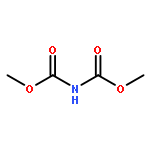
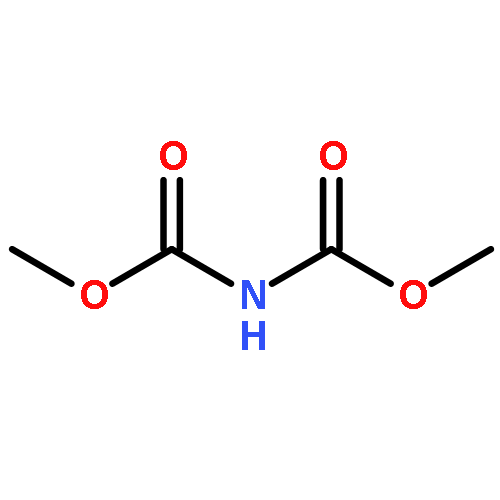
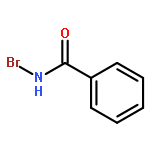
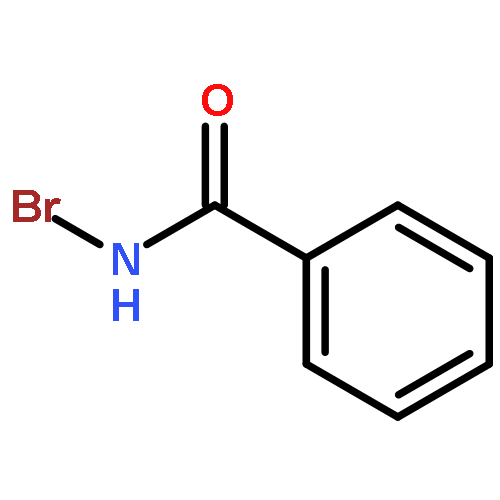
Co-reporter:Corey L. Jones, Evert Njomen, Benita Sjögren, Thomas S. Dexheimer, and Jetze J. Tepe
ACS Chemical Biology September 15, 2017 Volume 12(Issue 9) pp:2240-2240
Publication Date(Web):July 18, 2017
DOI:10.1021/acschembio.7b00489
The 20S proteasome is the main protease for the degradation of oxidatively damaged and intrinsically disordered proteins. When accumulation of disordered or oxidatively damaged proteins exceeds proper clearance in neurons, imbalanced pathway signaling or aggregation occurs, which have been implicated in the pathogenesis of several neurological disorders. Screening of the NIH Clinical Collection and Prestwick libraries identified the neuroleptic agent chlorpromazine as a lead agent capable of enhancing 20S proteasome activity. Chemical manipulation of chlorpromazine abrogated its D2R receptor binding affinity while retaining its ability to enhance 20S mediated proteolysis at low micromolar concentrations. The resulting small molecule enhancers of 20S proteasome activity induced the degradation of intrinsically disordered proteins, α-synuclein, and tau but not structured proteins. These small molecule 20S agonists can serve as leads to explore the therapeutic potential of 20S activation or as new tools to provide insight into the yet unclear mechanics of 20S-gate regulation.
Co-reporter:Tanner J. McDaniel, Theresa A. Lansdell, Amila A. Dissanayake, Lauren M. Azevedo, Jacob Claes, Aaron L. Odom, Jetze J. Tepe
Bioorganic & Medicinal Chemistry 2016 Volume 24(Issue 11) pp:2441-2450
Publication Date(Web):1 June 2016
DOI:10.1016/j.bmc.2016.04.005
Screening of a library of diverse heterocyclic scaffolds identified substituted quinolines as inhibitors of the human proteasome. The heterocyclic library was prepared via a novel titanium-catalyzed multicomponent coupling reaction, which rendered a diverse set of isoxazoles, pyrimidines, pyrroles, pyrazoles and quinolines. SAR of the parent lead compound indicated that hydrophobic residues on the benzo-moiety significantly improved potency. Lead compound 25 inhibits the chymotryptic-like proteolytic activity of the proteasome (IC50 5.4 μM), representing a new class of nonpeptidic, noncovalent proteasome inhibitors.
Co-reporter:Rahman Shah Zaib Saleem, Jetze J. Tepe
Tetrahedron Letters 2015 Volume 56(Issue 23) pp:3011-3013
Publication Date(Web):3 June 2015
DOI:10.1016/j.tetlet.2014.10.022
The synthesis of hymenialdisine has offered challenges that included the formation of the regio-isomers upon monobromination, atom-scrambling in the cyclization step, and low to moderate yields in the last steps. We herein present a short and concise (6 overall steps), high yielding route (44% overall yields) for this biologically interesting natural product that addresses these issues and avoids the use of any protecting groups.
Co-reporter:Dr. Philipp Beck;Theresa A. Lansdell;Nicole M. Hewlett;Dr. Jetze J. Tepe;Dr. Michael Groll
Angewandte Chemie International Edition 2015 Volume 54( Issue 9) pp:2830-2833
Publication Date(Web):
DOI:10.1002/anie.201410168
Abstract
The proteasome represents an invaluable target for the treatment of cancer and autoimmune disorders. The application of proteasome inhibitors, however, remains limited to blood cancers because their reactive headgroups and peptidic scaffolds convey unfavorable pharmacodynamic properties. Thus, the discovery of more drug-like lead structures is indispensable. In this study, we present the first structure of the proteasome in complex with an indolo-phakellin that exhibits a unique noncovalent binding mode unparalleled by all hitherto reported inhibitors. The natural product inspired pentacyclic alkaloid binds solely and specificially into the spacious S3 subpocket of the proteasomal β5 substrate binding channel, gaining major stabilization through halogen bonding with the protein backbone. The presented compound provides an ideal scaffold for the structure-based design of subunit-specific nonpeptidic proteasome-blockers.
Co-reporter:Dr. Philipp Beck;Theresa A. Lansdell;Nicole M. Hewlett;Dr. Jetze J. Tepe;Dr. Michael Groll
Angewandte Chemie 2015 Volume 127( Issue 9) pp:2872-2875
Publication Date(Web):
DOI:10.1002/ange.201410168
Abstract
Das Proteasom ist ein etabliertes Angriffsziel zur Behandlung von Krebs und Autoimmunerkrankungen. Die Anwendung von Proteasominhibitoren ist jedoch auf Blutkrebs beschränkt, da ihre peptidischen Grundgerüste und reaktiven Kopfgruppen ungünstige pharmakodynamische Eigenschaften mit sich bringen. Die Entdeckung neuer Leitstrukturen ist somit von grundlegender Bedeuteutung. Hier stellen wir die erste Kristallstruktur des Proteasoms im Komplex mit einem Indolo-Phakellin vor, das sich strukturell von allen bisheringen Inhibitoren signifikant unterscheidet und einen einzigartigen, nichtkovalenten Bindemodus aufweist. Das pentacyclische Alkaloid bindet ausschließlich und spezifisch in die S3-sub-Tasche des proteasomalen β5-Substratbindekanals und wird durch eine Halogenbrücke mit dem Proteinrückgrat stabilisiert. Der Ligand bietet ein ideales Grundgerüst für die strukturbasierte Entwicklung von Untereinheit-spezifischen nichtpeptidischen Proteasomhemmstoffen.
Co-reporter:Michael R. Kuszpit, Matthew B. Giletto, Corey L. Jones, Travis K. Bethel, and Jetze J. Tepe
The Journal of Organic Chemistry 2015 Volume 80(Issue 3) pp:1440-1445
Publication Date(Web):January 9, 2015
DOI:10.1021/jo502369d
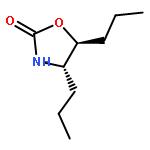
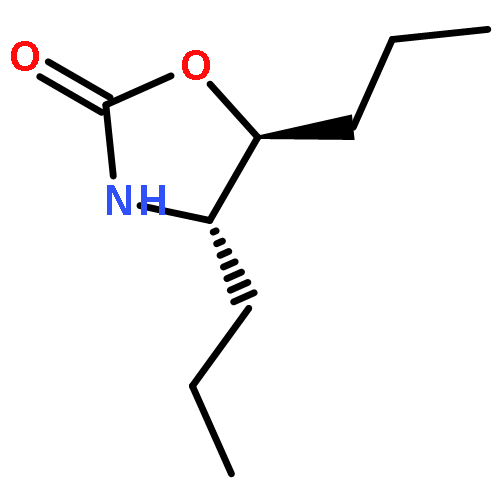
The hydroxyamination reagent Br-N-(CO2Me)2 underwent Markovnikov addition to various olefins in the presence of catalytic BF3·OEt2 and provides efficient access to aminoalcohols. The reaction provided the trans-1-bromo, 2-N-bis-carbamate adduct stereoisomer in all cases. The resulting adduct underwent cyclization to give an oxazolidinone, which could be readily hydrolyzed to an oxazolidin-2-one or an amino alcohol.
Co-reporter:Lauren M. Azevedo ; Theresa A. Lansdell ; Jacob R. Ludwig ; Robert A. Mosey ; Daljinder K. Woloch ; Dillon P. Cogan ; Gregory P. Patten ; Michael R. Kuszpit ; Jason S. Fisk ;Jetze J. Tepe
Journal of Medicinal Chemistry 2013 Volume 56(Issue 14) pp:5974-5978
Publication Date(Web):June 22, 2013
DOI:10.1021/jm400235r
The proteasome has emerged as the primary target for the treatment of multiple myeloma. Unfortunately, nearly all patients develop resistance to competitive-type proteasome inhibitors such as bortezomib. Herein, we describe the optimization of noncompetitive proteasome inhibitors to yield derivatives that exhibit nanomolar potency (compound 49, IC50 130 nM) toward proteasome inhibition and overcome bortezomib resistance. These studies illustrate the feasibility of the development of noncompetitive proteasome inhibitors as additives and/or alternatives to competitive proteasome inhibitors.
Co-reporter:Theresa A. Lansdell, Michelle A. Hurchla, Jingyu Xiang, Stacy Hovde, Katherine N. Weilbaecher, R. William Henry, and Jetze J. Tepe
ACS Chemical Biology 2013 Volume 8(Issue 3) pp:578
Publication Date(Web):November 30, 2012
DOI:10.1021/cb300568r
Multiple myeloma (MM) is a malignant disorder of differentiated B-cells for which standard care involves the inhibition of the proteasome. All clinically used proteasome inhibitors, including the chemotherapeutic drug bortezomib, target the catalytic active sites of the proteasome and inhibit protein proteolysis by competing with substrate binding. However, nearly all (∼97%) patients become intolerant or resistant to treatments within a few years, after which the average survival time is less than 1 year. We describe herein the inhibition of the human proteasome via a noncompetitive mechanism by the imidazoline scaffold, TCH-13. Consistent with a mechanism distinct from that of competitive inhibitors, TCH-013 acts additively with and overcomes resistance to bortezomib. Importantly, TCH-013 induces apoptosis in a panel of myeloma and leukemia cell lines, but in contrast, normal lymphocytes, primary bone marrow stromal cells (hBMSC), and macrophages are resistant to its cytotoxic effects. TCH-013 was equally effective in blocking MM cell growth in co-cultures of MM cells with hBMSC isolated from CD138 negative bone marrow (BM) samples of MM patients. The cellular activity translated well in vivo where TCH-013 delayed tumor growth in an MM xenograft model to a similar extent as bortezomib.
Co-reporter:Thu N. T. Nguyen, Rahman S. Z. Saleem, Micah J. Luderer, Stacy Hovde, R. William Henry, and Jetze J. Tepe
ACS Chemical Biology 2012 Volume 7(Issue 1) pp:172
Publication Date(Web):October 17, 2011
DOI:10.1021/cb200320c
DNA damage induced by ionizing radiation activates the ataxia telangiectasia mutated pathway, resulting in apoptosis or DNA repair. The serine/threonine checkpoint kinase (Chk2) is an important transducer of this DNA damage signaling pathway and mediates the ultimate fate of the cell. Chk2 is an advantageous target for the development of adjuvant drugs for cancer therapy, because inhibition of Chk2 allows normal cells to enter cell cycle arrest and DNA repair, whereas many tumors bypass cell cycle checkpoints. Chk2 inhibitors may thus have a radioprotective effect on normal cells. We report herein a class of natural product derived Chk2 inhibitors, exemplified by indoloazepine 1, that elicit a strong ATM-dependent Chk2-mediated radioprotection effect in normal cells and p53 wt cells, but not p53 mutant cells (>50% of all cancers). This study represents the first example of a radioprotective effect in human cells other than T-cells and implicates a functional ATM pathway as a requirement for IR-induced radioprotection by this class of Chk2 inhibitors. Several of the hymenialdisine-derived analogues inhibit Chk2 at nanomolar concentrations, inhibit autophosphorylation of Chk2 at Ser516 in cells, and increase the survival of normal cells following ionizing radiation.
Co-reporter:Theresa A. Lansdell, Nicole M. Hewlett, Amanda P. Skoumbourdis, Matthew D. Fodor, Ian B. Seiple, Shun Su, Phil. S. Baran, Ken S. Feldman, and Jetze J. Tepe
Journal of Natural Products 2012 Volume 75(Issue 5) pp:980-985
Publication Date(Web):May 16, 2012
DOI:10.1021/np300231f
We report herein that the oroidin-derived alkaloids palau’amine (1), dibromophakellin (2), and dibromophakellstatin (3) inhibit the proteolytic activity of the human 20S proteasome as well as the i20S immunoproteasome catalytic core. Palau’amine is found to prevent the degradation of ubiquitinylated proteins, including IκBα, in cell culture, which may be indicative of the potential mechanism by which these agents exhibit their exciting cytotoxic and immunosuppressive properties.
Co-reporter:Rahman Shah Zaib Saleem, Theresa A. Lansdell, Jetze J. Tepe
Bioorganic & Medicinal Chemistry 2012 Volume 20(Issue 4) pp:1475-1481
Publication Date(Web):15 February 2012
DOI:10.1016/j.bmc.2011.12.054
Natural products have been the subject of interest for drug discovery and as tools for understanding the underlying cellular pathways in various diseases. We present herein the synthesis and evaluation of new analogs of the marine sponge metabolite, debromohymenialdisine, as checkpoint kinase 2 (Chk2) inhibitors. We illustrate herein that slight modifications to the natural product scaffold can induce strong selectivity for Chk2 over Chk1. These Chk2 inhibitors can serve as drug templates or molecular tools to gain insight in Chk2 mediated radioprotection.
Co-reporter:Theresa A. Lansdell, Sandra O’Reilly, Tracey Woolliscroft, Lauren M. Azevedo, Daljinder K. Kahlon, Stacy Hovde, J. Justin McCormick, R. William Henry, Joseph A. Cornicelli, Jetze J. Tepe
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 14) pp:4816-4819
Publication Date(Web):15 July 2012
DOI:10.1016/j.bmcl.2012.05.056
The pathogenesis of rheumatoid arthritis is mainly driven by NF-κB-mediated production of cytokines, such as TNF-α. We report herein that the orally available imidazoline-based NF-κB inhibitor, TCH-013, was found to significantly reduce TNF-α signaling and attenuate collagen antibody induced arthritis in BALB/c mice.
Co-reporter:Theresa A. Lansdell, Sandra O’Reilly, Tracey Woolliscroft, Lauren M. Azevedo, Daljinder K. Kahlon, Stacy Hovde, J. Justin McCormick, R. William Henry, Joseph A. Cornicelli, Jetze J. Tepe
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 22) pp:6821-6824
Publication Date(Web):15 November 2012
DOI:10.1016/S0960-894X(12)01341-8
The pathogenesis of rheumatoid arthritis is mainly driven by NF-κB-mediated production of cytokines, such as TNF-α. We report herein that the orally available imidazoline-based NF-κB inhibitor, TCH-013, was found to significantly reduce TNF-α signaling and attenuate collagen antibody induced arthritis in BALB/c mice.
Co-reporter:Nicole M. Hewlett and Jetze J. Tepe
Organic Letters 2011 Volume 13(Issue 17) pp:4550-4553
Publication Date(Web):July 28, 2011
DOI:10.1021/ol201741r
(±)-Dibromophakellin has been synthesized in two steps from a known alkene intermediate. The key step in the synthesis is the NBS olefin activation to facilitate the addition of a guanidine molecule across the double bond.
Co-reporter:Ke Qu, Jason S. Fisk, Jetze J. Tepe
Tetrahedron Letters 2011 Volume 52(Issue 38) pp:4840-4842
Publication Date(Web):21 September 2011
DOI:10.1016/j.tetlet.2011.07.022
An efficient synthetic methodology of converting trans-4,5-diaryl-2-imidazolines to the corresponding cis-4,5-diaryl-2-imidazolines has been developed. This methodology features mild reaction conditions and a simple one-pot two-step procedure.
Co-reporter:Michael R. Kuszpit, William D. Wulff, and Jetze J. Tepe
The Journal of Organic Chemistry 2011 Volume 76(Issue 8) pp:2913-2919
Publication Date(Web):March 14, 2011
DOI:10.1021/jo200101q
We herein report a simple and convenient one-pot synthesis of highly substituted 2-imidazolines in a regiocontrolled and stereospecific matter through the ring expansion reaction of an imidoyl chloride with an aziridine, analogous to the Heine reaction.
Co-reporter:Rahman Shah Zaib Saleem and Jetze J. Tepe
The Journal of Organic Chemistry 2010 Volume 75(Issue 12) pp:4330-4332
Publication Date(Web):May 19, 2010
DOI:10.1021/jo100716m
We describe herein a convenient method for the synthesis of 1,2,4-triazolines using oxazolones and azodicarboxylates. Subsequent treatment of these 1,2,4-triazolines with NaOH provides efficient access to the corresponding triazoles.
Co-reporter:Daljinder K. Kahlon ; Theresa A. Lansdell ; Jason S. Fisk ; Christopher D. Hupp ; Timothy L. Friebe ; Stacy Hovde ; A. Daniel Jones ; Richard D. Dyer ; R. William Henry ;Jetze J. Tepe
Journal of Medicinal Chemistry 2009 Volume 52(Issue 5) pp:1302-1309
Publication Date(Web):February 16, 2009
DOI:10.1021/jm8013162
The mammalian nuclear transcription factor NF-κB is responsible for the transcription of multiple cytokines, including the pro-inflammatory cytokines tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6). Elevated levels of pro-inflammatory cytokines play an important role in the pathogenesis of inflammatory disorders such as rheumatoid arthritis (RA). Inhibition of the pro-inflammatory transcription factor NF-κB has therefore been identified as a possible therapeutic treatment for RA. We describe herein the synthesis and biological activity of a series of imidazoline-based scaffolds as potent inhibitors of NF-κB mediated gene transcription in cell culture as well as inhibitors of TNF-α and IL-6 production in interleukin 1 beta (IL-1β) stimulated human blood.
Co-reporter:Samantha M. Frawley Cass, Gavin E. Reid and Jetze J. Tepe
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 16) pp:3291-3299
Publication Date(Web):25 Jun 2009
DOI:10.1039/B906577F
We describe herein the synthesis and application of a range of diazo-functionalized materials as platforms for the solid phase enrichment of phosphorylated peptides from protein digests. Diazo-functionalized solid phase resins are employed to bind covalently and specifically with the phosphate moiety of a peptide substrate, allowing for non-phosphorylated peptides to be efficiently removed by filtration. The technique described herein is compatible with enrichment of phospho-serine, threonine and tyrosine residues and was used to successfully enrich phosphorylated substrates from β-casein and ovalbumin digests in low-picomolar quantities.
Co-reporter:Daljinder K. Kahlon, Theresa A. Lansdell, Jason S. Fisk, Jetze J. Tepe
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 8) pp:3093-3103
Publication Date(Web):15 April 2009
DOI:10.1016/j.bmc.2009.03.002
We herein describe the synthesis and anti-inflammatory properties of a small library of imidazoline-based NF-κB inhibitors. The structure–activity relationship of various substituents on an imidazoline core structure was evaluated for the ability to inhibit NF-κB mediated IL-6 production. Optimization of the scaffolds was pursued by correlating luciferase-based NF-κB reporter assays with inhibition of IL-6 production in IL-1β stimulated human blood. Several derivatives were found to inhibit NF-κB mediated IL-6 production in the nanomolar range in IL-1β stimulated human blood.
Co-reporter:Robert A. Mosey, Jetze J. Tepe
Tetrahedron Letters 2009 50(3) pp: 295-297
Publication Date(Web):
DOI:10.1016/j.tetlet.2008.10.154
Co-reporter:Christopher D. Hupp and Jetze J. Tepe
The Journal of Organic Chemistry 2009 Volume 74(Issue 9) pp:3406-3413
Publication Date(Web):March 30, 2009
DOI:10.1021/jo900264p
Reactions that create a quaternary stereocenter offer a wealth of synthetic utility and are often needed to provide access to the structural diversity of stereocenters found in natural products and biologically important molecules. We have developed a new 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI)-mediated oxazole rearrangement that affords quaternary 5,5-(aryl, allyl)-substituted hydantoins found in many biologically significant compounds. Furthermore, these quaternary hydantoins can be chemically manipulated to yield the corresponding quaternary imidazolones, which is a unique scaffold found in a compound from the tunicate Dendrodoa grossularia. Herein, we report the scope of this novel rearrangement and the proposed mechanism and showcase its utility through the total synthesis of a marine alkaloid from D. grossularia and two analogues.
Co-reporter:Robert A. Mosey, Jason S. Fisk, Jetze J. Tepe
Tetrahedron: Asymmetry 2008 Volume 19(Issue 24) pp:2755-2762
Publication Date(Web):12 December 2008
DOI:10.1016/j.tetasy.2008.11.033
Quaternary α-amino acids play vital roles in protein and synthetic chemistries. Stereoselective access to such molecules has been an intensive focus of research in recent years, and new methods are continuously being explored. The present mini-review gives an overview of stereoselective syntheses of quaternary α-amino acids produced from racemic oxazolones.Quaternary α-amino acids play vital roles in protein and synthetic chemistries. Stereoselective access to such molecules has been an intensive focus of research in recent years and new methods are continuously being explored. The present mini-review gives an overview of stereoselective syntheses of quaternary α-amino acids produced from racemic oxazolones.
Co-reporter:Samantha M. Frawley Cass, Gavin E. Reid and Jetze J. Tepe
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 16) pp:NaN3299-3299
Publication Date(Web):2009/06/25
DOI:10.1039/B906577F
We describe herein the synthesis and application of a range of diazo-functionalized materials as platforms for the solid phase enrichment of phosphorylated peptides from protein digests. Diazo-functionalized solid phase resins are employed to bind covalently and specifically with the phosphate moiety of a peptide substrate, allowing for non-phosphorylated peptides to be efficiently removed by filtration. The technique described herein is compatible with enrichment of phospho-serine, threonine and tyrosine residues and was used to successfully enrich phosphorylated substrates from β-casein and ovalbumin digests in low-picomolar quantities.

![Benzamide, N-bromo-N-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/902800/902775-65-9.png)
![Benzamide, N-bromo-N-[(4-methylphenyl)sulfonyl]-](http://img.cochemist.com/ccimg/902800/902775-65-9_b.png)
















![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6.png)
![L-Leucinamide,N-[(phenylmethoxy)carbonyl]-L-leucyl-N-[(1S)-1-formyl-3-methylbutyl]-](http://img.cochemist.com/ccimg/133500/133407-82-6_b.png)
![Pyrrolo[2,3-c]azepine-4,8(1H,5H)-dione,2-bromo-6,7-dihydro-](http://img.cochemist.com/ccimg/96600/96562-96-8.png)
![Pyrrolo[2,3-c]azepine-4,8(1H,5H)-dione,2-bromo-6,7-dihydro-](http://img.cochemist.com/ccimg/96600/96562-96-8_b.png)
![Pyrrolo[2,3-c]azepin-8(1H)-one,4-(2-amino-1,5-dihydro-5-oxo-4H-imidazol-4-ylidene)-2-bromo-4,5,6,7-tetrahydro-,(4Z)-](http://img.cochemist.com/ccimg/82100/82005-12-7.png)
![Pyrrolo[2,3-c]azepin-8(1H)-one,4-(2-amino-1,5-dihydro-5-oxo-4H-imidazol-4-ylidene)-2-bromo-4,5,6,7-tetrahydro-,(4Z)-](http://img.cochemist.com/ccimg/82100/82005-12-7_b.png)