Co-reporter:Tong Chen, Wenjiao Jiang, Huixin Zhang, Xintong You, Manling Liu, Linna Wang, Pengjun Xiang, Lijie Xu, Dongseng Zheng, Xuejiao Zhang, Hui Ji, Kun Hao and Tianhua Yan
RSC Advances 2016 vol. 6(Issue 24) pp:20081-20088
Publication Date(Web):12 Feb 2016
DOI:10.1039/C5RA21158A
The purpose of the present study was to evaluate gastroprotective effect of trillin on mucosal lesions induced by ethanol. Mice were orally administered with trillin (25 mg kg−1, 50 mg kg−1, 100 mg kg−1) or OME (20 mg kg−1). 1 hour later, the mice were intragastrically given ethanol (0.2 ml kg−1) and were sacrificed at 4 h after ethanol administration. Compared with the ethanol group, trillin treatment significantly showed increased levels of superoxide dismutase (SOD) in serum, decreased malonaldehyde (MDA) content in serum and decreased activity of myeloperoxidase (MPO) in stomach tissues, which suggested that trillin could prevent oxidative damage. Meanwhile, the serum levels of interleukin-6 (IL-6), interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) declined after pretreatment with trillin. In HE staining sections, ameliorative pathological changes of gastric lesions were markedly observed in the trillin group compared with those in the ethanol group. In the mechanistic study, the data showed that pretreatment with the trillin also effectively down-regulated protein expressions of p-NF-κBp65, p-IκBα and COX-2 in stomachs compared with those in the model group. Taken together, it could be concluded that trillin represented a potential therapeutic option to reduce the risk of gastric ulceration.
Co-reporter:Yong Ai; Yang Hu; Fenghua Kang; Yisheng Lai; Yanju Jia; Zhangjian Huang; Sixun Peng; Hui Ji; Jide Tian;Yihua Zhang
Journal of Medicinal Chemistry 2015 Volume 58(Issue 11) pp:4506-4520
Publication Date(Web):May 20, 2015
DOI:10.1021/jm5020023
γ-Lactam is an important structural motif in a large number of biologically active natural products and synthetic small pharmaceutical molecules. However, there is currently no effective approach to construct γ-lactam ring directly from natural rigid polycyclic amides. Herein, we report a facile methodology for synthesis of a new group of olean-28,13β-lactams (10a–j) from their corresponding amides, promoted by an easily available reagent 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ), through an intramolecular dehydrogenative C–N coupling reaction via a radical ion mechanism. Biological evaluation indicated that the most active lactam 10h displayed potent antiproliferative activity against human cancer cells but 13.84- to 16.92-fold less inhibitory activity on noncancer cells in vitro. In addition, 10h significantly inhibited the growth of implanted prostate cancer in vivo. Furthermore, 10h induced cell cycle arrest and apoptosis and down-regulated the AKT/mTOR signaling in DU-145 cells. Finally, 10h was more stable in rat plasma and human liver microsomes than CDDO-Me and had little hERG channel inhibitory activity. Collectively, 10h may be a potential antiprostate cancer agent for further investigation.
Co-reporter:Xiao Sheng, Kai Hua, Chunyu Yang, Xiaoli Wang, Hui Ji, Jinyi Xu, Zhangjian Huang, Yihua Zhang
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 17) pp:3535-3540
Publication Date(Web):1 September 2015
DOI:10.1016/j.bmcl.2015.06.090
Fourteen hybrids (10a–g, 11a–g) of 3-n-butylphthalide (NBP) and edaravone (Eda) analogues have been designed and synthesized as potential anti-ischemic stroke agents. In vitro biological studies showed that compounds 10d and 10g exhibited more potent anti-platelet aggregation than ticlopidine (Ticlid), aspirin (ASP) and NBP. Compound 10g more significantly prevented H2O2-mediated neuronal cell (PC12) death than NBP, Eda or NBP together with Eda. Meanwhile, 10g also possessed potent radical scavenging effects on hydroxyl radical (OH) and superoxide anion radical (O2−). Our findings may provide new insights into the development of these hybrids, like 10g, for the intervention of ischemic stroke.
Co-reporter:Pengjun Xiang;Tong Chen;Yi Mou;Hui Wu;Peng Xie;Guo Lu
Inflammation Research 2015 Volume 64( Issue 10) pp:799-808
Publication Date(Web):2015 October
DOI:10.1007/s00011-015-0863-4
The purpose of the present study was to evaluate the potential therapeutic effects of NZ on lipopolysaccharide (LPS)-induced RAW264.7 cells and explore its underlying mechanisms.The effect of NZ on NO generation in LPS-activated macrophage was measured by Griess assay. The concentrations of TNF-α, IL-18, IL-1β were analyzed with ELISA kits. The LPS-induced production of reactive oxygen species (ROS) was determined by flow cytometry. The protein expressions of TLR4, NF-κB and NLRP3 signaling pathway were investigated with Western blot analysis.It was shown that NZ significantly reduced the production of NO and the generation of pro-inflammatory cytokines in LPS-induced RAW264.7 cells. In addition, NZ markedly inhibited the up-regulation of toll-like receptor 4 (TLR4), myeloid differentiation factor 88 (MyD88) and the activation of nuclear factor kappa B (NF-κB) in LPS-stimulated RAW 264.7 macrophages. Of note, NZ suppressed the expression of the inflammasome component such as NOD-like receptor 3(NLRP3), apoptosis-associated speck-like protein containing CARD(ASC), as well as the levels of cytokines including Interleukin-18(IL-18) and Interleukin-1β(IL-1β).These results indicated that NZ inhibited the generations of NO and pro-inflammatory cytokines by suppressing TLR4/MyD88/NF-κB pathway, suggesting that NZ could be an effective candidate for ameliorating LPS-induced inflammatory responses.
Co-reporter:Tong Chen;Lu Xiao;Lingpeng Zhu;Shiping Ma;Tianhua Yan
Inflammation 2015 Volume 38( Issue 5) pp:1814-1822
Publication Date(Web):2015 October
DOI:10.1007/s10753-015-0159-4
The aim of the study was to investigate the anti-asthma effects of ginsenoside Rb1 (Rb1) and its possible mechanisms. A total of 50 mice were randomly assigned to five experimental groups: control, model, dexamethasone (2 mg/kg), and Rb1 (10 and 20 mg/kg). Airway resistance (RI) was measured; histological studies were evaluated by the hematoxylin and eosin (HE) staining; Th1/Th2, ovalbumin (OVA)-specific serum, and bronchoalveolar lavage fluid (BALF) IgE levels were evaluated enzyme-linked immunosorbent assay (ELISA); and T-bet/GATA3 proteins were evaluated by Western blot. Our study demonstrated that Rb1 inhibited OVA-induced increases in RI and eosinophil counts; interleukin (IL)-4 was recovered, and IFN-γlevel increased in bronchoalveolar lavage fluid. Histological studies demonstrated that Rb1 substantially inhibited OVA-induced eosinophilia in lung tissue. Western blot studies demonstrated that Rb1 substantially inhibited GATA3 and increased T-bet. These findings suggest that Rb1 may effectively ameliorate the progression of asthma and could be used as a therapy for patients with allergic asthma.
Co-reporter:Shengping Zhang, Jiani Tan, Zhonghui Lai, Ying Li, Junxia Pang, Jianhu Xiao, Zhangjian Huang, Yihua Zhang, Hui Ji, and Yisheng Lai
Journal of Chemical Information and Modeling 2014 Volume 54(Issue 6) pp:1785-1797
Publication Date(Web):May 24, 2014
DOI:10.1021/ci5002058
The NEDD8-activating enzyme (NAE) is an emerging target for cancer therapy, which regulates the degradation and turnover of a variety of cancer-related proteins by activating the cullin-RING E3 ubiquitin ligases. Among a limited number of known NAE inhibitors, the covalent inhibitors have demonstrated the most potent efficacy through their covalently linked adducts with NEDD8. Inspired by this unique mechanism, in this study, a novel combined strategy of virtual screening (VS) was adopted with the aim to identify diverse covalent inhibitors of NAE. To be specific, a docking-enabled pharmacophore model was first built from the possible active conformations of chosen covalent inhibitors. Meanwhile, a dynamic structure-based phamacophore was also established based on the snapshots derived from molecular dynamic simulation. Subsequent screening of a focused ZINC database using these pharmacophore models combined with covalent docking discovered three novel active compounds. Among them, compound LZ3 exhibited the most potent NAE inhibitory activity with an IC50 value of 1.06 ± 0.18 μM. Furthermore, a cell-based washout experiment proved the proposed covalent binding mechanism for compound LZ3, which confirmed the successful application of our combined VS strategy, indicating it may provide a viable solution to systematically discover novel covalent ligands.
Co-reporter:Xiaoli Wang, Linna Wang, Xiao Sheng, Zhangjian Huang, Tingting Li, Ming Zhang, Jinyi Xu, Hui Ji, Jian Yin and Yihua Zhang
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 31) pp:5995-6004
Publication Date(Web):20 Jun 2014
DOI:10.1039/C4OB00830H
In the present study, a series of hydrogen sulfide (H2S) releasing derivatives (8a–g and 9a–f) of 3-n-butylphthalide (NBP) were designed, synthesized and biologically evaluated. The most promising compound 8e significantly inhibited the adenosine diphosphate (ADP) and arachidonic acid (AA)-induced platelet aggregation in vitro, superior to NBP, ticlopidine hydrochloride and aspirin. Furthermore, 8e could slowly produce moderate levels of H2S in vitro, which could be beneficial for improving cardiovascular and cerebral circulation. Most importantly, 8e protected against the collagen and adrenaline induced thrombosis in mice, and exhibited greater antithrombotic activity than NBP and aspirin in rats. Overall, 8e could warrant further investigation for the treatment of thrombosis-related ischemic stroke.
Co-reporter:Ran You ; Wenyan Long ; Zhonghui Lai ; Lei Sha ; Kai Wu ; Xing Yu ; Yisheng Lai ; Hui Ji ; Zhangjian Huang ;Yihua Zhang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 5) pp:1984-1995
Publication Date(Web):February 1, 2013
DOI:10.1021/jm301652t
Fifteen novel derivatives of glycyrrhetinic acid (GA) were synthesized and evaluated for anti-inflammatory activities. It was found that the introduction of 1-en-3-one and 9(11),12-diene and 2,20-dinitrile functionalities into the scaffold of GA led to the discovery of potent compound 19 for inhibition of LPS-induced NO production. Furthermore, 19 effectively inhibited the protein and mRNA expression of inducible NO synthase (iNOS) and the mRNA expression of TNF-α, IL-6, and IL-1β in LPS-stimulated RAW 264.7 macrophages. Mechanistically, 19 exerted inhibitory effects on the activation of the three main MAPKs and phosphorylation and degradation of IκB-α, as well as the ratio of nuclear/cytosolic content of p65. Importantly, 19 significantly decreased the mortality rate in the mouse model of LPS-induced sepsis shock. It is noteworthy that inhibitory effect of 19 on NO production was not blocked by the glucocorticoid receptor antagonist mifepristone, indicating that it does not act through the glucocorticoid receptor.
Co-reporter:Xiaoli Wang ; Linna Wang ; Tingting Li ; Zhangjian Huang ; Yisheng Lai ; Hui Ji ; Xiaolong Wan ; Jinyi Xu ; Jide Tian ;Yihua Zhang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 7) pp:3078-3089
Publication Date(Web):March 19, 2013
DOI:10.1021/jm4001693
In search of novel anti-ischemic stroke agents with higher potency than a known drug 3-n-butylphthalide (NBP), a series of hybrids ((S)- and (R)-5a–f) from optically active ring-opened NBP derivative and isosorbide were synthesized for evaluating their anti-ischemic stroke activity. Compound (S)-5e displayed the strongest activity in inhibiting the adenosine diphosphate (ADP) and arachidonic acid (AA)-induced platelet aggregation in vitro, with 10.0- and 8.4-fold more effectiveness than (S)-NBP, respectively. Furthermore, (S)-5e was stable in artificial gastrointestinal fluids and could penetrate the blood–brain barrier (BBB) with an appreciate lipid/water partition coefficient relative to (S)-NBP. More importantly, oral treatment with (S)-5e protected from acute thrombosis and inhibited the ischemia/reperfusion-related brain injury in animals. Our findings suggest that (S)-5e may be promising for further evaluation for the intervention of ischemic stroke.
Co-reporter:Junjie Fu ; Ling Liu ; Zhangjian Huang ; Yisheng Lai ; Hui Ji ; Sixun Peng ; Jide Tian ;Yihua Zhang
Journal of Medicinal Chemistry 2013 Volume 56(Issue 11) pp:4641-4655
Publication Date(Web):April 25, 2013
DOI:10.1021/jm400393u
A series of hybrids from O2-(2,4-dinitrophenyl)diazeniumdiolate and oleanolic acid (OA) were designed, synthesized, and biologically evaluated as novel nitric oxide (NO)-releasing prodrugs that could be activated by glutathione S-transferase π (GSTπ) overexpressed in a number of cancer cells. It was discovered that the most active compound, 21, released high levels of NO selectively in HCC cells but not in the normal cells and exhibited potent antiproliferative activity in vitro as well as remarkable tumor-retarding effects in vivo. Compared with the reported GSTπ-activated prodrugs JS-K and PABA/NO, 21 exhibited remarkably improved stability in the absence of GSTπ. Importantly, the decomposition of 21 occurred in the presence of GSTπ and was much more effective than in glutathione S-transferase α. Additionally, 21 induced apoptosis in HepG2 cells by arresting the cell cycle at the G2/M phase, activating both the mitochondrion-mediated pathway and the MAPK pathway and enhancing the intracellular production of ROS.
Co-reporter:Xiaoli Wang, Linna Wang, Zhangjian Huang, Xiao Sheng, Tingting Li, Hui Ji, Jinyi Xu, Yihua Zhang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 7) pp:1985-1988
Publication Date(Web):1 April 2013
DOI:10.1016/j.bmcl.2013.02.035
A series of novel nitric oxide releasing derivatives of 6-amino-3-n-butylphthalide were designed, synthesized and evaluated as potential antiplatelet agents. Compound 10b significantly inhibited the adenosine diphosphate (ADP)-induced platelet aggregation in vitro, superior to 6-amino-3-n-butylphthalide, 3-n-butylphthalide (NBP) and ticlopidine. Meanwhile 10b released moderate levels of NO, which could be beneficial for improving cardiovascular and cerebral circulation. Furthermore, 10b had an enhanced aqueous solubility relative to NBP. These findings may provide new insights into the development of novel antiplatelet agents for the treatment of thrombosis-related ischemic stroke.The inhibitory activity of NO-releasing compound 10b on ADP-induced platelet aggregation was better than that of 6-amino-NBP (1), NBP and ticlopidine.
Co-reporter:Jing Wu ; Jingjing Ling ; Xuliang Wang ; Tingting Li ; Jingchao Liu ; Yisheng Lai ; Hui Ji ; Sixun Peng ; Jide Tian ;Yihua Zhang
Journal of Medicinal Chemistry 2012 Volume 55(Issue 16) pp:7173-7181
Publication Date(Web):July 24, 2012
DOI:10.1021/jm300681r
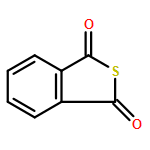
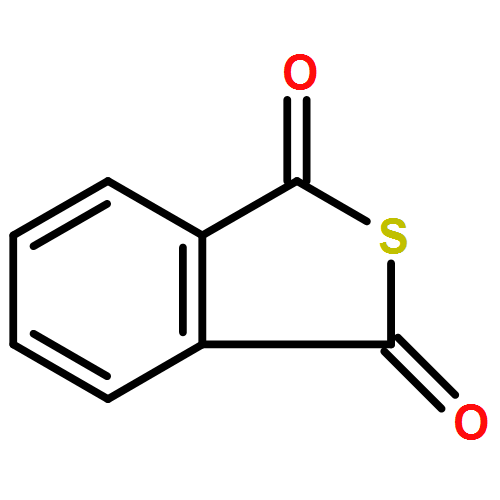
The development of novel antithrombotic agents with strong free radical scavenging activity is of great significance for the treatment of ischemic stroke. In the present study, 3-alkyl/arylalkyl-substituted benzo[c]thiophen-1(3H)-ones (5a–h) were designed and synthesized. The most active compound 5d significantly inhibited the adenosine diphosphate (ADP) induced and arachidonic acid (AA) induced in vitro platelet aggregation, superior to clinically used antiplatelet drug aspirin (ASP) and anti-ischemic stroke drugs 3-n-butylphthalide (NBP) and edaravone (Eda). More importantly, in comparison with both NBP and Eda, 5d exhibited stronger antithrombotic and free radical scavenging activities and better or comparable neuroprotective effects against ischemia/reperfusion (I/R) in rats by ameliorating neurobehavioral function, reducing infarct size and brain-water content, attenuating cerebral damage, and normalizing the levels of oxidative enzymes. Overall, our findings may provide an alternative strategy for the design of novel anti-ischemic stroke agents more potent than drugs like NBP and Eda.
Co-reporter:Xiaoli Wang, Qian Zhao, Xuliang Wang, Tingting Li, Yisheng Lai, Sixun Peng, Hui Ji, Jinyi Xu and Yihua Zhang
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 45) pp:9030-9040
Publication Date(Web):14 Sep 2012
DOI:10.1039/C2OB26511G
ZJM-289 is a potent racemic agent which inhibits both platelet aggregation and thrombosis superior to a known anti-ischemic stroke drug 3-n-butylphthalide (NBP). Herein, the enantiomers of ZJM-289, (S)-ZJM-289 and (R)-ZJM-289, were synthesized and evaluated for their biological activities. It was observed that the two enantiomers appeared to be almost as effective as ZJM-289 in inhibiting platelet aggregation in vitro and thrombus formation in vivo. Moreover, like ZJM-289, its enantiomers could regulate the ratio of thromboxane B2 (TXB2) and 6-keto-prostaglandin F1α, and enhanced levels of nitric oxide (NO), cAMP and cGMP, suggesting that the anti-platelet and antithrombotic activities of the enantiomers and ZJM-289 are associated with both the arachidonic acid cascade and cGMP–NO signal pathway. Furthermore, it was found that oral administration of the enantiomers and ZJM-289 for three days significantly reduced the infarct size, brain water content and neurological deficit in rats after cerebral ischemia reperfusion. Importantly, the two enantiomers equally improved blood flow in the ischemic stroke model and modulated endothelial function through releasing moderate levels of NO, which might, at least partially, contribute to their neuroprotection. Collectively, the present study demonstrates that the two enantiomers are as potent as ZJM-289 in inhibition of platelet aggregation and thrombosis and in neuroprotection, and (S)-ZJM-289 shows somewhat better effects than (R)-ZJM-289 and ZJM-289 in a few cases. These findings may provide new insights into the development of therapeutic agents like ZJM-289 for the intervention of thrombosis-related ischemic stroke.
Co-reporter:Jingjing Ling, Bo Wei, Guangping Lv, Hui Ji, Shaoping Li
Food Chemistry 2012 Volume 130(Issue 2) pp:229-235
Publication Date(Web):15 January 2012
DOI:10.1016/j.foodchem.2011.07.039
This study was to evaluate the anti-hyperlipidaemic and antioxidant effects of turmeric oil (TO), the supercritical fluid extract from turmeric, in hyperlipidaemic rats induced by a high-fat diet. TO significantly decreased (p < 0.05) the levels of serum total cholesterol, low-density lipoprotein cholesterol, triglyceride, and free fatty acid and increased (p < 0.05) that of high-density lipoprotein cholesterol in this model. It also markedly elevated (p < 0.05) the activities of superoxide dismutase and glutathione peroxidase and lowered (p < 0.05) maleic dialdehyde activity, to suppress oxidative reactions. Besides, histological morphology examination showed that TO prevented the damage of liver tissues induced by high-fat diet. Thus, the findings indicate that TO might provide protection against cardiovascular diseases.Highlights► We investigate the hypolipidaemic effect of turmeric oil using hyperlipidaemic rats. ► Turmeric oil attenuates oxidative stress accompanied by hyperlipidaemia. ► Turmeric oil prevents the damage of liver issues induced by high-fat diet.
Co-reporter:Yong Ling ; Xiaolei Ye ; Zhenzhen Zhang ; Yihua Zhang ; Yisheng Lai ; Hui Ji ; Sixun Peng ;Jide Tian
Journal of Medicinal Chemistry 2011 Volume 54(Issue 9) pp:3251-3259
Publication Date(Web):April 19, 2011
DOI:10.1021/jm1014814
Novel furoxan-based nitric oxide (NO) releasing derivatives (8a–p) of farnesylthiosalicylic acid (FTS) were synthesized. Compound 8l displayed the strongest inhibition on the proliferation of human hepatocellular carcinoma (HCC) cells in vitro, superior to FTS, sorafenib, and furoxan moiety, selectively induced high frequency of HCC cell apoptosis, and produced high levels of NO in HCC cells but not in nontumor liver cells. Furthermore, 8l exhibited low acute toxicity to mice and significantly inhibited the growth of HCC tumors in vivo and the Ras-related signaling in the tumors. Therefore, our novel findings may provide a new framework for the design of new NO-releasing furoxan/FTS hybrids for the intervention of human HCC.
Co-reporter:Xuliang Wang, Yang Li, Qian Zhao, Zhenli Min, Chao Zhang, Yisheng Lai, Hui Ji, Sixun Peng and Yihua Zhang
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 16) pp:5670-5681
Publication Date(Web):12 Apr 2011
DOI:10.1039/C1OB05478C
Novel nitric oxide (NO) releasing derivatives (7a–7l) of 3-n-butylphthalide (NBP) were designed and synthesized. Compound 7e inhibited the adenosine diphosphate (ADP), thrombin (TH) and arachidonic acid (AA)-induced in vitroplatelet aggregation, superior to NBP and aspirin, released moderate levels of NO, and improved aqueous solubility relative to NBP. Furthermore, 7e exhibited greater antithrombotic activity than NBP and aspirin in rats, and protected against collagen and adrenaline-induced thrombosis in mice. Therefore, NO-releasing NBP derivatives possessed potent antiplatelet aggregation and antithrombotic activity. Our findings may aid in the design of new therapeutic agents for the treatment of thrombosis-related ischemic stroke.
Co-reporter:Tingting Li;Qiaoling Yu;Deen Han;Ying Zhao;Di Zhao
Chromatographia 2011 Volume 73( Issue 11-12) pp:1111-1120
Publication Date(Web):2011 June
DOI:10.1007/s10337-011-2002-x
Picroside II is one of the main active constituents of Picrorhiza kurroa, which has hepatoprotective, anticholestatic, antioxidant, and immune-modulating activity. To gain an understanding of the biotransformation of picroside II in vivo, liquid chromatography–electrospray ionization ion-trap mass spectrometry (LC–ESI–IT–MS) was used to investigate the metabolism of picroside II in rats after intravenous administration of a single dose. This method could simultaneously determine picroside II and its metabolites in rat bile. The bile samples were purified by use of a C18 solid-phase extraction (SPE) cartridge and were separated on a Hypersil ODS2 C18 analytical column. Two phase II metabolites of picroside II in rat bile were characterized, and elucidation of their structures was performed by comparing changes in molecular masses (ΔM), retention times, and MS2 spectral patterns of metabolites with those of the parent drug. Two metabolites identified for the first time in this research were glucuronide and sulfate conjugates.
Co-reporter:Yong Ling, Xiaolei Ye, Hui Ji, Yihua Zhang, Yisheng Lai, Sixun Peng, Jide Tian
Bioorganic & Medicinal Chemistry 2010 Volume 18(Issue 10) pp:3448-3456
Publication Date(Web):15 May 2010
DOI:10.1016/j.bmc.2010.03.077
Novel furoxan-based nitric oxide (NO)-releasing derivatives (11a–p) of farnesylthiosalicylic acid (FTA) were synthesized. Compounds 11d, 11f, 11k, and 11m–o displayed anti-tumor activities superior to FTA and sorafenib in most cancer cells tested. Analysis of six compounds revealed that 11d, 11f, 11n, 11o, and 11p, but not 11a that had low anti-tumor activity, produced high levels of NO, associated with their strong anti-tumor activity. Furthermore, the anti-tumor activity of 11f was partially mimicked by the furoxan moiety, but reduced by pre-treatment with hemoglobin. Importantly, treatment with 11f inhibited Ras-related signaling in cancer cells. Apparently, the high anti-tumor activity of 11f was attributed to the synergic effect of high levels of NO production and inhibition of Ras-related signaling in cancer cells. Our findings suggest that the furoxan/FTA hybrids may hold greater promise as therapeutic agents for the intervention of human cancers.A series of novel furoxan-based nitric oxide (NO)-releasing derivatives of farnesylthiosalicylic acid (FTA) were synthesized and evaluated for their cytotoxicity against human cancer cells, NO-releasing ability and Ras inhibitory activity.
Co-reporter:Yisheng Lai, Lihong Shen, Zhenzhen Zhang, Wenqing Liu, Yihua Zhang, Hui Ji, Jide Tian
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 22) pp:6416-6420
Publication Date(Web):15 November 2010
DOI:10.1016/j.bmcl.2010.09.070
A series of novel furoxan-based nitric oxide (NO)-releasing derivatives of glycyrrhetinic acid (GA) were designed, synthesized, and evaluated for their in vitro cytotoxicity against human hepatocellular carcinoma (HCC) and non-tumor liver cells. Five furoxan/GA hybrids, 7b–d, 7f, and 7g, displayed potent cytotoxicity against HCC cells (IC50: 0.25–1.10 μM against BEL-7402 cells and 1.32–6.78 μM against HepG2 cells), but had a little effect on the growth of LO2 cells, indicating that these compounds had selective cytotoxicity against HCC cells. Furthermore, these compounds produced high concentrations of NO in HCC cells, but low in LO2 cells and treatment with hemoglobin partially reduced the cytotoxicity of the hybrid in HCC cells. Apparently, the high concentrations of NO produced by NO donor moieties and the bioactivity of GA synergistically contribute to the cytotoxicity, but the NO is a major player against HCC cells in vitro. Potentially, our findings may aid in the design of new chemotherapeutic reagents for the intervention of human HCC at clinic.A series of novel furoxan-based nitric oxide (NO)-releasing derivatives of glycyrrhetinic acid were synthesized and evaluated for their cytotoxicity against human hepatocellular carcinoma (HCC) cells and NO-releasing ability. Five hybrids displayed selective cytotoxicity against HCC cells with a little effect on the growth of LO2 cells.
Co-reporter:Yang Shen;Yong-Qi Li;Shao-Ping Li;Lin Ma;Li-Ju Ding
Journal of Natural Medicines 2010 Volume 64( Issue 3) pp:336-345
Publication Date(Web):2010 July
DOI:10.1007/s11418-010-0416-7
To examine the effects of Panax notoginseng saponins (PNS), the main active components of Panax notoginseng, on ovariectomy-induced osteoporosis in rats. A total of 72 six-month-old female rats were randomly assigned to sham-operated group and five ovariectomized (OVX) groups: OVX with distilled water (5 ml/kg/day, p.o.), OVX with graded doses of PNS (75, 150, 300 mg/kg/day, p.o.), and OVX with nilestriol (1 mg/kg/week, p.o.). Animals were sacrificed after a 13-week treatment course. Compared with the OVX group, PNS administration prevented OVX-induced decrease in bone mineral density (BMD) of lumbar vertebrae and total femur, and significantly increased bone structural biomechanical properties. Improvements of BMD and biomechanical properties were accompanied by the beneficial changes of PNS on trabecular microarchitecture in the tibial metaphysis. PNS at the highest dose significantly prevent decrease in trabecular bone volume over bone total volume, trabecular number, trabecular thickness, connectivity density, and increase in trabecular separation and structure model index in OVX rats. The bone-modulating effects of PNS may be due to the increased bone formation and decreased bone resorption, as was evidenced by the elevated level of serum alkaline phosphatase and decreased level of urinary deoxypyridinoline. PNS treatment is able to enhance BMD, bone strength, and prevent the deterioration of trabecular microarchitecture without hyperplastic effect on uterus. Therefore, PNS might be a potential alternative medicine for the prevention and treatment of postmenopausal osteoporosis.
Co-reporter:Xiaolei Ye;Wu Zhou;Yongqi Li;Yihua Sun;Yihua Zhang;Hui Ji;Yisheng Lai
Cancer Chemotherapy and Pharmacology 2010 Volume 66( Issue 2) pp:277-285
Publication Date(Web):2010/07/01
DOI:10.1007/s00280-009-1161-z
Inflammation plays a crucial role in the development of lung cancer. Accumulated studies have proved that non-steroidal anti-inflammatory drugs (NSAIDs) which block inflammation by their actions on arachidonic acid (AA) metabolism have a potential role in cancer chemotherapy and chemoprevention. The aim of our study was to investigate whether darbufelone, a novel anti-inflammatory drug, has anticancer effects in lung cancer.Human non-small cell lung cancer cell lines were treated with darbufelone at various doses and time points for analysis of cell viability, cell cycle, and apoptosis in vitro. The in vivo effect of darbufelone was assessed in Lewis lung carcinoma mice model.Darbufelone inhibited the proliferation of non-small cell lung cancer cell lines in a dose-dependent manner, and induced cell cycle arrest at G0/G1 phase through up-regulation of p27 expression. Treatment with darbufelone also induced apoptosis by activating caspase-3 and caspase-8. Lewis lung carcinoma growth was also significantly inhibited by darbufelone treatment at daily dose of 80 mg/kg.Taken together, these studies suggested that darbufelone, an anti-inflammation drug, might represent a novel therapeutic approach for lung cancer treatment.
Co-reporter:Ying Zhao;Desheng Zhai;Hui He;Tingting Li
European Journal of Clinical Pharmacology 2009 Volume 65( Issue 6) pp:579-584
Publication Date(Web):2009 June
DOI:10.1007/s00228-009-0619-6
Our objective was to study the effects of polymorphic the CYP3A5 (allele *1 and *3), MDR1 [single nucleotide polymorphisms (SNPs) G2677T, C3435T] and CACNA1C (SNPs rs2239128, rs2239050, rs2238032) genes on nimodipine oral disposition and response in healthy Chinese subjects.Pharmacokinetics and pharmacodynamics data were obtained from a bioequivalence study, and the same 20 subjects were genotyped for CYP3A, MDR1 and CACNA1C. An additional 41 healthy Chinese subjects were recruited to obtain an indication of the distribution of CACNA1C polymorphisms in the Chinese population. Racial differences in the frequency of CACNA1C alleles were assessed. The phenotype differences between genotypes were analyzed.The allelic frequencies of rs2239050 and rs2238032 in our Chinese cohort were different from those in a Caucasian population (p < 0.01). Subjects with mutant alleles (*3/*3) of the CYP3A5 gene had a decreased oral clearance of nimodipine, with a higher lnCmax or \(\ln {\text{AUC}}_{0 - \infty } \) compared with those subjects with the heterozygote (*1/*3) or wild type (*1/*1) gene. The CACNA1C rs2239128 C and rs2239050 G SNPs were associated with a stronger efficacy compared with their respective alleles, rs2239128 T and rs2239050 C. MDR1 polymorphisms showed no significance in terms of nimodipine disposition.The polymorphic CYP3A5 (allele *1 and *3) and CACNA1C genes have effects on nimodipine oral disposition and response in healthy Chinese subjects. The homozygous variant of CYP3A5 (*3/*3) was associated with significantly increased nimodipine exposure. CACNA1C SNPs rs2239128 C and rs2239050 G were associated with a stronger efficacy.
Co-reporter:Tao Wang, Yi Hua Zhang, Shan Yu, Hui Ji, Yi Sheng Lai, Si Xun Peng
Chinese Chemical Letters 2008 Volume 19(Issue 8) pp:928-930
Publication Date(Web):August 2008
DOI:10.1016/j.cclet.2008.05.044
In search of novel anticancer agents, a series of thalidomide analogs (6a–j) were designed and synthesized. Cytotoxicity of these compounds against human hepatoma cells (HepG2) was evaluated by MTT method. Compounds 6d, 6h and 6i showed significant cytotoxic activities comparable to or stronger than control 5-fluorouracil.
Co-reporter:Li Chen, Qian Zhao, Xu-Liang Wang, Ran You, ... Yi-Sheng Lai
Vascular Pharmacology (November–December 2011) Volume 55(Issues 5–6) pp:135-142
Publication Date(Web):1 November 2011
DOI:10.1016/j.vph.2011.07.003
Nonsteroidal anti-inflammatory drugs (NSAIDs) are previously found to possess prostaglandin and leukotriene-independent anti-inflammatory effect. The aim of the present study was to investigate the prostaglandin and leukotriene-independent anti-inflammatory effect of an imidazolone COX/5-LOX inhibitor ZLJ-6 and the underlying mechanism. Pretreatment human umbilical vein endothelial cells (HUVECs) with ZLJ-6 (3, 10 and 30 μM) concentration-dependently decreased TNF-α-induced monocyte–endothelial interactions in both static and dynamic conditions whereas no effect was found after pretreatment with the COX-2 inhibitor celecoxib (30 μM), 5-LOX inhibitor zileuton (30 μM) and the combination of them. ZLJ-6 also attenuated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cytoadhesion molecule-1 (VCAM-1) on TNF-α-induced HUVECs. A further analysis indicated that ZLJ-6 attenuated TNF-α-induced nuclear translocation of NF-κB, IκB phosphorylation, IκB kinase β (IKKβ) activity, and subsequent NF-κB-DNA complex formation, suggesting that NF-κB pathway was involved in TNF-α-induced inflammation. However, ZLJ-6 did not affect TNF-α-induced extracellular signal-regulated kinases (ERK1/2), c-Jun N-terminal kinases (JNK) and p38 phosphorylation. Taken together, our results indicated that ZLJ-6 potently inhibited TNF-α-induced monocyte–endothelial interactions and adhesion molecule (E-selectin, ICAM-1 and VCAM-1) expression and these effects were mediated by NF-κB signaling pathway rather than its primary pharmacological target COX-2 or 5-LOX.Download high-res image (310KB)Download full-size image
Co-reporter:Tong Chen, Yi Mou, Jiani Tan, Linlin Wei, Yixue Qiao, Tingting Wei, Pengjun Xiang, Sixun Peng, Yihua Zhang, Zhangjian Huang, Hui Ji
International Immunopharmacology (March 2015) Volume 25(Issue 1) pp:55-64
Publication Date(Web):1 March 2015
DOI:10.1016/j.intimp.2015.01.011
•We first explained that CDDO-Me ameliorated LPS-induced acute lung injury through NF-κB and MAPK pathways.CDDO-Me, initiated in a phase II clinical trial, is a potential useful therapeutic agent for cancer and inflammatory dysfunctions, whereas the therapeutic efficacy of CDDO-Me on LPS-induced acute lung injury (ALI) has not been reported as yet. The purpose of the present study was to explore the protective effect of CDDO-Me on LPS-induced ALI in mice and to investigate its possible mechanism. BalB/c mice received CDDO-Me (0.5 mg/kg, 2 mg/kg) or dexamethasone (5 mg/kg) intraperitoneally 1 h before LPS stimulation and were sacrificed 6 h later. W/D ratio, lung MPO activity, number of total cells and neutrophils, pulmonary histopathology, IL-6, IL-1β, and TNF-α in the BALF were assessed. Furthermore, we estimated iNOS, IL-6, IL-1β, and TNF-α mRNA expression and NO production as well as the activation of the three main MAPKs, AkT, IκB-α and p65. Pretreatment with CDDO-Me significantly ameliorated W/D ratio, lung MPO activity, inflammatory cell infiltration, and inflammatory cytokine production in BALF from the in vivo study. Additionally, CDDO-Me had beneficial effects on the intervention for pathogenesis process at molecular, protein and transcriptional levels in vitro. These analytical results provided evidence that CDDO-Me could be a potential therapeutic candidate for treating LPS-induced ALI.
Co-reporter:Su-Su Tang, Hao Hong, Lan Chen, Zhen-lin Mei, Miao-jin Ji, Guo-qing Xiang, Ning Li, Hui Ji
Neurobiology of Aging (March 2014) Volume 35(Issue 3) pp:590-599
Publication Date(Web):1 March 2014
DOI:10.1016/j.neurobiolaging.2013.09.036
Accumulation of amyloid-β (Aβ) is thought to be associated with the progressive neuronal death observed in Alzheimer's disease, but the mechanisms underlying neurotoxicity triggered by Aβ remain elusive. In the current study, we investigated the roles of cysteinyl leukotriene receptor 1 (CysLT1R) in Aβ1–42-induced neurotoxicity in vitro or in vivo. In vitro exposure of mouse primary neurons to Aβ1–42 caused a gradual increases in CysLT1R expression. In vivo bilateral intrahippocampal injection of Aβ1–42 also elicited time-dependent increases of CysLT1R expression in the hippocampus and cortex of mice. The CysLT1R antagonist pranlukast not only reversed Aβ1–42-induced upregulation of CysLT1R, but also suppressed Aβ1–42-triggered neurotoxicity evidenced by enhanced nuclear factor-kappa B p65, activated caspase-3, decreased B-cell lymphoma-2 and cell viability and impaired memory. Furthermore, chronic treatment with pranlukast produced similar beneficial effects on memory behavior and hippocampal long-term potentiation to memantine or donepezil in intrahippocampal Aβ1–42-injected mice. Our data indicate that CysLT1R is involved in Aβ1–42-induced neurotoxicity, and that blockade of CysLT1R, such as application of CysLT1R antagonist, could be a novel and promising strategy for the treatment of Alzheimer's disease.
Co-reporter:Tong Chen, Jin Gao, Pengjun Xiang, Yongde Chen, Jing Ji, Peng Xie, Hui Wu, Wei Xiao, Yidan Wei, Shumin Wang, Li Lan, Hui Ji, Tianhua Yan
International Immunopharmacology (June 2015) Volume 26(Issue 2) pp:338-348
Publication Date(Web):1 June 2015
DOI:10.1016/j.intimp.2015.04.001
•Platycodin D inhibited liver injury in alloxan-induced diabetic mice.•Platycodin D increased Treg cells in alloxan-induced diabetic mice.•Platycodin D inhibited Th17 cells in alloxan-induced diabetic mice.Platycodin D is a major pharmacological constituent of Platycodi Radix with immunomodulatory activity. The present study was designed to investigate how platycodin D (PLD) reveals liver injury in diabetic mice and its mechanism. Fifty mice were divided into five groups randomly: control group, model group, rosiglitazone (ROG, 10 mg/kg) group, PLD (50 mg/kg) group, and PLD (100 mg/kg) group. Diabetes was induced with the injection of alloxan monohydrate (150 mg/kg) subcutaneously, and animals with blood glucose level of ≥ 250 mg/dl were considered as diabetic mice. After the first day of diabetes induction, the treatments were performed for 8 weeks. Then the animals were anaesthetized, and blood and liver samples were also collected for further assay. PLD significantly decreased the serum levels of glucose, insulin, interleukin-6 (IL-6), interleukin-1β, tumor necrosis factor-α (TNF-α), and interleukin (IL)-17A and increased IL-10 level in serum. PLD effectively downregulated aspartate transaminase (AST), alanine aminotransferase (ALT), total cholesterol (TC), and triglycerides (TG) in liver. PLD also attenuated liver histological change. In addition, PLD significantly attenuated IL-17A and IL-10 levels in vitro, flow cytometry (FCM) studies also showed that PLD remarkably inhibited Th17 cells and significantly increased Treg cells in liver tissues and spleen cells. Western blot demonstrated PLD inhibited the phosphorylation of JAK and STAT-3 and the expression of RORγt and increased the expression of Foxp3. The findings showed that PLD exerts beneficial effects on alloxan-induced liver injury in mice.
Co-reporter:Qian Zhao, Chao Zhang, Xuliang Wang, Li Chen, Hui Ji, Yihua Zhang
Neurochemistry International (January 2012) Volume 60(Issue 2) pp:134-144
Publication Date(Web):1 January 2012
DOI:10.1016/j.neuint.2011.11.013
Pharmacological compounds that release nitric oxide (NO) have been recognized as the potential therapeutic agents for acute stroke. (S)-ZJM-289 is a novel NO-releasing derivative of 3-n-butylphthalide (NBP) with enhanced anti-platelet and anti-thrombotic actions. The present study was performed to investigate the neuroprotective effects and related mechanisms of (S)-ZJM-289 on ischemic neuronal injury in vitro and in vivo. Primary cortical neuronal cultures were exposured to oxygen–glucose deprivation followed by recovery (OGD/R), a model of ischemia-like injury, and treated with (S)-ZJM-289 before OGD. In vitro results showed that (S)-ZJM-289 attenuated OGD/R-induced neuronal injury, which was associated with the maintenance of mitochondrial integrity and function by alleviating intracellular calcium overload and reactive oxygen species (ROS) accumulation, preventing mitochondrial membrane depolarization and preserving respiratory chain complexes activities. Moreover, (S)-ZJM-289 treatment suppressed mitochondrial release of cytochrome c (cyt c) and nuclear translocation of apoptosis-inducing factor (AIF), thereby blocking mitochondria-mediated cell death, which may be partially mediated by up-regulation of Hsp70. The neuroprotection by (S)-ZJM-289 was also studied using a model of middle cerebral artery occlusion (MCAO). Oral administration of (S)-ZJM-289 at the onset of reperfusion for 3 d significantly reduced the brain infarct size, improved neurological deficit and prevented neuronal loss and apoptosis. In current study, (S)-ZJM-289 appears to be more potent in ischemic neuroprotection than NBP, in particular at the lower doses, which may be due to the synergistic action of NBP and NO. These findings point to that (S)-ZJM-289 could be an attractive alternative to NBP in preventing the process of ischemia/reperfusion (I/R) injury.Highlights► (S)-ZJM-289 is a novel NO-releasing derivative of 3-n-butylphthalide (NBP). ► It enhances neuroprotection by the synergistic action of NBP and NO. ► It provides neuronal protection by regulating mitochondrial function. ► It blocks mitochondrial cell death pathways and promotes cell survival signals. ► It could be an attractive alternative to NBP in treatment for stroke.
Co-reporter:Qi Jiang, Min Yi, Qianqian Guo, Ciman Wang, Huimin Wang, Shanshan Meng, Chao Liu, Yeliu Fu, Hui Ji, Tong Chen
International Immunopharmacology (December 2015) Volume 29(Issue 2) pp:370-376
Publication Date(Web):1 December 2015
DOI:10.1016/j.intimp.2015.10.027
•We first argued that polydatin could be a potential useful therapeutic agent for LPS-induced acute lung injury in mice.•We first explained that polydatin ameliorated LPS-induced acute lung injury through TLR4-MyD88-NF-κB pathway.•Polydatin exhibited protective effects on LPS-induced ALI and BEAS-2B cells via TLR4-MyD88-NF-κB pathway.The purpose of this study was to investigate the protective effect of PD against lipopolysaccharide (LPS)-induced acute lung injury (ALI) and explore its potential mechanism. In vivo, PD and dexamethasone were intraperitoneally administered 1 h before LPS stimulation. Then, mice were sacrificed at 6 h post-LPS stimulation. Neutrophil number, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6) and interleukin-1β (IL-1β) in bronchoalveolar lavage fluid (BALF) were determined, as well as lung wet to dry ratio (W/D) and polymorphonuclear (MPO) activity. The protein expressions of Toll like receptor 4 (TLR4), myeloid differentiating factor 88 (MyD88), IL-1R-associated kinases 1 (IRAK1), IRAK4, inhibitor of nuclear factor kappa-B kinase (IKK)α, p-IKKα, IKKβ, p-IKKβ, inhibitor of NF-κB (IκBα), p-IκBα and NF-κB in lung tissues were assessed. Besides, we detected the IL-6, IL-1β, IL-8, TNF-α levels and TLR4, MyD88, NF-κB protein expressions in LPS-induced BEAS-2B cells. Consequently, PD significantly inhibited the levels of W/D, MPO, neutrophils number, TNF-α, IL-6, IL-1β and reversed TLR4-MyD88-NF-κB signaling pathway in lung tissues. In vitro assays, PD effectively negatively mediated the inflammatory cytokines and ameliorated the high expressions of TLR4, MyD88, NF-κB caused by LPS simulation in Human bronchial epithelial BEAS-2B cells. This study indicated that PD played a protective role in LPS-induced ALI and BEAS-2B cells. The results supported further study of PD as potential candidate for acute lung injury.
Co-reporter:Bao-qin Lin, Pei-bo Li, Yong-gang Wang, Wei Peng, Zhong Wu, Wei-wei Su, Hui Ji
Pulmonary Pharmacology & Therapeutics (April 2008) Volume 21(Issue 2) pp:259-263
Publication Date(Web):1 April 2008
DOI:10.1016/j.pupt.2007.05.001
The expectorant activity of naringenin was studied. Mucus secretion was evaluated in mice by measuring the tracheal output of phenol red. Mucociliary movement function was investigated using a migration method of carbon granules in unanesthetized pigeons. And the effect of naringenin on the secretion of mucin and lysozyme was performed in the rat tracheal ring explants. Naringenin could significantly increase the secretion of phenol red from mouse tracheas at the doses of 30–67 mg/kg (i.g.) (P<0.05). Naringenin, at the dose of 90 mg/kg, increased the tracheal mucociliary velocity (TMV) to 144.4% of control (P<0.01). 100 μM naringenin could enhance the basal lysozyme secretion, but had no effect on the basal mucin secretion from the rat tracheal ring explants. Treatment with naringenin at higher concentration (10 μmol/l) could inhibit the 100 ng/ml lipopolysaccharide (LPS)-induced mucin increase. These data suggest, therefore, that naringenin has the expectorant activity.
Co-reporter:Xuliang Wang, Yang Li, Qian Zhao, Zhenli Min, Chao Zhang, Yisheng Lai, Hui Ji, Sixun Peng and Yihua Zhang
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 16) pp:NaN5681-5681
Publication Date(Web):2011/04/12
DOI:10.1039/C1OB05478C
Novel nitric oxide (NO) releasing derivatives (7a–7l) of 3-n-butylphthalide (NBP) were designed and synthesized. Compound 7e inhibited the adenosine diphosphate (ADP), thrombin (TH) and arachidonic acid (AA)-induced in vitroplatelet aggregation, superior to NBP and aspirin, released moderate levels of NO, and improved aqueous solubility relative to NBP. Furthermore, 7e exhibited greater antithrombotic activity than NBP and aspirin in rats, and protected against collagen and adrenaline-induced thrombosis in mice. Therefore, NO-releasing NBP derivatives possessed potent antiplatelet aggregation and antithrombotic activity. Our findings may aid in the design of new therapeutic agents for the treatment of thrombosis-related ischemic stroke.
Co-reporter:Xiaoli Wang, Linna Wang, Xiao Sheng, Zhangjian Huang, Tingting Li, Ming Zhang, Jinyi Xu, Hui Ji, Jian Yin and Yihua Zhang
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 31) pp:NaN6004-6004
Publication Date(Web):2014/06/20
DOI:10.1039/C4OB00830H
In the present study, a series of hydrogen sulfide (H2S) releasing derivatives (8a–g and 9a–f) of 3-n-butylphthalide (NBP) were designed, synthesized and biologically evaluated. The most promising compound 8e significantly inhibited the adenosine diphosphate (ADP) and arachidonic acid (AA)-induced platelet aggregation in vitro, superior to NBP, ticlopidine hydrochloride and aspirin. Furthermore, 8e could slowly produce moderate levels of H2S in vitro, which could be beneficial for improving cardiovascular and cerebral circulation. Most importantly, 8e protected against the collagen and adrenaline induced thrombosis in mice, and exhibited greater antithrombotic activity than NBP and aspirin in rats. Overall, 8e could warrant further investigation for the treatment of thrombosis-related ischemic stroke.
Co-reporter:Xiaoli Wang, Qian Zhao, Xuliang Wang, Tingting Li, Yisheng Lai, Sixun Peng, Hui Ji, Jinyi Xu and Yihua Zhang
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 45) pp:NaN9040-9040
Publication Date(Web):2012/09/14
DOI:10.1039/C2OB26511G
ZJM-289 is a potent racemic agent which inhibits both platelet aggregation and thrombosis superior to a known anti-ischemic stroke drug 3-n-butylphthalide (NBP). Herein, the enantiomers of ZJM-289, (S)-ZJM-289 and (R)-ZJM-289, were synthesized and evaluated for their biological activities. It was observed that the two enantiomers appeared to be almost as effective as ZJM-289 in inhibiting platelet aggregation in vitro and thrombus formation in vivo. Moreover, like ZJM-289, its enantiomers could regulate the ratio of thromboxane B2 (TXB2) and 6-keto-prostaglandin F1α, and enhanced levels of nitric oxide (NO), cAMP and cGMP, suggesting that the anti-platelet and antithrombotic activities of the enantiomers and ZJM-289 are associated with both the arachidonic acid cascade and cGMP–NO signal pathway. Furthermore, it was found that oral administration of the enantiomers and ZJM-289 for three days significantly reduced the infarct size, brain water content and neurological deficit in rats after cerebral ischemia reperfusion. Importantly, the two enantiomers equally improved blood flow in the ischemic stroke model and modulated endothelial function through releasing moderate levels of NO, which might, at least partially, contribute to their neuroprotection. Collectively, the present study demonstrates that the two enantiomers are as potent as ZJM-289 in inhibition of platelet aggregation and thrombosis and in neuroprotection, and (S)-ZJM-289 shows somewhat better effects than (R)-ZJM-289 and ZJM-289 in a few cases. These findings may provide new insights into the development of therapeutic agents like ZJM-289 for the intervention of thrombosis-related ischemic stroke.