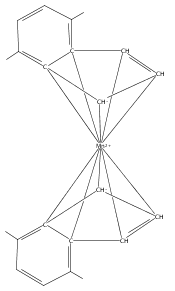
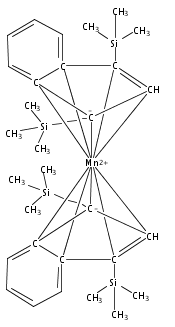
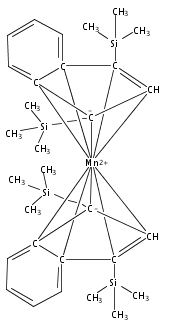
Co-reporter:Laura K. Engerer, Christin N. Carlson, Timothy P. Hanusa, William W. Brennessel, and Victor G. Young Jr.
Organometallics 2012 Volume 31(Issue 17) pp:6131-6138
Publication Date(Web):August 15, 2012
DOI:10.1021/om300478v
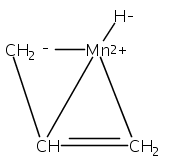
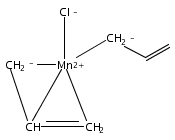
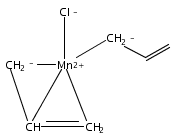
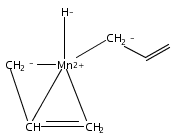
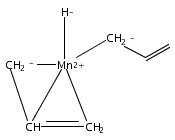
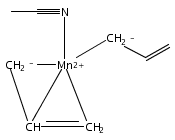
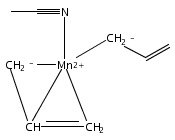
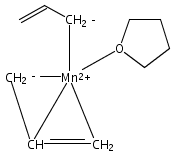
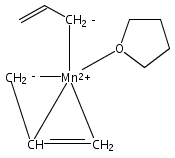
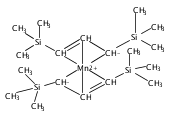
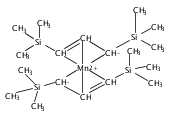
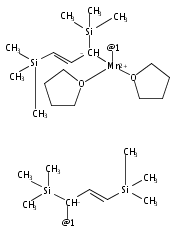
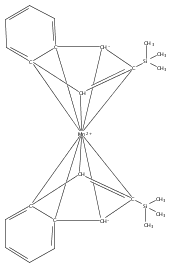
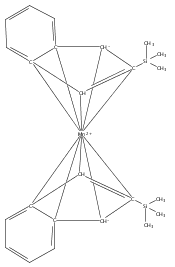
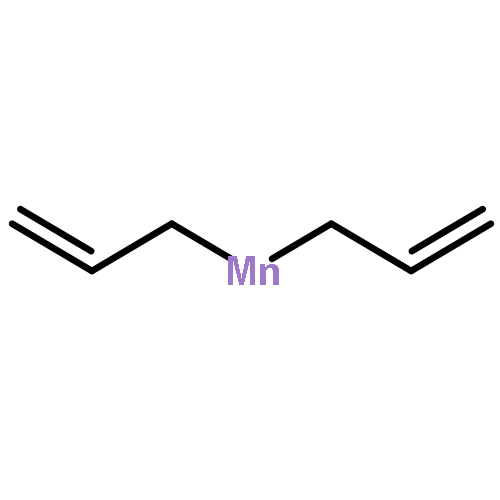
Reaction of two equivalents of K[1,3-(SiMe3)2C3H3] (= K[A′]) with MnCl2 in THF produces the allyl complex A′2Mn(thf)2; if the reaction is conducted in ether, the solvent-free heterometallic manganate species K2MnA′4 is isolated instead. With the related allyl K[1,1′,3-(SiMe3)3C3H2] (= K[A″]), reaction with MnCl2 in THF/TMEDA produces the corresponding adduct A″2Mn(tmeda). In the solid state, both A′2Mn(thf)2 and A″2Mn(tmeda) are monomeric complexes with σ-bonded allyl ligands (Mn–C = 2.174(2) and 2.189(2) Å, respectively). In contrast, K2MnA′4 is a two-dimensional coordination polymer, in which two of the allyl ligands on the Mn cation are σ-bonded (Mn–C = 2.197(6), 2.232(7) Å) and the third is π-bonded (Mn–C = 2.342(7)–2.477(7) Å). Both σ-allyls are π-coordinated to potassium cations, promoting the polymer in two directions; the π-allyl ligand is terminal. Density functional theory (DFT) calculations indicate that isolated high-spin (C3R2H3)2Mn (R = H, SiMe3) complexes would possess π-bound ligands. A mixed hapticity (π-allyl)(σ-allyl)MnE structure would result with the addition of either a neutral ligand (e.g., THF, MeCN) or one that is charged (Cl, H). Both allyl ligands in a bis(allyl)manganese complex are expected to adopt a σ-bonded mode if two THF ligands are added, as is experimentally observed in A′2Mn(thf)2. The geometry of allyl–Mn(II) bonding is readily modified; DFT results predict that [(C3H5)Mn]+ and (C3H5)MnCl should be σ-bonded, but the allyl in (C3H5)MnH is found to exhibit a symmetrical π-bonded arrangement. Some of this behavior is reminiscent of that found in bis(allyl)magnesium chemistry.
Co-reporter:Eric J. Bierschenk ; Nicholas R. Wilk ; Jr.
Inorganic Chemistry 2011 Volume 50(Issue 23) pp:12126-12132
Publication Date(Web):November 4, 2011
DOI:10.1021/ic201718a
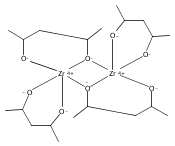
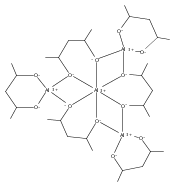
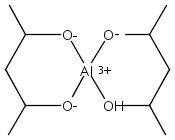
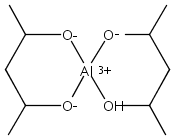
When 2,4-pentanediol (2,4-H2pd) is deprotonated, the resulting dianion (2,4-pd) serves as a type of “hybrid” ligand, i.e., an alkoxide that possesses structural features of a β-diketonate. 2,4-Pentanediol reacts with Al(O-s-Bu)3 and Zr(O-i-Pr)4 to form chelated multinuclear complexes. The aluminum-containing product is first isolated as the insoluble [Al(2,4-pd)(2,4-Hpd)]n; on sublimation, a hydrocarbon-soluble mixture of polymetallic species is generated. Mass spectral evidence suggests that both Al4(2,4-pd)6 and Al5(2,4-pd)7(2,4-Hpd) are present. The zirconium complex is isolated as an adduct, [Zr(2,4-pd)2]2·(2,4-H2pd). The pentanediolates decompose on heating to form Al2O3 and ZrO2. Unlike the mononuclear Al(acac)3 and Zr(acac)4 derivatives (acac = acetylacetonate), the formation of aggregates with the 2,4-pd ligand suggests that the latter has more coordinative flexibility. The geometries of several model aluminum complexes with oxygen donor ligands were studied with density functional theory methods. The optimized structures were used with the gauge, including atomic orbital (GIAO) method to calculate their 27Al NMR magnetic shielding values for comparison with experiment.
Co-reporter:Laura K. Engerer and Timothy P. Hanusa
The Journal of Organic Chemistry 2011 Volume 76(Issue 1) pp:42-49
Publication Date(Web):December 14, 2010
DOI:10.1021/jo101307z
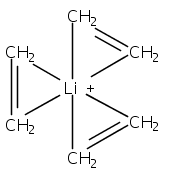
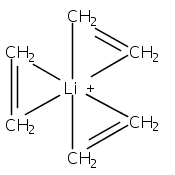
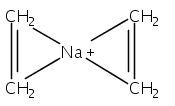
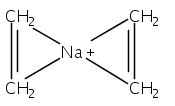
Although cation−π interactions commonly involve aromatic or heteroaromatic rings as the source of π-electrons, isolated and nonconjugated olefins are equally effective donors of π-electron density. Previous comparisons of these π-electron sources have indicated that the net energy of the binding interactions is not a simple additive function of the number of π-bonds involved. For instance, the enthalpy of binding (ΔH°) of Li+, Na+, or K+ cations to two ethylene molecules or to one benzene molecule is approximately the same, despite the 4:6 ratio of π-electrons involved. This present density functional theory study indicates that geometric factors can partially account for the proportionally greater interaction energies of olefins, but whether they are symmetrically placed around the cation or grouped on one hemisphere has little effect on the binding energy. Instead, flexible ligands that permit olefinic π-electrons to be oriented more favorably toward the metal than those in rigid aromatic rings can be correlated with greater bonding. For Li+ complexes, this appears to be an appreciable factor, although it is less significant with Na+ and K+ complexes. For all three cations, stronger polarization interactions with olefins compared to arenes contribute to the strength of cation−π interactions involving olefinic π-bonds.
Co-reporter:Phillip Jochmann, Thomas P. Spaniol, Stephen C. Chmely, Timothy P. Hanusa, and Jun Okuda
Organometallics 2011 Volume 30(Issue 19) pp:5291-5296
Publication Date(Web):September 14, 2011
DOI:10.1021/om200749f
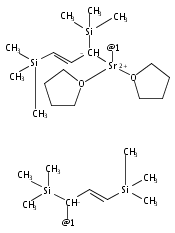
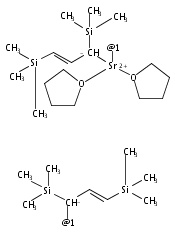
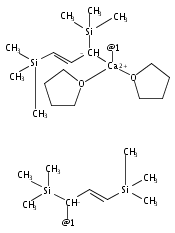
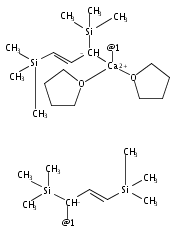
The synthesis, characterization, and decomposition pathway of the 18-crown-6 adduct of bis(allyl)calcium [Ca(η1-C3H5)(η3-C3H5)(18-crown-6)] (2) are reported. The solid-state structure of adduct 2 features one σ- and one π-bonded allyl ligand at the metal center. 2 is the first structurally characterized example of a mononuclear calcium complex bearing a purely σ-bound allyl ligand. DFT calculations indicate that [Ca(η1-C3H5)(η3-C3H5)(18-crown-6)] and [Ca(η1-C3H5)2(18-crown-6)] conformations are close in energy. In THF solution, η3-allyl ligands are observed exclusively at both ambient temperature and −95 °C. 2 undergoes rapid cleavage of the crown ether to give vinyl-terminated alcoholates of different chain length with a half-life of t1/2 = 2.5 h.
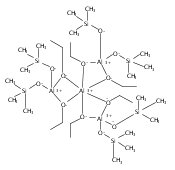
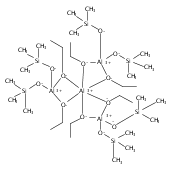
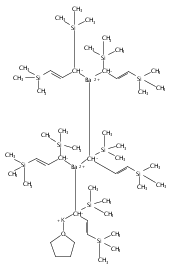
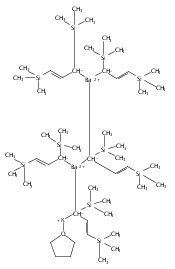
Co-reporter:Keith T. Quisenberry, Rosemary E. White, Timothy P. Hanusa and William W. Brennessel
New Journal of Chemistry 2010 vol. 34(Issue 8) pp:1579-1584
Publication Date(Web):09 Apr 2010
DOI:10.1039/C0NJ00084A
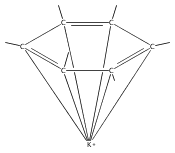
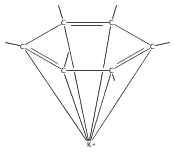
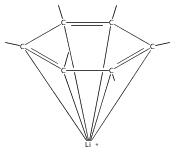
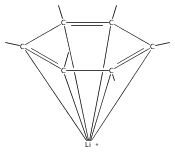
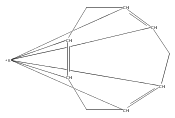
Reaction of two equivalents of K[1,3-(SiMe3)2C3H3] (= KA′) with SrI2 in thf produces the allyl complex SrA′2(thf)2. In the solid state, the monomeric complex is isostructural with its calcium counterpart; the allyl ligands are bound in a symmetrical trihapto manner to the metals, with an average Sr–C distance of 2.801(5) Å. The reaction of KA′ and BaI2 does not result in the expected bis(allyl)barium complex, but rather in a heterometallic barium/potassium species, K(thf)Ba2A′5. The allyls are in fast exchange in solution, and in the solid state, the complex forms a polymeric chiral chain. Each barium center is coordinated by one terminal and two bridging allyl ligands; the potassium center is coordinated by two bridging allyl ligands and one thf molecule. On reaction with iodine, C–C coupling occurs with CaA′2(thf)2, SrA′2(thf)2 and K(thf)Ba2A′5 to give the 1,5-hexadiene, [(SiMe3)2C3H3]2. CaA′2(thf)2, SrA′2(thf)2 and K(thf)Ba2A′5 are initiators for methyl methacrylate polymerization; the analogous magnesium complex is not active.
Co-reporter:Stephen C. Chmely
European Journal of Inorganic Chemistry 2010 Volume 2010( Issue 9) pp:1321-1337
Publication Date(Web):
DOI:10.1002/ejic.200900813
Abstract
The allyl anion [C3H5]– is often found in combination with other ligands in organometallic complexes. Compounds in which allyl groups are the only or principal type of ligand, however, are often coordinatively unsaturated and prone to decomposition. It is possible to increase the thermal and sometimes oxidative stability of such complexes by adding sterically bulky substituents (e.g., –SiMe3) to the allyl group. Main group, transition metal, and f-element compounds constructed with bulky allyl ligands display a wide range ofmonomeric, oligomeric, and polymeric structures. Some of these complexes have no counterparts with unsubstitutedallyl groups, and others are active polymerization catalysts. Research in this area has served to expand the range of structural types and bonding that can be incorporated into metal allyl chemistry.
Co-reporter:C. Heather McMillen, Cameron K. Gren, Timothy P. Hanusa, Arnold L. Rheingold
Inorganica Chimica Acta 2010 Volume 364(Issue 1) pp:61-68
Publication Date(Web):15 December 2010
DOI:10.1016/j.ica.2010.07.079
Co-reporter:StephenC. Chmely;TimothyP. Hanusa ;WilliamW. Brennessel Dr.
Angewandte Chemie 2010 Volume 122( Issue 34) pp:6006-6010
Publication Date(Web):
DOI:10.1002/ange.201001866
Co-reporter:Jeffrey A. Crisp, Ryan M. Meier, Jason S. Overby, Timothy P. Hanusa, Arnold L. Rheingold and William W. Brennessel
Organometallics 2010 Volume 29(Issue 10) pp:2322-2331
Publication Date(Web):April 23, 2010
DOI:10.1021/om100162j
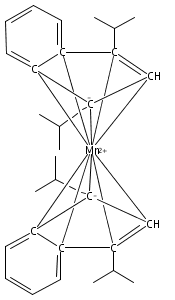
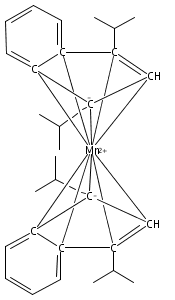
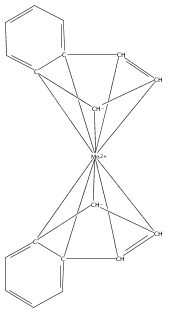
Bis(cyclopentadienyl) complexes (Cp2M) of the divalent first-row transition metals V−Ni have been known and used for over 50 years. For almost as long, an analogous series of compounds has been known with the indenyl ligand (i.e., Ind2M), with the conspicuous exception of M = Mn. Bis(indenyl) complexes of manganese(II), Ind′2MnLn, have now been synthesized by halide metathesis from MnCl2 and an appropriate potassium indenide. Depending on the indenyl ligand substituents and the presence of coordinated bases, a variety of structural motifs and bonding modes of the indenyl ligand are found in the resulting complexes. Single-crystal X-ray structures obtained for [2-(SiMe3)C9H6]2Mn, [1,3-(SiMe3)2C9H5]2Mn, and [1,3-(i-Pr)2C9H5]2Mn confirm that they possess classic η5-bound sandwich structures. In contrast, the unsubstituted parent complex recrystallizes from THF as a disolvate with two differently bonded indenyl ligands: i.e., (η3-C9H7)(η1-C9H7)Mn(thf)2. Without the coordinated solvent, density functional theory calculations suggest that the complex would have two slipped η5-bound ligands. When methyl groups are present on the benzo portion of the indenyl ligand, specifically in the 4,7-positions, the corresponding manganese complex is isolated as a cyclic octomer, {(4,7-Me2C9H5)2Mn}8, containing both bridging and terminal indenyl ligands. In the presence of 1,4-dioxane, however, attempted synthesis of (4,7-Me2C9H5)2Mn results in the isolation of the [K(dioxane)1.5][Mn(4,7-Me2C9H5)3] salt, in which each manganese atom is surrounded by a paddlewheel of three η2-bound 4,7-dimethylindenyl ligands. Cation−π bonding to the potassium and the presence of coordinated dioxane molecules generates a layered structure for the salt. Magnetic susceptibility measurements on the compounds indicate the presence of high-spin Mn(II) centers in all cases. These compounds demonstrate the high degree of conformational flexibility in the Mn(II)−indenyl bond.
Co-reporter:Stephen C. Chmely, Timothy P. Hanusa, and Arnold L. Rheingold
Organometallics 2010 Volume 29(Issue 21) pp:5551-5557
Publication Date(Web):August 9, 2010
DOI:10.1021/om100474m
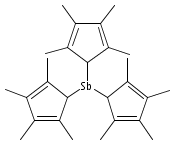
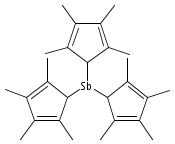
Arsenic and antimony triiodide react with 3 equiv of Na[C5Me4H] in THF to form the yellow tris(tetramethylcyclopentadienyl) derivatives M(C5Me4H)3 in 61% and 76% yields, respectively. When equimolar amounts of SbI3 and Na[C5Me4H] react in THF, the orange diiodo species Sb(C5Me4H)I2 is produced in 87% yield. The effect on the complexes of the methylated cyclopentadienyl rings varies from minimal to substantial. For example, the properties of the antimony compound Sb(C5Me4H)3 are similar to those of the unsubstituted complex Sb(C5H5)3; in contrast, As(C5Me4H)3 and Sb(C5Me4H)I2 are considerably more thermally stable than their parent versions. The complexes are fluxional on the 1H NMR time scale at room temperature. A single-crystal X-ray structure of As(C5Me4H)3 reveals three σ-bound rings with strongly localized bonding. The As−C distances are nearly identical, averaging 2.033(3) Å, and the C−As−C angles range from 96.4° to 109.3°. The antimony structure is similar, with average Sb−C bonds of 2.238(6) Å and C−Sb−C angles from 93.6° to 107.7°. The diiodo complex Sb(C5Me4H)I2 displays an η3-bound ring and forms a coordination polymer in the solid state, with ordering driven by intermolecular I···I′ and Me···Me′ van der Waals attractions. The structures of As(C5H5)3, As(C5Me4H)3, and Sb(C5H5)3 were examined with density functional theory calculations; these indicate that the energy barrier for haptotropic rearrangements is roughly the same in the arsenic compounds, 12−15 kcal mol−1, but is about half that for the antimony complex.
Co-reporter:StephenC. Chmely;TimothyP. Hanusa ;WilliamW. Brennessel Dr.
Angewandte Chemie International Edition 2010 Volume 49( Issue 34) pp:5870-5874
Publication Date(Web):
DOI:10.1002/anie.201001866
Co-reporter:StephenC. Chmely;TimothyP. Hanusa ;WilliamW. Brennessel Dr.
Angewandte Chemie 2010 Volume 122( Issue 39) pp:
Publication Date(Web):
DOI:10.1002/ange.201090126
No abstract is available for this article.
Co-reporter:Stephen C. Chmely ; Christin N. Carlson ; Timothy P. Hanusa ;Arnold L. Rheingold
Journal of the American Chemical Society 2009 Volume 131(Issue 18) pp:6344-6345
Publication Date(Web):April 21, 2009
DOI:10.1021/ja900998t
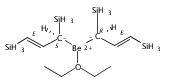
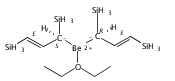
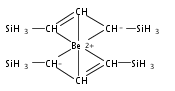
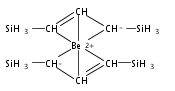
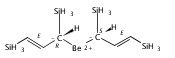
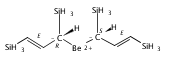
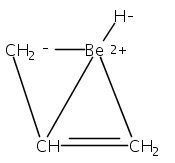
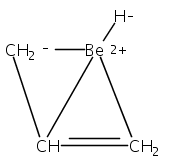
Magnesium allyl complexes are regularly isolated with classical, σ-bonded ligands, and this has been thought to be their preferred mode of bonding. Density funtional theory calculations confirm that such bonding is the most stable mode when coordinated bases are present, but in their absence, π-bonded forms are expected to be lower in energy. The isolation of the unsolvated [Mg{C3(SiMe3)2H3}2]2 complex supports this prediction, as it is a dinuclear species in which two allyl ligands bridge the metals and display cation−π interactions with them.
Co-reporter:Adam M. Johns ; Stephen C. Chmely
Inorganic Chemistry 2009 Volume 48(Issue 4) pp:1380-1384
Publication Date(Web):January 12, 2009
DOI:10.1021/ic8012766
Ca[N(SiMe3)2]2 (1) is isolated in nearly quantitative yield from the room temperature reaction of Ca(CH2Ph)2(THF) and HN(SiMe3)2 in toluene. A commonly used preparation of 1 involving the reaction of potassium bis(trimethylsilyl)amide, K[N(SiMe3)2] (2), with CaI2 can produce material that contains substantial amounts of potassium, probably in the form of a calciate such as K[Ca{N(SiMe3)2}3]. The favorable formation of K[Ca{N(SiMe3)2}3] from 1 and 2 was confirmed with density functional theory calculations. Deliberate doping of solutions of 1 with 2 initially causes only an upfield shift in the single 1H NMR resonance observed for 1; not until K/Ca ratios exceed 1:1 is the presence of the added potassium obvious by the appearance of an additional peak in the spectrum.
Co-reporter:M. Brett Meredith, C. Heather McMillen, Jonathan T. Goodman, Timothy P. Hanusa
Polyhedron 2009 28(12) pp: 2355-2358
Publication Date(Web):
DOI:10.1016/j.poly.2009.04.037
Co-reporter:Erik D. Brady, Stephen C. Chmely, Kumudini C. Jayaratne, Timothy P. Hanusa and Victor G. Young Jr.
Organometallics 2008 Volume 27(Issue 7) pp:1612-1616
Publication Date(Web):February 28, 2008
DOI:10.1021/om700635c
Treatment of the phosphonium salt [Me(t-Bu)P(C5Me4H)2]I with 2 equiv of KH produces the coordination polymer {K[Me(t-Bu)P(C5Me4)2](thf)}∞. If the phosphonium salt reacts with a single equivalent of KH followed by Ca[N(SiMe3)2]2, the calcium complex [Me(t-Bu)P(C5Me4)2]CaN(SiMe3)2 is produced, which is structurally related to Cp′2LnX organolanthanides. Density functional theory studies of the H3E (E = C, Si, P)-substituted cyclopentadienyl ring indicate that the energy required for out-of-plane bending of the substituent decreases in the order H3C > H3Si > H3P.
Co-reporter:M. Brett Meredith, Jeffrey A. Crisp, Erik D. Brady, Timothy P. Hanusa, Gordon T. Yee, Maren Pink, William W. Brennessel and Victor G. Young Jr.
Organometallics 2008 Volume 27(Issue 21) pp:5464-5473
Publication Date(Web):September 27, 2008
DOI:10.1021/om800473z
The magnetic properties of monomeric bis(indenyl)chromium(II) complexes are sensitive to the relative orientation of the ligands (staggered, eclipsed, or gauche), regardless of whether such a conformation is imposed by sterically bulky substituents or is a consequence of crystal-packing forces. The staggered monomethylated compound (2-MeC9H6)2Cr has previously been found to display a high-spin state in the solid state and solution, whereas the monomeric (1-MeC9H6)2Cr is eclipsed in the solid state and exhibits spin-crossover behavior over a wide temperature range. The present work examines the synthesis, structures, and magnetic properties of polymethylated indenyl complexes containing two, three, and five substituents on the indenyl ligand. Methyl groups on the benzo portion appear to be critical in supporting a low-spin ground state; the order of the preference to exist as a low-spin complex is approximately (2,4,5,6,7-Me5C9H2)2Cr > (2,4,7-Me3C9H4)2Cr ≈ (4,7-Me2C9H5)2Cr > (1,2,3-Me3C9H4)2Cr > (1-MeC9H6)2Cr ≥ (2-MeC9H6)2Cr. The (2,4,7-Me3C9H4)2Cr complex crystallizes with two conformations in the unit cell: one with an eclipsed geometry and the other staggered. When the temperature is raised from 173 to 293 K, the staggered form undergoes approximately 3 times the bond length increase as the eclipsed form. The difference is attributed to the symmetry-driven preference of the staggered configuration for the high-spin state.
Co-reporter:Melanie J. Harvey;Keith T. Quisenberry;Timothy P. Hanusa;Victor G. Young Jr.
European Journal of Inorganic Chemistry 2003 Volume 2003(Issue 18) pp:
Publication Date(Web):12 SEP 2003
DOI:10.1002/ejic.200300284
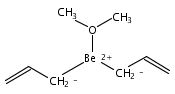
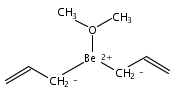
Bis[1,2,4-tris(trimethylsilyl)cyclopentadienyl] complexes [1,2,4-(SiMe3)3C5H2]2Ae [(Cp3Si)2Ae; Ae = Ca (1), Sr (2), Ba (3)] are isolated from the 2:1 reaction of K[Cp3Si] and AeI2 in diethyl ether. Under the same reaction conditions used for the heavier metallocenes, the corresponding (Cp3Si)2Be compound was not formed, although calculations suggest that the beryllocene would be sterically feasible. Compound 1 crystallizes as a bent monomer from hexanes (ring centroid−Ca−ring centroid = 166.7°) with two η5-Cp3Si ligands on the calcium atom [Ca−C(av) = 2.63(2) Å]. Metallocene 2, the first crystallographically characterized example of an unsolvated strontocene, is also isolated as a bent monomer (159.4°) from hexanes with two η5-Cp3Si ligands on the strontium atom [Sr−C(av) = 2.819(2) Å]. Compound 3 crystallizes on sublimation as a coordination dimer in which each barium atom is flanked by two η5-Cp3Si rings [Ba−C(av) = 3.01(2) Å] and one methyl group from a neighboring Cp′ ring; the intermolecular Ba···C(methyl)′ distance is 3.275(6) Å. The amount of bending observed in 1−3 and other Group-2 metallocenes varies with the metals, the steric bulk of the cyclopentadienyl ligands, and crystal packing effects, and is not easily predictable. (© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2003)
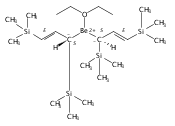
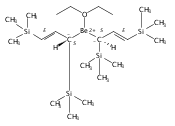
Co-reporter:Christopher J Kuehl, Cheslan K Simpson, Kevin D John, Alfred P Sattelberger, Christin N Carlson, Timothy P Hanusa
Journal of Organometallic Chemistry 2003 Volume 683(Issue 1) pp:149-154
Publication Date(Web):7 October 2003
DOI:10.1016/S0022-328X(03)00558-8
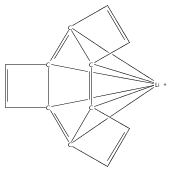
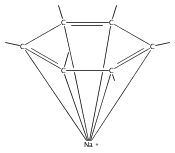
A new class of allyl-lanthanide salts of the type [K(thf)4][(C3H3(SiMe3)2)3LnI] (Ln=Ce, Pr, Nd, Gd, Tb, Dy, Er) have been prepared and isolated by reaction of three equivalents of the 1,3-bis(trimethylsilyl)allyl anion with LnI3. The neutral complex [C3H3(SiMe3)2]3Nd(thf) has been isolated from the reaction of the triflate complex Nd(O3SCF3)3 with three equivalents of the 1,3-bis(trimethylsilyl)allyl anion. These complexes have been structurally characterized using single crystal X-ray diffraction.A new class of allyl-lanthanide salts of the type [K(thf)4][(C3H3(SiMe3)2)3LnI] (Ln=Ce, Pr, Nd, Gd, Tb, Dy, Er) has been prepared and isolated by reaction of three equivalents of the 1,3-bis(trimethylsilyl)allyl anion with LnI3. The neutral complex [C3H3(SiMe3)2]3Nd(thf) has been isolated from the reaction of the triflate complex Nd(O3SCF3)3 with three equivalents of the 1,3-bis(trimethylsilyl)allyl anion.
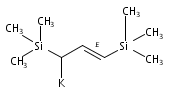
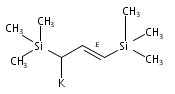
Co-reporter:Christin N Carlson, J.Dominic Smith, Timothy P Hanusa, William W Brennessel, Victor G Young Jr.
Journal of Organometallic Chemistry 2003 Volume 683(Issue 1) pp:191-199
Publication Date(Web):7 October 2003
DOI:10.1016/S0022-328X(03)00603-X
Reaction of two equivalents of K[1-(SiMe3)C3H4], K[1,3-(SiMe3)2C3H3], or K[1,1′,3-(SiMe3)3C3H2] with CrCl2 in THF at −78 °C produces the red complexes {[1-(SiMe3)C3H4]2Cr}2 (1), [1,3-(SiMe3)2C3H3]2Cr (2) and [1,1′,3-(SiMe3)3C3H2]2Cr (3), respectively. They are thermally stable compounds, remarkably so for the monomeric 2 and 3, which possess formal 12-electron counts. Single crystal X-ray structures confirm the dimeric nature of {[1-(SiMe3)C3H4]2Cr}2, which is constructed around a Cr2 core with CrCr′=1.9784(7) Å. In the monomeric complexes, the allyl ligands are bound in a trihapto manner to the metals, with CrC distances of 2.193(2)–2.257(2) Å in 2 and 2.223(5)–2.319(5) Å in 3. The allyl ligands adopt staggered conformations, with a 10.4° angle between the C3 planes in (2) and parallel ligands in (3). The trimethylsilyl groups in both complexes are in a syn, anti conformation. The monomeric complexes are high-spin, with four unpaired electrons. The steric shielding provided to the metal by the trimethylsilyl groups is probably responsible for the lack of reactivity of 2 with bulky donors such as PPh3, although it forms a monoadduct with the more sterically compact PMe3. Density functional theory calculations were performed on (C3H5)2Cr, and comparisons made with the structures of the trimethylsilylated derivatives.Use of trimethylsilylated allyl ligands affords a new family of homoleptic chromium complexes. The dimeric {[1-(SiMe3)C3H4]2Cr}2 has a structure much like the parent [(C3H5)2Cr]2, but the bis- and tris-trimethylsilyated ligands generate monomeric high-spin bis(π-allyl)chromium(II) complexes [(SiMe3)nC3H5−n]2Cr that are thermally stable at room temperature even with their extremely low 12-electron counts.
Co-reporter:Melanie J. Harvey, Timothy P. Hanusa and Maren Pink
Dalton Transactions 2001 (Issue 7) pp:1128-1130
Publication Date(Web):08 Mar 2001
DOI:10.1039/B009359I
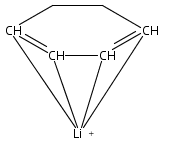
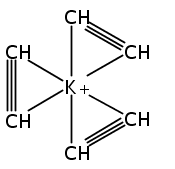
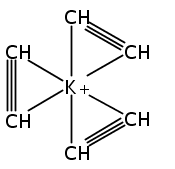
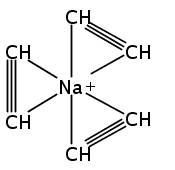
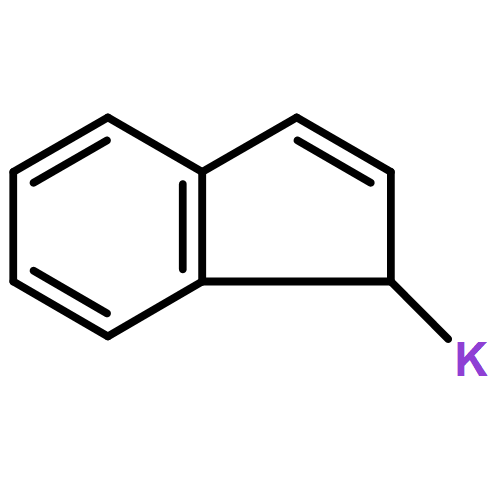
Unsolvated 1,2,4-tris(trimethylsilyl)cyclopentadienyl potassium (K[Cp3Si]) crystallizes as a base-free “super-sandwich” of alternating potassium ions and Cp′ rings. The polymeric chains in the structure are not connected as in other unsolvated KCp′ structures, but uniquely consist of independent columns. This type of structure has previously been observed in base-free lithium cyclopentadienyl complexes; however, K[Cp3Si] is the first base-free potassium cyclopentadienyl complex that crystallizes without interactions between neighboring chains.
Co-reporter:Melanie J. Harvey, Timothy P. Hanusa, Victor G. Young Jr.
Journal of Organometallic Chemistry 2001 Volume 626(1–2) pp:43-48
Publication Date(Web):30 April 2001
DOI:10.1016/S0022-328X(01)00658-1
The mono(cyclopentadienyl)calcium iodide complex [{(Cp3T)CaI(THF)x}n] (Cp3T=[C5(t-Bu)3H2]−) is generated from the 1:1 reaction of K[Cp3T] and CaI2 in THF. No redistribution into [(Cp3T)2Ca] and CaI2(THF)n is observed in THF solution. The mono(ring) compound crystallizes from THF as a monomer with a distorted piano stool geometry; the coordination environment around the calcium consists of a pentahapto [Cp3T]− ligand, an iodide, and two THF molecules. It is the first structurally authenticated monomeric mono(ring) halide complex of a heavy alkaline-earth metal. From a toluene–THF mixture, the mono(ring) complex crystallizes as an iodide-bridged dimer, with a pentahapto [Cp3T]− ligand and one terminal THF on each metal atom. The CaI and CaI′ distances are nearly equal at 3.087(2) and 3.101(2) Å.
Co-reporter:Melanie J. Harvey, Timothy P. Hanusa and Maren Pink
Chemical Communications 2000 (Issue 6) pp:489-490
Publication Date(Web):07 Mar 2000
DOI:10.1039/A908674I
The triethylborohydride anion
[HBEt3]− transfers intact from
Na[HBEt3] to a calcium center to form the monomeric
organocalcium complex
[Ca(HBEt3){1,2,4-C5(SiMe3)3
H2}(thf)2], which is stable in solution
and the solid state; the [HBEt3]− anion is
coordinated in a multidentate fashion to the metal, which likely
contributes to the failure of attempts to abstract the triethylborane
moiety from the compound.
Co-reporter:Melanie J. Harvey;Victor G. Young Jr.
Angewandte Chemie 1999 Volume 111(Issue 1‐2) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19990115)111:1/2<241::AID-ANGE241>3.0.CO;2-6
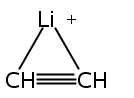
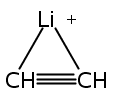
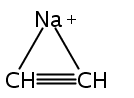
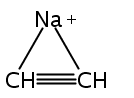
Zweiη3-Allylliganden in anti-Konfiguration weist die Titelverbindung auf (Struktur im Kristall siehe Bild). Zwar haben die Ca-C-Bindungen etwa die Länge, die man für Cyclopentadienylkomplexe erwartet, doch ähneln die H-Atom-Positionen im Bis(trimethylsilyl)allyl-Anion eher denen anderer Hauptgruppen- und Übergangsmetall-π-Allyl-Komplexe.
Co-reporter:Melanie J. Harvey;Victor G. Young, Jr.
Angewandte Chemie International Edition 1999 Volume 38(Issue 1‐2) pp:
Publication Date(Web):18 JAN 1999
DOI:10.1002/(SICI)1521-3773(19990115)38:1/2<217::AID-ANIE217>3.0.CO;2-Q
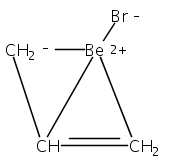
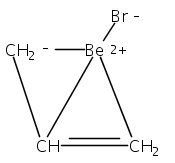
Two η3-allyl ligands in an anti configuration are present in the title compound (structure depicted). Even though the Ca−C bonds are about the length expected for cyclopentadienyl complexes, the bis(trimethylsilyl)allyl anion displays hydrogen-atom distortions similar to those found in other main group and transition metal π-allyl complexes.
Co-reporter:Nicholas C. Boyde, Nicholas R. Rightmire, Eric J. Bierschenk, Grant W. Steelman, Timothy P. Hanusa and William W. Brennessel
Dalton Transactions 2016 - vol. 45(Issue 46) pp:NaN18642-18642
Publication Date(Web):2016/11/02
DOI:10.1039/C6DT03199D
Despite their usefulness in catalytic and materials chemistry, the mixed cyclopentadienyl/alkoxide complexes of Ti, Zr, and Hf (Cp2M(OR)2) have few reliable synthetic routes available to them. We describe the use of mechanical ball milling to promote halide metathesis from Cp2MCl2, and compare these results to those obtained in hexanes and THF. Even without solvent, ring lability is extensive with titanium complexes, and alkoxide compounds with 0–3 Cp rings are isolated. The ball milling reactions are much faster than those in solution, but the distributions of products are similar to those obtained in hexanes, although different from those in THF. The range of compounds obtained from Zr and Hf starting materials is more limited, as Cp ring exchange does not occur.
Co-reporter:Nicholas R. Rightmire and Timothy P. Hanusa
Dalton Transactions 2016 - vol. 45(Issue 6) pp:NaN2362-2362
Publication Date(Web):2016/01/05
DOI:10.1039/C5DT03866A
Solvent-based syntheses have long been normative in all areas of chemistry, although mechanochemical methods (specifically grinding and milling) have been used to good effect for decades in organic, and to a lesser but growing extent, inorganic coordination chemistry. Organometallic synthesis, in contrast, represents a relatively underdeveloped area for mechanochemical research, and the potential benefits are considerable. From access to new classes of unsolvated complexes, to control over stoichiometries that have not been observed in solution routes, mechanochemical (or ‘M-chem’) approaches have much to offer the synthetic chemist. It has already become clear that removing the solvent from an organometallic reaction can change reaction pathways considerably, so that prediction of the outcome is not always straightforward. This Perspective reviews recent developments in the field, and describes equipment that can be used in organometallic synthesis. Synthetic chemists are encouraged to add mechanochemical methods to their repertoire in the search for new and highly reactive metal complexes and novel types of organometallic transformations.
.jpg)






















![Potassium, [1,1,3-tris(trimethylsilyl)-2-propenyl]-](http://img.cochemist.com/ccimg/629700/629663-15-6.png)
![Potassium, [1,1,3-tris(trimethylsilyl)-2-propenyl]-](http://img.cochemist.com/ccimg/629700/629663-15-6_b.png)


![Tetracyclo[8.2.0.02,5.06,9]dodeca-1,3,5,7,9,11-hexaene](/data/chemimg/2713800/69038-28-4.png)
![Tetracyclo[8.2.0.02,5.06,9]dodeca-1,3,5,7,9,11-hexaene](/data/chemimg/2713800/69038-28-4_b.png)















![Potassium, [1,3-bis(trimethylsilyl)-2-propenyl]-](http://img.cochemist.com/ccimg/221100/221047-77-4.png)
![Potassium, [1,3-bis(trimethylsilyl)-2-propenyl]-](http://img.cochemist.com/ccimg/221100/221047-77-4_b.png)