Co-reporter:Cui Liu, Yue Li, Bing-Yu Han, Li-Dong Gong, Li-Nan Lu, Zhong-Zhi Yang, and Dong-Xia Zhao
Journal of Chemical Theory and Computation May 9, 2017 Volume 13(Issue 5) pp:2098-2098
Publication Date(Web):April 12, 2017
DOI:10.1021/acs.jctc.6b01206
DNA damage caused by oxidized bases can lead to aging and cancer in living beings. Luckily, a repair enzyme is able to repair the oxidized bases. The key step is to accurately recognize the oxidized bases, which mainly rely on complex hydrogen bond interactions. We have calibrated the charge parameters and torsional parameters of the ABEEMσπ polarization force field (ABEEMσπ PFF) to accurately describe the intermolecular and intramolecular interactions. Taking the experiment and quantum chemical method as the benchmark, a series of properties of base pair-amino acid residue systems, DNA and DNA–protein interaction systems were calculated and compared with those of other force fields. We have done a tremendous amount of tasks in testing, calibrations, and analyses. The ABEEMσπ PFF not only explicitly gives the position and the partial charge of lone-pair electrons but also introduces a function kHB to fit special electrostatic interactions in hydrogen bond interaction regions. Therefore, it can accurately simulate the polarization effect and charge transfer of hydrogen bond interactions, especially for charged systems and sulfur-containing systems, such as the binding energy between amino acid and base pairs (24–28 kcal/mol), which is induced by charge transfer. The RMSD of ABEEMσπ PFF is 1.18 kcal/mol, whereas the RMSD of Amber OL15 is 8.21 kcal/mol. The relative positions of the amino acid residue have significantly changed, and the hydrogen bonds were broken when simulated by fixed charge force fields. In addition, owing to refitting the reasonable torsional parameters, the geometric structures optimized by ABEEMσπ PFF were well consistent with those of the M06-2X/6-311++G** method, but the simulations by fixed force fields have a large rotation of methyl and distortion of the plane of the base pair. After extensive MD simulation with four test DNAs and a DNA–protein system, we conclude that ABEEMσπ PFF shows better agreement when compared to experimental structures, which illustrates the reliability of our model and the transferability of the parameters.
Co-reporter:Shuang Xu, Dong-xia Zhao, Li-dong Gong, Cui Liu, Zhong-Zhi Yang
Chemical Physics Letters 2015 Volume 618() pp:147-152
Publication Date(Web):2 January 2015
DOI:10.1016/j.cplett.2014.10.073
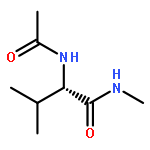
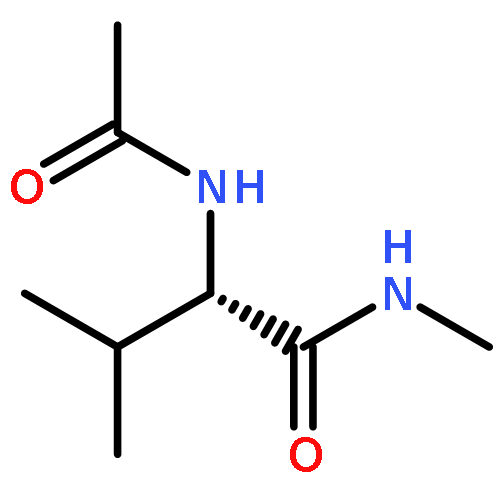
•ABEEMσπ PFF can search out all the conformations of Val-polypeptides as B3LYP does.•The total energies of Val-polypeptides from ABEEMσπ PFF are close to those of MP2.•The results of ABEEMσπ PFF are much better than those of the usual FF.•Polarization effect is important in conformational analysis.ABEEMσπ polarizable force field (PFF) with fluctuating charges works well for searching stable conformations of Val-dipeptide and Val-tripeptide, showing importance of the polarization. The results demonstrate that ABEEMσπ PFF is able to search out all the 6 types of stable conformations of Val-dipeptide and 34 types of stable conformations of Val-tripeptide that are just all exactly found by the calculations of ab initio B3LYP/6-311++G(d,p) and MP2/6-311++G(d,p) methods. In contrast, the force fields with the fixed-charges (FC), such as ABEEMσπ-FC, AMBER-FC 99sb and OPLS/AA-FC force fields can only search out less or much less numbers of stable conformations for them.
Co-reporter:Cui Liu, Yang Wang, Dongxia Zhao, Lidong Gong, Zhongzhi Yang
Journal of Molecular Graphics and Modelling 2014 Volume 47() pp:62-76
Publication Date(Web):February 2014
DOI:10.1016/j.jmgm.2013.10.008
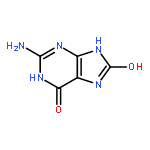
•We have developed the ABEEMσπ polarizable force field.•The performance of this method is better than those of the common force fields.•The mutations induced by oxidized guanines have been investigated by this method.•The accuracy of this method is close to that of the MP2 method.The integrity of the genetic information is constantly threatened by oxidizing agents. Oxidized guanines have all been linked to different types of cancers. Theoretical approaches supplement the assorted experimental techniques, and bring new sight and opportunities to investigate the underlying microscopic mechanics. Unfortunately, there is no specific force field to DNA system including oxidized guanines. Taking high level ab initio calculations as benchmark, we developed the ABEEMσπ fluctuating charge force field, which uses multiple fluctuating charges per atom. And it was applied to study the energies, structures and mutations of base pairs containing oxidized guanines. The geometries were obtained in reference to other studies or using B3LYP/6-31+G* level optimization, which is more rational and timesaving among 24 quantum mechanical methods selected and tested by this work. The energies were determined at MP2/aug-cc-pVDZ level with BSSE corrections. Results show that the constructed potential function can accurately simulate the change of H-bond and the buckled angle formed by two base planes induced by oxidized guanine, and it provides reliable information of hydrogen bonding, stacking interaction and the mutation processes. The performance of ABEEMσπ polarizable force field in predicting the bond lengths, bond angles, dipole moments etc. is generally better than those of the common force fields. And the accuracy of ABEEMσπ PFF is close to that of the MP2 method. This shows that ABEEMσπ model is a reliable choice for further research of dynamics behavior of DNA fragment including oxidized guanine.The mutations induced by oxidized guanine (such as 8-oxo-G) using ABEEMσπ fluctuating charge force field, which partitions a molecule into atoms, σ bonds, lone pair electrons, and π bonds. This figure only shows these atoms, σ bonds, and lone pair electrons of 8-oxo-G. Four common oxidized guanines, which are shown in this figure, have been investigated.