Co-reporter:Daniel L. Reger;Elizabeth A. Foley;Andrea E. Pascui;Mark D. Smith;Agnieszka Wojciechowska;Julia Jezierska;Sebastian A. Stoian;Andrew Ozarowski
Inorganic Chemistry March 6, 2017 Volume 56(Issue 5) pp:2884-2901
Publication Date(Web):February 20, 2017
DOI:10.1021/acs.inorgchem.6b02933
A series of monochloride-bridged, dinuclear metallacycles of the general formula [M2(μ-Cl)(μ-L)2](ClO4)3 have been prepared using the third-generation, ditopic bis(pyrazolyl)methane ligands L = m-bis[bis(1-pyrazolyl)methyl]benzene (Lm), M = Cu(II), Zn(II), and L = m-bis[bis(3,5-dimethyl-1-pyrazolyl)methyl]benzene (Lm*), M = Fe(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II). These complexes were synthesized from the direct reactions of M(ClO4)2·6H2O, MCl2, and the ligand, Lm or Lm*, in the appropriate stoichiometric amounts. Three analogous complexes of the formula [M2(μ-Cl)(μ-L)2](BF4)3, L = Lm, M = Cu(II), and L = Lm*, M = Co(II), Cu(II), were prepared from the reaction of [M2(μ-F)(μ-L)2](BF4)3 and (CH3)3SiCl. The bromide-bridged complex [Cu2(μ-Br)(μ-Lm*)2](ClO4)3 was prepared by the first method. Three acyclic complexes, [Co2(μ-Lm)μ-Cl4], [Co2(μ-Lm*)Cl4], and [Co2(μ-Lm*)Br4], were also prepared. The structures of all [M2(μ-X)(μ-L)2]3+ (X = Cl–, Br–) complexes have two ditopic bis(pyrazolyl)methane ligands bridging two metals in a metallacyclic arrangement. The fifth coordination site of the distorted trigonal bipyramidal metal centers is filled by a bridging halide ligand that has an unusual linear or nearly linear M–X–M angle. The NMR spectra of [Zn2(μ-Cl)(μ-Lm*)2](ClO4)3 and especially [Cd2(μ-Cl)(μ-Lm*)2](ClO4)3 demonstrate that the metallacycle structure is maintained in solution. Solid state magnetic susceptibility data for the copper(II) compounds show very strong antiferromagnetic exchange interactions, with −J values of 536 cm–1 for [Cu2(μ-Cl)(μ-Lm)2](ClO4)3·xCH3CN, 720 cm–1 for [Cu2(μ-Cl)(μ-Lm*)2](ClO4)3, and 945 cm–1 for [Cu2(μ-Br)(μ-Lm*)2](ClO4)3·2CH3CN. Smaller but still substantial antiferromagnetic interactions are observed with other first row transition metals, with −J values of 98 cm–1 for [Ni2(μ-Cl)(μ-Lm*)2](ClO4)3, 55 cm–1 for [Co2(μ-Cl)(μ-Lm*)2](ClO4)3, and 34 cm–1 for [Fe2(μ-Cl)(μ-Lm*)2](ClO4)3. EPR spectra of [Cu2(μ-Cl)(μ-Lm*)2](BF4)3 confirm the dz2 ground state of copper(II). In addition, the sign of the zero-field splitting parameter D was determined to be positive for [Cu2(μ-F)(μ-Lm*)2](BF4)3. Electronic spectra of the copper(II) complexes as well as Mössbauer spectra of the iron(II) complexes were also studied in relation with the EPR spectra and magnetic properties, respectively. Density functional theory calculations were performed using ORCA, and exchange integral values were obtained that parallel but are slightly higher than the experimental values by about 30%.
Co-reporter:Daniel L. Reger, Andrew P. Leitner, and Mark D. Smith
Crystal Growth & Design 2016 Volume 16(Issue 1) pp:527-536
Publication Date(Web):December 14, 2015
DOI:10.1021/acs.cgd.5b01575
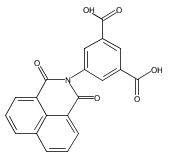
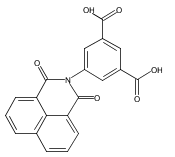
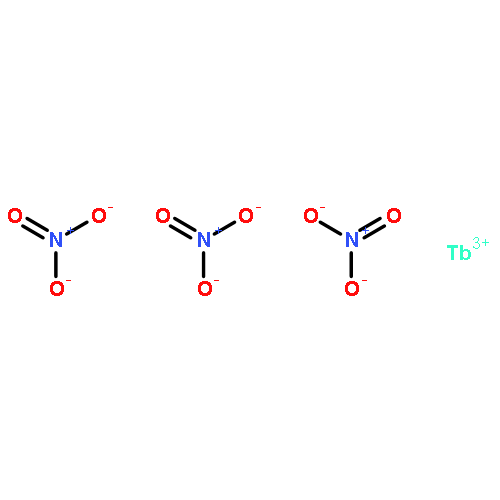
The new ligand 5-(1,8-naphthalimido)isophthalate (L1352–), containing two carboxylate donor groups and the 1,8-naphthalimide supramolecular tecton, has been used under solvothermal conditions to prepare a series of group 2, lanthanide, and actinide metal complexes: [Ca4(L135)4(H2O)8]·(H2O)9.5(DMF)2.6 (1), Ba(L135)(H2O)1.5(DMF)0.5 (2), La2(L135)3(DMF)4 (3), Ce2(L135)3(DMF)4 (4), Eu2(L135)3(DMF)4 (5), Tb2(L135)3(DMF)4 (6), [UO2(L135)(DMF)]·(py)0.5(EtOH)0.5 (7), and Th(L135)(NO3)2(DMF)2]·(DMF)2 (8). The solid state structure of the calcium complex 1 is based on helical rod-shaped secondary building-units (SBUs) of edge-shared polyhedra bridged by oxygen atoms from the carboxylate groups. The crystals are racemic, with the one-dimensional (1D) helical rods organized by π···π stacking interactions of the naphthalimide group into a three-dimensional (3D) supramolecular metal-organic framework (SMOF) structure. Although the structure of the barium complex 2 also contains rod-shaped SBUs, the rods are linked through the aryl backbone of the ditopic L1352– ligands into two-dimensional (2D) sheets. The sheets are further engaged in naphthalimide π···π stacking interactions to build a 3D SMOF. The lanthanide(III) complexes 3–6 are isostructural, based on binuclear SBUs linked through the ligands into a square-shaped, 2D grid pattern, with π-stacking interactions linking adjacent sheets to generate a 3D SMOF. The uranium(VI) complex 7 contains 7-coordinate pentagonal bipyramidal uranyl cations bridged through the ligands into 1D ribbons. The solid state structure of the thorium(IV) complex 8 consists of 10-coordinate thorium cations, also bridged through the ligands into 1D ribbons. Both of these actinide structures are organized into 2D supramolecular sheets by π-stacking interactions. Compounds 1, 2, 3, 6, and 8 exhibit solid-state luminescence dominated by the naphthalimide chromophore in the ligand. The group 2 complexes are slightly red-shifted, and the lanthanum complex 3 and the thorium complex 8 are slightly blue-shifted with respect to the ligand. The terbium compound, 6, is greatly blue-shifted by ∼75 nm, and naphthalimide sensitization of the metal emission occurs for the europium complex 5. The cerium(III) and uranyl(VI) compounds 4 and 7 have no solid state emission.
Co-reporter:Daniel L. Reger, Andrea E. Pascui, Mark D. Smith, Julia Jezierska, and Andrew Ozarowski
Inorganic Chemistry 2015 Volume 54(Issue 4) pp:1487-1500
Publication Date(Web):January 20, 2015
DOI:10.1021/ic502485p
The reactions of Cu(ClO4)2 with NaCN and the ditopic ligands m-bis[bis(1-pyrazolyl)methyl]benzene (Lm) or m-bis[bis(3,5-dimethyl-1-pyrazolyl)methyl]benzene (Lm*) yield [Cu2(μ-CN)(μ-Lm)2](ClO4)3 (1) and [Cu2(μ-CN)(μ-Lm*)2](ClO4)3 (3). In both, the cyanide ligand is linearly bridged (μ-1,2) leading to a separation of the two copper(II) ions of ca. 5 Å. The geometry around copper(II) in these complexes is distorted trigonal bipyramidal with the cyanide group in an equatorial position. The reaction of [Cu2(μ-F)(μ-Lm)2](ClO4)3 and (CH3)3SiN3 yields [Cu2(μ-N3)(μ-Lm)2](ClO4)3 (2), where the azide adopts end-on (μ-1,1) coordination with a Cu–N–Cu angle of 138.0° and a distorted square pyramidal geometry about the copper(II) ions. Similar chemistry in the more sterically hindered Lm* system yielded only the coordination polymer [Cu2(μ-Lm*)(μ-N3)2(N3)2]. Attempts to prepare a dinuclear complex with a bridging iodide yield the copper(I) complex [Cu5(μ-I4)(μ-Lm*)2]I3. The complexes 1 and 3 show strong antiferromagnetic coupling, −J = 135 and 161 cm–1, respectively. Electron paramagnetic resonance (EPR) studies coupled with density functional theory (DFT) calculations show that the exchange interaction is transmitted through the dz2 and the bridging ligand s and px orbitals. High field EPR studies confirmed the dz2 ground state of the copper(II) ions. Single-crystal high-field EPR has been able to definitively show that the signs of D and E are positive. The zero-field splitting is dominated by the anisotropic exchange interactions. Complex 2 has −J = 223 cm–1 and DFT calculations indicate a predominantly dx2–y2 ground state.
Co-reporter:Daniel L. Reger, Andrew Leitner, and Mark D. Smith
Crystal Growth & Design 2015 Volume 15(Issue 11) pp:5637-5644
Publication Date(Web):October 12, 2015
DOI:10.1021/acs.cgd.5b01387
The reactions of the lithium salt of (S)-2-(1,8-naphthalimido)-3-hydroxypropanoate (Lser–), an enantiopure carboxylate ligand containing a 1,8-naphthalimide π···π stacking supramolecular tecton and an alcohol functional group, with La(NO3)3, Ce(NO3)3, SmCl3, Eu(NO3)3, Gd(NO3)3, Tb(NO3)3, and Dy(NO3)3 under solvothermal conditions (water/ethanol) produced single crystals (characterized by single-crystal X-ray crystallography) of [La3(Lser)8(OH)(H2O)]·(H2O,EtOH)x (1), [Ce3(Lser)8(OH)(H2O)]·(H2O,EtOH)x (2), [Sm3(Lser)8(OEt)]·(H2O,EtOH)x (3), [Eu3(Lser)8(OEt)]·(H2O,EtOH)x (4), [Gd3(Lser)8(OEt)]·(H2O,EtOH)x (5), [Tb3(Lser)8(OEt)]·(H2O,EtOH)x (6), and [Dy3(Lser)8(OEt)]·(H2O,EtOH)x (7), respectively. Mixed-metal complexes [Ce2.3Tb0.7(Lser)8(OH)]·(H2O,EtOH)x (8), [Gd0.4Tb2.6(Lser)8(OEt)]·(H2O,EtOH)x (9), and [Ce1.4Gd0.3Tb1.3(Lser)8(OH)]·(H2O,EtOH)x (10) were prepared by using two or more types of lanthanides in the solvothermal reactions (additional mixed-metal complexes were prepared and characterized by ICP-MS). Single crystals of compounds 1–10 are isostructural: trinuclear, carboxylate-bonded helicates organized by the non-covalent, π···π stacking interactions of the 1,8-naphthalimide groups into intertwined M helices, with a pitch of 56 Å, that are further arranged into a three-dimensional supramolecular framework by additional π···π stacking interactions. Magnetic measurements of several compounds were as expected for the metal(s) present, indicating no significant interactions between metals within the helicates. The Ce complex 2 showed weak antiferromagnetic ordering below 50 K. All of the complexes, with the exception of 2, showed luminescence based on the 1,8-naphthalimide group. Complex 2 has no emission, and complexes with mixed Ce/Tb ratios showed significant quenching of the naphthalimide-based luminescence, as quantitated with solid-state, absolute quantum yield measurements of these mixed-metal and the pure metal complexes. Lanthanide-based luminescence was only observed for the Eu complex 4.
Co-reporter:Daniel L. Reger, Andrew Leitner, Mark D. Smith
Journal of Molecular Structure 2015 1091() pp: 31-36
Publication Date(Web):
DOI:10.1016/j.molstruc.2015.01.046
Co-reporter:Daniel L. Reger, Andrew Leitner, Perry J. Pellechia, and Mark D. Smith
Inorganic Chemistry 2014 Volume 53(Issue 18) pp:9932-9945
Publication Date(Web):August 26, 2014
DOI:10.1021/ic501581c
The reactions of the potassium salts of the ligands (S)-2-(1,8-naphthalimido)propanoate (KLala), (S)-2-(1,8-naphthalimido)-3-hydroxypropanoate (KLser), and (R)-2-(1,8-naphthalimido)propanoate (KLala*), enantiopure carboxylate ligands containing a 1,8-naphthalimide π···π stacking supramolecular tecton, and, in the case of Lser–, an alcohol functional group with calcium or strontium nitrate under solvothermal conditions produce crystalline [Ca(Lala)2(H2O)]·(H2O) (1); [Ca(Lser)2]·(H2O)2 (2); [Sr(Lala)2(H2O)]·(H2O)3 (3); [Sr(Lala*)2(H2O)]·(H2O)3 (3*); and [Sr(Lser)2(H2O)] (5). Placing 3 under vacuum removes the interstitial waters to produce [Sr(Lala)2(H2O)] (4) in a single-crystal to single-crystal transformation; introduction of water vapor to 4 leads to the reformation of crystalline 3. Each of these new complexes has a solid-state structure based on homochiral rod secondary building unit (SBUs) central cores. Supramolecular π···π stacking interactions between 1,8-naphthalimide rings link adjacent rod SBUs into three-dimensional structures for 1, 3, 4, and 5 and two-dimensional structure for 2. Compounds 1 and 3 have open one-dimensional channels along the crystallographic c axis that are occupied by disordered solvent. For 3, these channels close and open in the reversible single-crystal conversion to 4; the π···π stacking interactions of the naphthalimide rings facilitate this process by rotating and slipping. Infrared spectroscopy demonstrated that the rehydration of 4 with D2O leads to 3d8, and the process of dehydration and rehydration of 3d8 with H2O leads to 3, thus showing exchange of the coordinated water in this process. These forms of 3 and 4 were characterized by 1H, 2H, and 13C solid-state NMR spectroscopy, and thermal and luminescence data are reported on all of the complexes.
Co-reporter:Daniel L. Reger, Andrea E. Pascui, Elizabeth A. Foley, Mark D. Smith, Julia Jezierska, and Andrew Ozarowski
Inorganic Chemistry 2014 Volume 53(Issue 4) pp:1975-1988
Publication Date(Web):January 30, 2014
DOI:10.1021/ic4017905
The reactions of M(ClO4)2·xH2O and the ditopic ligands m-bis[bis(1-pyrazolyl)methyl]benzene (Lm) or m-bis[bis(3,5-dimethyl-1-pyrazolyl)methyl]benzene (Lm*) in the presence of triethylamine lead to the formation of monohydroxide-bridged, dinuclear metallacycles of the formula [M2(μ-OH)(μ-Lm)2](ClO4)3 (M = Fe(II), Co(II), Cu(II)) or [M2(μ-OH)(μ-Lm*)2](ClO4)3 (M = Co(II), Ni(II), Cu(II)). With the exception of the complexes where the ligand is Lm and the metal is copper(II), all of these complexes have distorted trigonal bipyramidal geometry around the metal centers and unusual linear (Lm*) or nearly linear (Lm) M–O–M angles. For the two solvates of [Cu2(μ-OH)(μ-Lm)2](ClO4)3, the Cu–O–Cu angles are significantly bent and the geometry about the metal is distorted square pyramidal. All of the copper(II) complexes have structural distortions expected for the pseudo-Jahn–Teller effect. The two cobalt(II) complexes show moderate antiferromagnetic coupling, −J = 48–56 cm–1, whereas the copper(II) complexes show very strong antiferromagnetic coupling, −J = 555–808 cm–1. The largest coupling is observed for [Cu2(μ-OH)(μ-Lm*)2](ClO4)3, the complex with a Cu–O–Cu angle of 180°, such that the exchange interaction is transmitted through the dz2 and the oxygen s and px orbitals. The interaction decreases, but it is still significant, as the Cu–O–Cu angle decreases and the character of the metal orbital becomes increasingly dx2–y2. These intermediate geometries and magnetic interactions lead to spin Hamiltonian parameters for the copper(II) complexes in the EPR spectra that have large E/D ratios and one g matrix component very close to 2. Density functional theory calculations were performed using the hybrid B3LYP functional in association with the TZVPP basis set, resulting in reasonable agreement with the experiments.
Co-reporter:Daniel L. Reger, Andrea E. Pascui, Perry J. Pellechia, Mark D. Smith, Julia Jezierska, and Andrew Ozarowski
Inorganic Chemistry 2014 Volume 53(Issue 9) pp:4325-4339
Publication Date(Web):April 21, 2014
DOI:10.1021/ic403013d
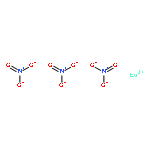
The reactions of M(ClO4)2·xH2O (M = Ni(II) or Cd(II)) and m-bis[bis(1-pyrazolyl)methyl]benzene (Lm) in the presence of triethylamine lead to the formation of hydroxide-bridged cubane compounds of the formula [M4(μ3-OH)4(μ-Lm)2(solvent)4](ClO4)4, where solvent = dimethylformamide, water, acetone. In the solid state the metal centers are in an octahedral coordination environment, two sites are occupied by pyrazolyl nitrogens from Lm, three sites are occupied by bridging hydroxides, and one site contains a weakly coordinated solvent molecule. A series of multinuclear, two-dimensional and variable-temperature NMR experiments showed that the cadmium(II) compound in acetonitrile-d3 has C2 symmetry and undergoes an unusual dynamic process at higher temperatures (ΔGLm‡ = 15.8 ± 0.8 kcal/mol at 25 °C) that equilibrates the pyrazolyl rings, the hydroxide hydrogens, and cadmium(II) centers. The proposed mechanism for this process combines two motions in the semirigid Lm ligand termed the “Columbia Twist and Flip:” twisting of the pyrazolyl rings along the Cpz–Cmethine bond and 180° ring flip of the phenylene spacer along the CPh–Cmethine bond. This dynamic process was also followed using the spin saturation method, as was the exchange of the hydroxide hydrogens with the trace water present in acetonitrile-d3. The nickel(II) analogue, as shown by magnetic susceptibility and electron paramagnetic resonance measurements, has an S = 4 ground state, and the nickel(II) centers are ferromagnetically coupled with strongly nonaxial zero-field splitting parameters. Depending on the Ni–O–Ni angles two types of interactions are observed: J1 = 9.1 cm–1 (97.9 to 99.5°) and J2 = 2.1 cm–1 (from 100.3 to 101.5°). “Broken symmetry” density functional theory calculations performed on a model of the nickel(II) compound support these observations.
Co-reporter:Andrew Ozarowski
Inorganic Chemistry 2013 Volume 52(Issue 21) pp:12741-12748
Publication Date(Web):October 11, 2013
DOI:10.1021/ic402016m
The nuclear magnetic resonance (NMR) spectra of single-anion bridged, dinuclear copper(II) metallacycles [Cu2(μ-X)(μ-L)2](A)3 (Lm = m-bis[bis(1-pyrazolyl)methyl]benzene: X = F–, A = BF4–; X = Cl–, OH–, A = ClO4–; Lm* = m-bis[bis(3,5-dimethyl-1-pyrazolyl)methyl]benzene: X = CN–, F–, Cl–, OH–, Br–, A = ClO4–) have relatively sharp 1H and 13C NMR resonances with small hyperfine shifts due to the strong antiferromagnetic superexchange interactions between the two S = 1/2 metal centers. The complete assignments of these spectra, except X = CN–, have been made through a series of NMR experiments: 1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC, T1 measurements and variable-temperature 1H NMR. The T1 measurements accurately determine the Cu···H distances in these molecules. In solution, the temperature dependence of the chemical shifts correlate with the population of the paramagnetic triplet (S = 1) and diamagnetic singlet (S = 0) states. This correlation allows the determination of antiferromagnetic exchange coupling constants, −J (Ĥ = −JŜ1Ŝ2), in solution for the Lm compounds 338(F–), 460(Cl–), 542(OH–), for the Lm* compounds 128(CN–), 329(F–), 717(Cl–), 823(OH–), and 944(Br–) cm–1, respectively. These values are of similar magnitudes to those previously measured in the solid state (−Jsolid = 365, 536, 555, 160, 340, 720, 808, and 945 cm–1, respectively). This method of using NMR to determine −J values in solution is an accurate and convenient method for complexes with strong antiferromagnetic superexchange interactions. In addition, the similarity between the solution and solid-state −J values of these complexes confirms the information gained from the T1 measurements: the structures are similar in the two states.
Co-reporter:Daniel L. Reger, Andrew Leitner, and Mark D. Smith, T. Thao Tran and P. Shiv Halasyamani
Inorganic Chemistry 2013 Volume 52(Issue 17) pp:10041-10051
Publication Date(Web):August 14, 2013
DOI:10.1021/ic401327h
The reactions of (S)-2-(1,8-naphthalimido)propanoic acid (HLala) and (S)-2-(1,8-naphthalimido)-3-hydroxypropanoic acid (HLser), protonated forms of ligands that contain a carboxylate donor group, an enantiopure chiral center, and a 1,8-naphthalimide π···π stacking supramolecular tecton and in the case of HLser an alcohol functional group, with the appropriate alkali metal hydroxide followed by a variety of crystallization methods leads to the formation of crystalline K(Lala)(MeOH) (1), K(Lala)(H2O) (2), Na(Lala)(H2O) (3), KLser (4), CsLser (5), and CsLala (6). Each of these new complexes has a solid state structure based on six-coordinate metals linked into homochiral helical rod secondary building unit (SBU) central cores. In addition to the bonding of the carboxylate and solvent (in the case of Lser the ligand alcohol) to the metals, both oxygens on the 1,8-naphthalimide act as donor groups. One naphthalimide oxygen bonds to the same helical rod SBU as the carboxylate group of that ligand forming a chelate ring. The other naphthalimide oxygen bonds to adjacent SBUs. In complexes 1–3, this inter-rod link has a square arrangement bonding four other rods forming a three-dimensional enantiopure metal–organic framework (MOF) structure, whereas in 4–6 this link has a linear arrangement bonding two other rods forming a two-dimensional, sheet structure. In the latter case, the third dimension is supported exclusively by interdigitated π···π stacking interactions of the naphthalimide supramolecular tecton, forming enantiopure supramolecular MOF solids. Compounds 1–3 lose the coordinated solvent when heating above 100 °C. For 1, the polycrystalline powder reverts to 1 only by recrystallization from methanol, whereas compounds 2 and 3 undergo gas/solid, single-crystal to single-crystal transformations to form dehydrated compounds 2* and 3*, and rehydration occurs when crystals of these new complexes are left out in air. The reversible single-crystal to single-crystal transformation of 2 involves the dissociation/coordination of a terminal water ligand, but the case of 3 is remarkable considering that the water that is lost is the only bridging ligand between the metals in the helical rod SBU and a carboxylate oxygen that is a terminal ligand in 3 moves into a bridging position in 3* to maintain the homochiral helical rods. Both 2* and 3* contain five-coordinate metals. There are no coordinated solvents in compounds 4–6, in two cases by designed ligand modification, which allows them to have high thermal stability. Compounds 1–3 did not exhibit observable Second Harmonic Generation (SHG) efficiency at an incident wavelength of 1064 nm, but compounds 4–6 did exhibit modest SHG efficiency for MOF-like compounds in the range of 30 × α-SiO2.
Co-reporter:Daniel L. Reger, Andrea E. Pascui, Perry J. Pellechia, and Mark D. Smith
Inorganic Chemistry 2013 Volume 52(Issue 19) pp:11638-11649
Publication Date(Web):September 9, 2013
DOI:10.1021/ic402073d
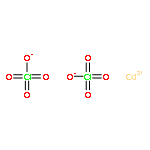
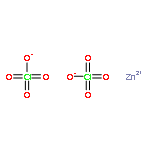
The reactions of M(ClO4)2·6H2O [M = Zn(II), Cd(II)] and the ligands m-bis[bis(1-pyrazolyl)methyl]benzene, Lm, or m-bis[bis(3,5-dimethyl-1-pyrazolyl)methyl]benzene, Lm*, in the presence of a base yield the hydroxide bridged dinuclear metallacycles [M2(μ-OH)(μ-L)2](ClO4)3, L = Lm, M = Zn(II) (1); L = Lm*, M = Zn(II) (2), Cd(II) (3). In the solid state, the coordination environment of the metals is distorted trigonal bipyramidal with the bridging hydroxide in an equatorial position and M-O-M angles greater than 161°. The observation of two equal intensity resonances for each type of pyrazolyl-ring hydrogen in the 1H NMR for all three complexes coupled with the determination of the hydrodynamic radius based on the diffusion coefficient of 1 that matches that observed in the crystal structure, demonstrate this structure is retained in solution. Additional proof of the dinuclear structures in solution is given by the 113Cd NMR spectrum of [Cd2(μ-OH)(μ-Lm*)2](ClO4)3 showing 111/113Cd satellites (J111Cd-113Cd = 173 Hz). Complex 1 is dynamic in solution, with the resonances for each type of pyrazolyl-ring hydrogen broadening and averaging at higher temperatures. Detailed variable temperature studies show that ΔGpz⧧ = 15.2(±0.2) kcal/mol, ΔHpz⧧ = 6.6(±0.1) kcal/mol, and ΔSpz⧧ = −28.8(±0.4) cal/mol·K at 25 °C for this process. The same ΔG⧧ value for the dynamic process was also determined by saturation transfer experiments. The most plausible mechanism for this dynamic process, which exchanges the axial and equatorial positions of the pyrazolyl rings in the trigonal bipyramidal arrangement, involves Berry pseudorotation at both metal sites using the bridging oxygen atom as the pivot ligand, coupled with the ring flip of the ligand’s phenylene spacer by 180°, a rearrangement process we termed the “Columbia Twist and Flip”. This process was shown to be influenced by trace amounts of water in the solvent, with a linear relationship between the water concentration and ΔGpz⧧; increasing the water concentration lowers ΔGpz⧧. Spin saturation transfer experiments demonstrated the exchange of the hydrogens between the water in the solvent and the bridging hydroxide group, with ΔGOH⧧ = 16.8(±0.2) kcal/mol at 25 °C, a value larger than the barrier of ΔGpz⧧ = 15.2(±0.2) kcal/mol for the “Columbia Twist and Flip”. Compounds 2 and 3 do not show dynamic behavior involving the pyrazolyl-rings in solution because of steric crowding caused by the methyl group substitution, but do show the exchange between the water in the solvent and the bridging hydroxide group.
Co-reporter:Daniel L. Reger ; Andrea E. Pascui ; Mark D. Smith ; Julia Jezierska ;Andrew Ozarowski
Inorganic Chemistry 2012 Volume 51(Issue 15) pp:7966-7968
Publication Date(Web):July 26, 2012
DOI:10.1021/ic301321r
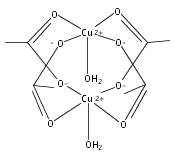
Centrosymmetric [Cu2(μ-X)(μ-Lm*)2](ClO4)3 (X = F–, Cl–, Br–, OH–, Lm* = m-bis[bis(3,5-dimethyl-1-pyrazolyl)methyl]benzene)], the first example of a series of bimetallic copper(II) complexes linked by a linearly bridging mononuclear anion, have been prepared and structurally characterized. Very strong antiferromagnetic exchange coupling between the copper(II) ions increases along the series F– < Cl– < OH– < Br–, where −J = 340, 720, 808, and 945 cm–1. DFT calculations explain this trend by an increase in the energy along this series of the antibonding antisymmetric combination of the p orbital of the bridging anion interacting with the copper(II) dz2 orbital.
Co-reporter:Daniel L. Reger, Andrea E. Pascui, Mark D. Smith, Julia Jezierska, and Andrew Ozarowski
Inorganic Chemistry 2012 Volume 51(Issue 21) pp:11820-11836
Publication Date(Web):October 9, 2012
DOI:10.1021/ic301757g
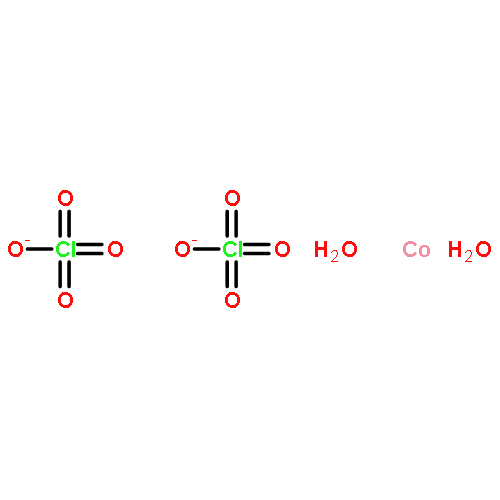
The reaction of M(BF4)2·xH2O, where M is Fe(II), Co(II), Ni(II), Cu(II), Zn(II), and Cd(II), with the new ditopic ligand m-bis[bis(3,5-dimethyl-1-pyrazolyl)methyl]benzene (Lm*) leads to the formation of monofluoride-bridged dinuclear metallacycles of the formula [M2(μ-F)(μ-Lm*)2](BF4)3. The analogous manganese(II) species, [Mn2(μ-F)(μ-Lm*)2](ClO4)3, was isolated starting with Mn(ClO4)2·6H2O using NaBF4 as the source of the bridging fluoride. In all of these complexes, the geometry around the metal centers is trigonal bipyramidal, and the fluoride bridges are linear. The 1H, 13C, and 19F NMR spectra of the zinc(II) and cadmium(II) compounds and the 113Cd NMR of the cadmium(II) compound indicate that the metallacycles retain their structure in acetonitrile and acetone solution. The compounds with M = Mn(II), Fe(II), Co(II), Ni(II), and Cu(II) are antiferromagnetically coupled, although the magnitude of the coupling increases dramatically with the metal as one moves to the right across the periodic table: Mn(II) (−6.7 cm–1) < Fe(II) (−16.3 cm–1) < Co(II) (−24.1 cm–1) < Ni(II) (−39.0 cm–1) ≪ Cu(II) (−322 cm–1). High-field EPR spectra of the copper(II) complexes were interpreted using the coupled-spin Hamiltonian with gx = 2.150, gy = 2.329, gz = 2.010, D = 0.173 cm–1, and E = 0.089 cm–1. Interpretation of the EPR spectra of the iron(II) and manganese(II) complexes required the spin Hamiltonian using the noncoupled spin operators of two metal ions. The values gx = 2.26, gy = 2.29, gz = 1.99, J = −16.0 cm–1, D1 = −9.89 cm–1, and D12 = −0.065 cm–1 were obtained for the iron(II) complex and gx = gy = gz = 2.00, D1 = −0.3254 cm–1, E1 = −0.0153, J = −6.7 cm–1, and D12 = 0.0302 cm–1 were found for the manganese(II) complex. Density functional theory (DFT) calculations of the exchange integrals and the zero-field splitting on manganese(II) and iron(II) ions were performed using the hybrid B3LYP functional in association with the TZVPP basis set, resulting in reasonable agreement with experiment.
Co-reporter:Daniel L. Reger ; Andrew P. Leitner ;Mark D. Smith
Inorganic Chemistry 2012 Volume 51(Issue 19) pp:10071-10073
Publication Date(Web):September 11, 2012
DOI:10.1021/ic301228j
Two trifunctional ligands built from enantiopure amino acids and containing a 1,8-naphthalimide group have been used to prepare two new complexes of potassium that have extended structures based on homochiral-rod secondary building units. One structure is a three-dimensional metal–organic framework (MOF), while the other is a two-dimensional solid that is organized into a supramolecular MOF by strong π···π-stacking interactions of the naphthalimide groups in the third dimension.
Co-reporter:Daniel L. Reger;Andrea E. Pascui ;Mark D. Smith
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 29) pp:4593-4604
Publication Date(Web):
DOI:10.1002/ejic.201200118
Abstract
The equal molar reaction of Cu(BF4)2·3H2O with the ditopic, bis(pyrazolyl)methane ligand p-[CH(pz)2]2C6H4 (Lp) where pz is a pyrazolyl ring, in methanol produces [Cu(μ-Lp)(CH3OH)](BF4)2·(CH3OH)0.62 (1) and a similar reaction in water/methanol yields [Cu(μ-Lp)(CH3OH)]2(SiF6)(BF4)2·(CH3OH)2 (2). The water/ethanol solvent system yields both [Cu2(μ-Lp)(H2O)6](SiF6)2·(H2O)4 (3) and [Cu(μ-Lp)(H2O)](BF4)2·(CH3CH2OH)2 (4), and if the ratio of Cu(BF4)2·3H2O and Lp is changed to 2:1, only 3 forms. Recrystallization of the solid formed from the reaction of THF solutions of Cu(BF4)2·3H2O with Lp from DMSO yields [Cu2(μ-Lp)(DMSO)6](BF4)4·(DMSO)2·C6H6·(H2O)0.5 (5) and a recrystallization of compound 1 from DMSO yields [Cu2(μ-Lp)(DMSO)6](BF4)4·(DMSO)2·C6H6 (6). Complexes 1, 2, and 4 are coordination polymers in which the N4O-coordinated, square-pyramidal copper(II) ions are oriented in an anti conformation withrespect to the phenylene spacer of Lp. Complexes 3, 5 and 6 are dinuclear with N2O3-coordinated, square-pyramidal copper(II) ions again oriented in an anti configuration with respect to the bridging Lp. The square pyramidal geometries show axial elongation, a result of pseudo Jahn–Teller electronic effects. In the cases of the coordination polymers, the axially coordinated solvent molecule is lost at low temperatures upon heating of the crystals. The three compounds in each structural type have nearly the same overall configurations. Cooperative effects of O–H···F, O–H···O and C–H···F hydrogen bonding interactions organize the supramolecular structures and influence the crystal packing of complexes 1–4. The C–H···F interactions have metrics that indicate they are unusually strong, due to the enhanced polarization of the C–H bond and charge assistance from the anionic fluorine atom.






















![2-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)benzoic acid](http://img.cochemist.com/ccimg/56900/56813-55-9.png)
![2-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)benzoic acid](http://img.cochemist.com/ccimg/56900/56813-55-9_b.png)





![1H-Benz[de]isoquinoline-2(3H)-aceticacid, 1,3-dioxo-](/data/chemimg/1131900/51411-04-2.png)
![1H-Benz[de]isoquinoline-2(3H)-aceticacid, 1,3-dioxo-](/data/chemimg/1131900/51411-04-2_b.png)

![4-(1,3-Dioxo-1H,3H-benzo[de]isoquinolin-2-yl)-benzoic acid](http://img.cochemist.com/ccimg/136000/135980-47-1.png)
![4-(1,3-Dioxo-1H,3H-benzo[de]isoquinolin-2-yl)-benzoic acid](http://img.cochemist.com/ccimg/136000/135980-47-1_b.png)
![4-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)butanoic acid](/data/chemimg/986000/88909-96-0.png)
![4-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)butanoic acid](/data/chemimg/986000/88909-96-0_b.png)