Co-reporter:Miho Otsuka, Yukie Mori, Keiko Takano
Chemical Physics Letters 2016 Volume 647() pp:95-102
Publication Date(Web):March 2016
DOI:10.1016/j.cplett.2016.01.028
Highlights
- •
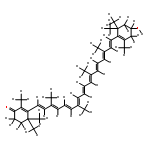
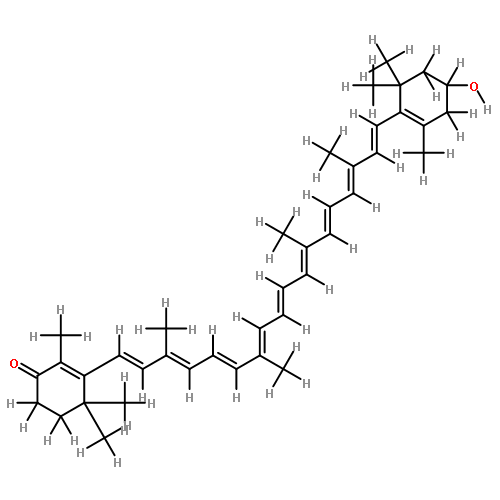
Singlet excited states of a carbonyl carotenoid were studied by RASPT2 calculations.
- •
The S1 state is 2Ag− and it gains a partial ICT character at twisted geometries.
- •
The origin of ICT character is mixing with 1Bu+ having a large dipole moment.
- •
Hydrogen-bonding interactions with the protein enhance the ICT character.
Co-reporter:Miho Otsuka, Noriko Tsuchida, Yousuke Ikeda, Natacha Lambert, Rina Nakamura, Yuichiro Mutoh, Youichi Ishii, and Keiko Takano
Organometallics 2015 Volume 34(Issue 16) pp:3934-3943
Publication Date(Web):August 3, 2015
DOI:10.1021/om501280c
DFT calculations on the transformation reaction of the internal alkynes [CpM(PhC≡CC6H4R-p)(dppe)]+ (M = Fe, Os; R = OMe, CO2Et, Cl) to the corresponding vinylidenes were carried out. It was found that the isomerization of all complexes of group 8 elements studied in the present work, as well as the Ru complex that had already been reported in our previous study, proceeds via a direct 1,2-aryl shift. For the Fe complex, two types of direct 1,2-aryl shifts (paths 1 and 2), which depend on the orientation of the alkyne/vinylidene parts, were found, and the activation free energy of path 2 is smaller than that of path 1. As for the Os complex, path 1 and another direct 1,2-shift, path 3, were obtained, and path 3 has smaller activation free energy, which is the case for the Ru complex. Therefore, the isomerization reaction of internal alkynes to vinylidenes proceeds through path 2 for the Fe complexes and path 3 for the Ru and Os complexes. The 1,2-migration reactions via path 2 for the Fe complex and path 3 for the Os complex were found to be nucleophilic, which is based on an orbital interaction corresponding to an electron transfer from a carbon on the migrating group to the atom being migrated, as well as for the Ru complexes. To evaluate the stability of the alkyne and vinylidene complexes, orbital interaction energies between an organometallic complex part and an internal alkyne or a vinylidene moiety were calculated by natural bond orbital (NBO) analysis. It was revealed that the Os complex has the strongest interaction, followed by the Ru and Fe complexes. Namely, both the internal alkyne complexes and the vinylidene complexes are more stabilized in a heavier metal complex. The activation free energy for migration of the aryl or phenyl group is actually the lowest for the Fe complex among the three metals. These findings contribute to the development of the synthetic strategy of vinylidenes from internal alkynes.
Co-reporter:Yuka Koyama, Kaori Ueno-Noto, Keiko Takano
Computational Biology and Chemistry 2014 Volume 49() pp:36-44
Publication Date(Web):April 2014
DOI:10.1016/j.compbiolchem.2014.01.013
•Affinities of HIV-1 antibody 2G12 with monosaccharides were examined.•QM methods with explicit and implicit water models were applied.•d-Fructose's higher binding affinity over d-mannose was clarified.•Stronger binding of d-fructose over d-mannose was due to the solvation effects.•Significant pair interactions among d-fructose, amino acids, and waters were found.In order to develop potential ligands to HIV-1 antibody 2G12 toward HIV-1 vaccine, binding mechanisms of the antibody 2G12 with the glycan ligand of d-mannose and d-fructose were theoretically examined. d-Fructose, whose molecular structure is slightly different from d-mannose, has experimentally shown to have stronger binding affinity to the antibody than that of d-mannose. To clarify the nature of d-fructose's higher binding affinity over d-mannose, we studied interaction between the monosaccharides and the antibody using ab initio fragment molecular orbital (FMO) method considering solvation effect as implicit model (FMO-PCM) as well as explicit water model. The calculated binding free energies of the glycans were qualitatively well consistent with the experimentally reported order of their affinities with the antibody 2G12. In addition, the FMO-PCM calculation elucidated the advantages of d-fructose over d-mannose in the solvation energy as well as the entropic contribution term obtained by MD simulations. The effects of explicit water molecules observed in the X-ray crystal structure were also scrutinized by means of FMO methods. Significant pair interaction energies among d-fructose, amino acids, and water molecules were uncovered, which indicated contributions from the water molecules to the strong binding ability of d-fructose to the antibody 2G12. These FMO calculation results of explicit water model as well as implicit water model indicated that the strong binding of d-fructose over d-mannose was due to the solvation effects on the d-fructose interaction energy.
Co-reporter:Yuka Koyama, Kaori Ueno-Noto, Keiko Takano
Chemical Physics Letters 2013 Volume 578() pp:144-149
Publication Date(Web):18 July 2013
DOI:10.1016/j.cplett.2013.06.009
•We assess glycan moieties’ role in the interaction dynamically and energetically.•Man D1 is the crucial glycan moiety in the ligand recognition.•The notable functions of Man 4 and Man 4′ are pointed out.•The significance of the branched structure is theoretically indicated.In HIV-1 infection, human antibody 2G12 is capable of recognizing the high-mannose glycans on the HIV-1 surface glycoprotein, gp120. To investigate the ligand binding mechanisms of antibody 2G12 with glycans aiming for the contribution to the medications, we carried out classical molecular dynamics (MD) simulations and ab initio fragment molecular orbital (FMO) calculations on the antibody 2G12 complex with its high-mannose ligand. We found that Mannose D1 of the ligand had the largest binding affinity with the antibody, which was well consistent with experimental reports. Furthermore, significant roles of Mannose 4 and 4′ in the ligand binding were theoretically indicated.
Co-reporter:Miho Otsuka ; Noriko Tsuchida ; Yousuke Ikeda ; Yusuke Kimura ; Yuichiro Mutoh ; Youichi Ishii
Journal of the American Chemical Society 2012 Volume 134(Issue 42) pp:17746-17756
Publication Date(Web):September 20, 2012
DOI:10.1021/ja308018b
Internal alkyne-to-vinylidene isomerization in the Ru complexes ([CpRu(η2-PhC≡CC6H4R-p)(dppe)]+ (Cp = η5-C5H5; dppe = Ph2PCH2CH2PPh2; R = OMe, Cl, CO2Et)) has been investigated using a combination of quantum mechanics and molecular mechanics methods (QM/MM), such as ONIOM(B3PW91:UFF), and density functional theory (DFT) calculations. Three kinds of model systems (I–III), each having a different QM region for the ONIOM method, revealed that considering both the quantum effect of the substituent of the aryl group in the η2-alkyne ligand and that of the phenyl groups in the dppe ligand is essential for a correct understanding of this reaction. Several plausible mechanisms have been analyzed by using DFT calculations with the B3PW91 functional. It was found that the isomerization of three complexes (R = OMe, CO2Et, and Cl) proceeds via a direct 1,2-shift in all cases. The most favorable process in energy was path 3, which involves the orientation change of the alkyne ligand in the transition state. The activation energies were calculated to be 13.7, 15.0, and 16.4 kcal/mol, respectively, for the three complexes. Donor–acceptor analysis demonstrated that the aryl 1,2-shift is a nucleophilic reaction. Furthermore, our calculation results indicated that an electron-donating substituent on the aryl group stabilizes the positive charge on the accepting carbon rather than that on the migrating aryl group itself at the transition state. Therefore, unlike the general nucleophilic reaction, the less-electron-donating aryl group has an advantage in the migration.
Co-reporter:Noriko Tsuchida, Miwa Isoi, Hiroshi Nakazawa, Keiko Takano
Journal of Organometallic Chemistry 2012 697(1) pp: 41-50
Publication Date(Web):
DOI:10.1016/j.jorganchem.2011.10.014
Co-reporter:Miho Otsuka, Hirotoshi Mori, Hitomi Kikuchi, Keiko Takano
Computational and Theoretical Chemistry 2011 Volume 973(1–3) pp:69-75
Publication Date(Web):15 October 2011
DOI:10.1016/j.comptc.2011.07.002
Structures, chemical bonding nature, and vibrational properties of polyiodide anion species (In-; n = 3, 5, 7) are investigated by means of density functional theory calculations (B3PW91/MCPtzp+). All polyiodide anion species are predicted to have single or branched chain structures. Natural bond orbital (NBO) analyses of In- (n = 5, 7) isomers clearly show that the polyiodide species consists of I2 and linear I3- units. A comparison of theoretically predicted Raman spectra with experimental reports revealed the existence of In- (n = 5, 7) isomers with three-center four-electron (3c–4e) bonds.Graphical abstractHighlights► Polyiodide anion cluster. ► Cluster structures. ► Chemical bonding nature. ► 3c–4e bonds. ► Raman spectra.
Co-reporter:Megumi Kayanuma, Haruko Hosoi, Akiko Furuya, Yuichi Masuda, Keiko Takano
Chemical Physics Letters 2010 Volume 494(4–6) pp:139-143
Publication Date(Web):19 July 2010
DOI:10.1016/j.cplett.2010.05.082
Abstract
A theoretical study of 1,3-dinitrobenzene (DNB) radical anion, which exhibits an intramolecular electron transfer reaction in solution, is reported. The geometries of 1,3-DNB and 1,4-DNB radical anions have been optimized as isolated species to reveal their intrinsic properties by using CASSCF theory with the aug-cc-pVDZ basis set. Single-point energy calculations have been carried out at the CASPT2 and CCSD(T) levels. It is demonstrated that the 1,3-DNB radical anion is slightly more stable as an electronically localized structure in the absence of solvent, whereas the 1,4-DNB radical anion has a delocalized structure.



![1-[4-(3-METHYL-BUTOXY)-PHENYL]-ETHANONE](http://img.cochemist.com/ccimg/30300/30237-26-4.png)
![1-[4-(3-METHYL-BUTOXY)-PHENYL]-ETHANONE](http://img.cochemist.com/ccimg/30300/30237-26-4_b.png)