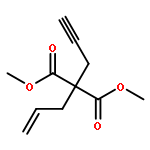
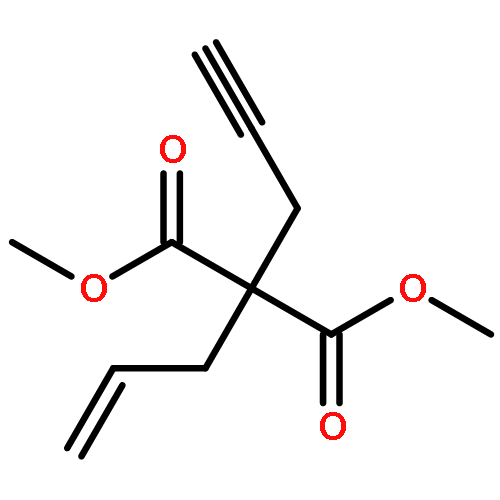
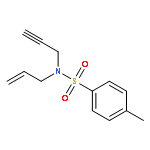
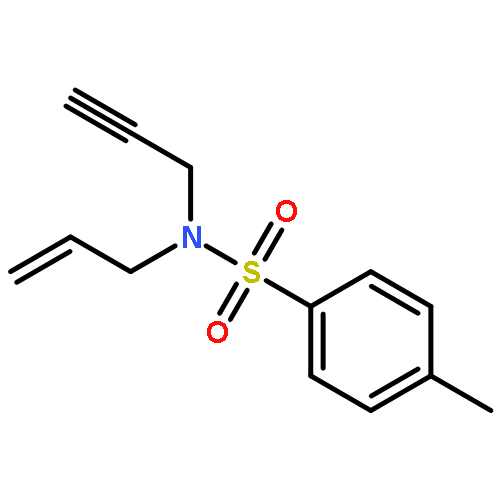
Co-reporter:Matthew J. Barrett;Ghulam F. Khan;Paul W. Davies
Chemical Communications 2017 vol. 53(Issue 42) pp:5733-5736
Publication Date(Web):2017/05/23
DOI:10.1039/C7CC02244A
Alkynyl sulfoxides are shown to act as α-sulfinyl metallocarbene synthons under oxidative gold catalysis, enabling reactions that are not available from diazo-precursors. This strategy is exemplified in the synthesis of fused α-sulfinyl cyclopropanes.
Co-reporter:Matthew N. Pennell;Michael P. Kyle;Samantha M. Gibson;Louise Male;Peter G. Turner;Tom D. Sheppard
Advanced Synthesis & Catalysis 2016 Volume 358( Issue 9) pp:1519-1525
Publication Date(Web):
DOI:10.1002/adsc.201600101
Co-reporter:Matthew J. Barrett, Paul W. Davies and Richard S. Grainger
Organic & Biomolecular Chemistry 2015 vol. 13(Issue 32) pp:8676-8686
Publication Date(Web):07 Jul 2015
DOI:10.1039/C5OB01241D
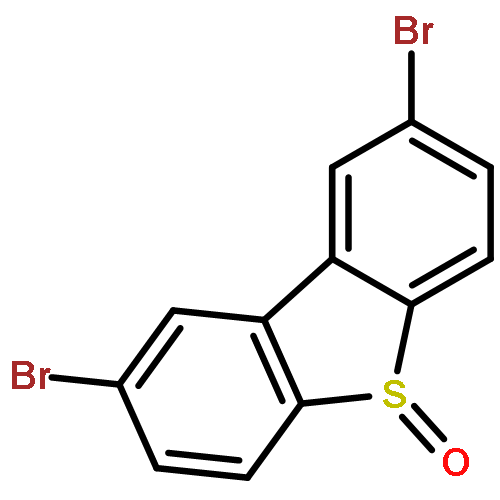
A protocol has been developed for direct Csp3–Csp2 bond formation at the 4- and 6-positions of dibenzothiophenes using a gold(I) catalyst with terminal alkynes and dibenzothiophene-S-oxides. The sulfoxide acts as a traceless directing group to avoid the need to prefunctionalise at carbon. The iterative use of this protocol is possible and has been employed in the preparation of novel macrocyclic structures. In addition, a cascade process shows how oxyarylations can be combined with other processes resulting in complex, highly efficient transformations.
Co-reporter:Richard S. Grainger, Kevin R. Munro
Tetrahedron 2015 Volume 71(Issue 41) pp:7795-7835
Publication Date(Web):14 October 2015
DOI:10.1016/j.tet.2015.06.053
Figure optionsDownload full-size imageDownload as PowerPoint slide
Co-reporter:Dr. Marie Betou;Dr. Louise Male; Jonathan W. Steed;Dr. Richard S. Grainger
Chemistry - A European Journal 2014 Volume 20( Issue 21) pp:6505-6517
Publication Date(Web):
DOI:10.1002/chem.201304982
Abstract
In an approach to the biologically important 6-azabicyclo[3.2.1]octane ring system, the scope of the tandem 4-exo-trig carbamoyl radical cyclization—dithiocarbamate group transfer reaction to ring-fused β-lactams is evaluated. β-Lactams fused to five-, six-, and seven-membered rings are prepared in good to excellent yield, and with moderate to complete control at the newly formed dithiocarbamate stereocentre. No cyclization is observed with an additional methyl substituent on the terminus of the double bond. Elimination of the dithiocarbamate group gives α,β- or β,γ-unsaturated lactams depending on both the methodology employed (base-mediated or thermal) and the nature of the carbocycle fused to the β-lactam. Fused β-lactam diols, obtained from catalytic OsO4-mediated dihydroxylation of α,β-unsaturated β-lactams, undergo semipinacol rearrangement via the corresponding cyclic sulfite or phosphorane to give keto-bridged bicyclic amides by exclusive N-acyl group migration. A monocyclic β-lactam diol undergoes Appel reaction at a primary alcohol in preference to semipinacol rearrangement. Preliminary investigations into the chemo- and stereoselective manipulation of the two carbonyl groups present in a representative 7,8-dioxo-6-azabicyclo[3.2.1]octane rearrangement product are also reported.
Co-reporter:Carlotta Figliola, Louise Male, Peter N. Horton, Mateusz B. Pitak, Simon J. Coles, Sarah L. Horswell, and Richard S. Grainger
Organometallics 2014 Volume 33(Issue 17) pp:4449-4460
Publication Date(Web):August 18, 2014
DOI:10.1021/om500683p
Eight dithiolato-, diselenolato-, and mixed S,Se-Fe2(CO)6 complexes based on peri-substituted naphthalene and phenanthrene dichalcogenides are prepared by oxidative insertion of Fe3(CO)12 into the dichalcogen bonds of 2,7-dimethoxynaphtho[1,8-cd][1,2]dithiole, three naphtho[1,8-cd][1,2]diselenoles, two naphtho[1,8-cd][1,2]thiaselenoles, phenanthro[1,10-cd][1,2]dithiole, and phenanthro[1,10-cd][1,2]diselenole. Complexes are characterized by 1H, 13C NMR, UV/vis, and IR spectroscopy and by X-ray analysis. The effect of replacing sulfur with selenium, incorporating electron-donating groups (2,7-di-tert-butyl, 2,7-dimethoxy) on the naphthalene ring system, and changing the degree of conjugation in the aromatic backbone (naphthalene vs phenanthrene) on the reduction potential is evaluated by cyclic voltammetry. The electrocatalytic activity of these [FeFe]-hydrogenase synthetic mimics for proton reduction in the presence of increasing concentrations of p-TsOH is investigated. Diselenolate-based [FeFe]-complexes show enhanced catalytic activity for proton reduction compared with their sulfur congeners.
Co-reporter:Thomas B. Parsons, Neil Spencer, Chi W. Tsang and Richard S. Grainger
Chemical Communications 2013 vol. 49(Issue 23) pp:2296-2298
Publication Date(Web):01 Feb 2013
DOI:10.1039/C3CC39062D
The first synthesis of kottamide E, a marine natural product containing a 5,6-dibromoindole linked via a (Z)-enamide to an unusual 1,2-dithiolane-containing amino acid, is reported.
Co-reporter:Kevin R. Munro, Louise Male, Neil Spencer and Richard S. Grainger
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 39) pp:6856-6862
Publication Date(Web):30 Aug 2013
DOI:10.1039/C3OB41390J
α-Hydroxyalkylidene carbenes, generated from thermolysis of α,β-epoxy-N-aziridinylimines, undergo diastereotopic group selective 1,5 C–H insertion reactions on 2,4-dimethyl-8-oxabicyclo[3.2.1]oct-6-ene ring systems. Protection of a tertiary alcohol at C-3 of the bridged oxabicycle as a trimethylsilyl ether reverses the sense of diastereoselectivity. 1,5 C–H insertion into a methine adjacent to an OBn group, 1,5 O–R insertion into a tertiary alcohol (R = H) or silylether (R = TMS) at C-3 to form spirocyclic dihydrofurans, 1,2-rearrangement to an alkyne and fragmentation to a ketone are competing major pathways for 2-benzyloxy-substituted 8-oxabicyclo[3.2.1]oct-6-ene systems. Dihydrofuran formation is shown to be a result of substitution on the oxabicyclic ring system through comparison with other methods of alkylidene carbene formation.
Co-reporter:Richard S. Grainger, Marie Betou, Louise Male, Mateusz B. Pitak, and Simon J. Coles
Organic Letters 2012 Volume 14(Issue 9) pp:2234-2237
Publication Date(Web):April 18, 2012
DOI:10.1021/ol300605y
The 6-azabicyclo[3.2.1]octane ring system, prevalent in a range of biologically active molecules, is prepared through a novel semipinacol rearrangement utilizing a cyclic phosphorane or sulfite intermediate. The rearrangement proceeds with exclusive N-acyl group migration of a β-lactam ring and results in carbonyl functionality at the 7- and bridging 8-position of the bicycle. Precursor ring-fused β-lactam diols are prepared through a sequence of 4-exo trig carbamoyl radical cyclization, regioselective dithiocarbamate group elimination, and dihydroxylation.
Co-reporter:Benedicte Defaut, Thomas B. Parsons, Neil Spencer, Louise Male, Benson M. Kariuki and Richard S. Grainger
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 25) pp:4926-4932
Publication Date(Web):21 May 2012
DOI:10.1039/C2OB25384D
A concise, stereoselective synthesis of the trans-hydrindane core of the marine natural product dictyoxetane is reported, starting from a Robinson annelation derived bicyclic enone. A phosphorane-mediated, pinacol-like rearrangement of a cis-diol, via a formal 1,2-hydride shift, is used to establish the requisite trans ring junction. 31P NMR supports the formation of the intermediate phosphorane, generated in situ from the reaction of a diol with Ph3PCl2.
Co-reporter:Claire McMaster, Robert N. Bream and Richard S. Grainger
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 24) pp:4752-4758
Publication Date(Web):15 May 2012
DOI:10.1039/C2OB25434D
Reductive desulfurisation of dithiocarbamates is conveniently achieved using H3PO2–Et3N–ACCN in refluxing dioxane. Fused and spirocyclic β-lactams, prepared through 4-exo trig carbamoyl radical cyclisation–dithiocarbamate group transfer reactions, are reduced without fragmentation of the strained 4-membered ring. Diethyl tetraacetyl-D-glucopyranosyl dithiocarbamate is selectively reduced with or without acyloxy group migration depending on reaction conditions and choice of reductant. Deuterium incorporation from D3PO2–Et3N is observed for a system involving a nucleophilic radical intermediate, but not in the case of the electrophilic radical obtained through acyloxy group migration on a glucose derivative.
Co-reporter:Richard S. Grainger ; Bhaven Patel ; Benson M. Kariuki ; Louise Male ;Neil Spencer
Journal of the American Chemical Society 2011 Volume 133(Issue 15) pp:5843-5852
Publication Date(Web):March 28, 2011
DOI:10.1021/ja108865w
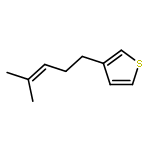
Three peri-substituted trisulfide-2-oxides are prepared by treatment of 1,8-naphthalene dithiols with thionyl chloride and pyridine. The 1,2,3-trithiane-2-oxide ring adopts a sofa conformation in the solid state, with a pseudoaxial oxygen and evidence of ring strain (peri-interaction). Heating the trisulfide-2-oxides in the presence of a diene results in formal sulfur monoxide (SO) transfer to form unsaturated cyclic sulfoxides, along with a recyclable 1,8-naphthalene disulfide. The presence of o-methoxy or o-tert-butyl substituents on the naphthalene ring lowers the temperature and increases the rate at which SO transfer occurs. Trapping experiments and kinetic studies are consistent with the generation of triplet SO, followed by in situ trapping by diene. Transfer of SO also occurs upon irradiation at room temperature, but yields of sulfoxide are lower. Dehydration of the sulfoxides under Pummerer conditions gives thiophenes, including the naturally occurring thioperillene. Two dienes form thiophenes directly under the SO transfer conditions. The methodology is applied in a formal synthesis of the antiplatelet medication Plavix.
Co-reporter:Bhaven Patel, Julie Carlisle, Steven E. Bottle, Graeme R. Hanson, Benson M. Kariuki, Louise Male, John C. McMurtrie, Neil Spencer and Richard S. Grainger
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 7) pp:2336-2344
Publication Date(Web):14 Feb 2011
DOI:10.1039/C0OB00976H
Acyclic bissulfonylnitroxides have never been isolated, and degrade through fragmentation. In an approach to stabilising a bissulfonylnitroxide radical, the cyclic, peri-substituted N,N-bissulfonylhydroxylamine, 2-hydroxynaphtho[1,8-de][1,3,2]dithiazine 1,1,3,3-tetraoxide (1), has been prepared by formal nitrogen insertion into the sulfur–sulfur bond of a sulfinylsulfone, naphtho[1,8-cd][1,2]dithiole 1,1,2-trioxide. The heterocyclic ring of 1 is shown to adopt a sofa conformation by X-ray crystallography, with a pseudo-axial hydroxyl group. N,N-Bissulfonylhydroxylamine 1 displays high thermal, photochemical and hydrolytic stability compared to acyclic systems. EPR analysis reveals formation of the corresponding bissulfonylnitroxide 2 upon oxidation of 1 with the Ce(IV) salts CAN and CTAN. Although 2 does not undergo fragmentation, it cannot be isolated, since hydrogen atom abstraction to reform 1 occurs in situ. The stability and reactivity of 1 and 2 are compared with the known cyclic benzo-fused N,N-bissulfonylhydroxylamine, N-hydroxy-O-benzenedisulfonimide (6), for which the X-ray data, and EPR of the corresponding nitroxide 10, are also reported for the first time.
Co-reporter:Thomas B. Parsons, Cédric Ghellamallah, Louise Male, Neil Spencer and Richard S. Grainger
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 14) pp:5021-5023
Publication Date(Web):13 May 2011
DOI:10.1039/C1OB05522D
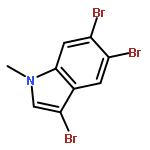
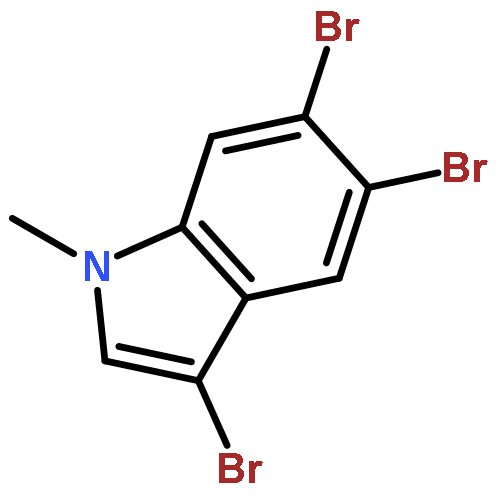
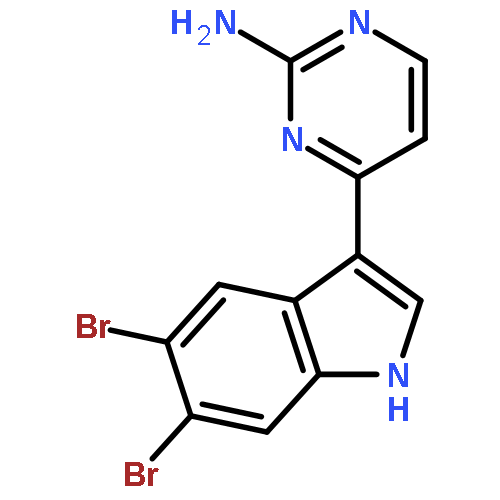
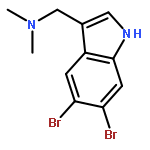
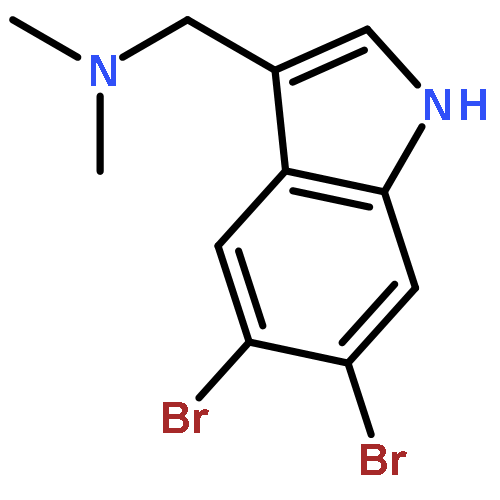
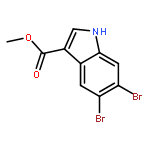
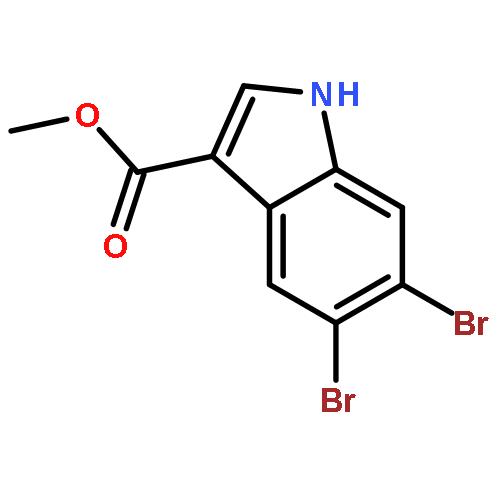
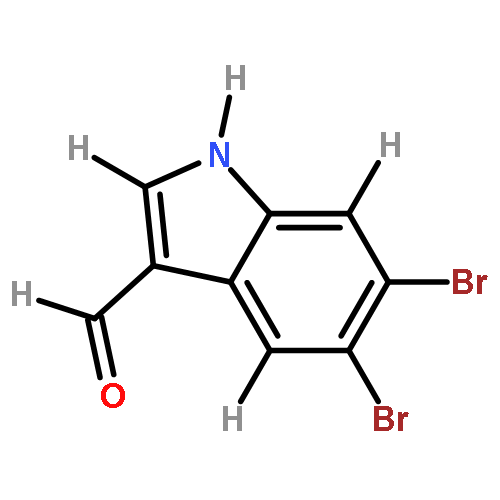
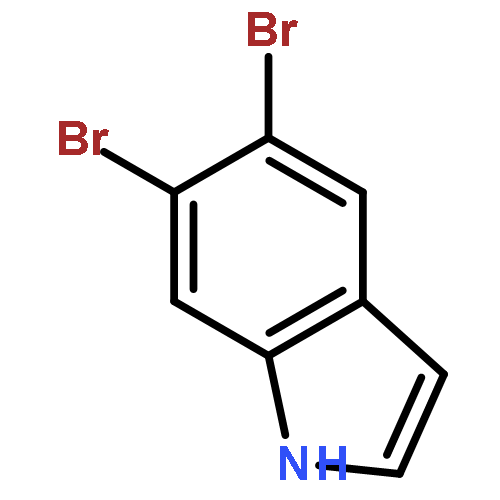
Treatment of methyl indole-3-carboxylate with bromine in acetic acid gives methyl 5,6-dibromoindole-3-carboxylate regioselectively, from which the parent 5,6-dibromoindole can be accessed via a one-pot, microwave-mediated ester hydrolysis and decarboxylation. Application of these building blocks in syntheses of natural and non-natural 5,6-dibromoindole derivatives, including meridianin F and 5,6-dibromo-2′-demethylaplysinopsin, is reported.
Co-reporter:Stig Allenmark;Susanne Olsson;Bhaven Patel
European Journal of Organic Chemistry 2011 Volume 2011( Issue 22) pp:4089-4092
Publication Date(Web):
DOI:10.1002/ejoc.201100419
Abstract
The trans isomer of 2,7-di-tert-butylnaphtho[1,8-cd][1,2]dithiole 1,2-dioxide, a vic-disulfoxide of the highest thermal stability reported to date, has been resolved into its enantiomers by preparative chiral liquid chromatography. This represents the first resolution achieved of a vic-disulfoxide. The absolute configuration of the isolated enantiomers has been determined to be (+)-(S,S), (–)-(R,R) from a comparison of experimental and calculated ECD and VCD spectra. The long-wavelength ECD band at about 330 nm had a positive sign in the (+)-(S,S)-form and a negative sign in the (–)-(R,R)-enantiomer. A photocatalyzed slow decrease in optical activity was observed in solution, most likely due to racemization by homolytic (O)S–S(O) bond cleavage and bissulfinyl radical recombination.
Co-reporter:Tyrone C. Casey, Julie Carlisle, Patrizia Tisselli, Louise Male, Neil Spencer, and Richard S. Grainger
The Journal of Organic Chemistry 2010 Volume 75(Issue 21) pp:7461-7464
Publication Date(Web):October 11, 2010
DOI:10.1021/jo101531b
Pyrrolidine enamines derived from three 1,3-dioxan-5-ones undergo α,α′-annelation reactions with methyl α-(bromomethyl)acrylate to produce bridged 2,4-dioxabicyclo[3.3.1]nonane ring systems with complete stereocontrol. Stereochemical outcomes have been rationalized based on steric and stereoelectronic interactions in intermediate boat-like conformations of the 1,3-dioxane ring and subsequent kinetic protonation to set an axial ester group on the cyclohexanone ring. Base-mediated ester epimerization provides the stereochemical array found in the highly oxygenated cyclohexane ring of phyllaemblic acid and glochicoccins B and D.
Co-reporter:RichardS. Grainger Dr.;Bhaven Patel;BensonM. Kariuki Dr.
Angewandte Chemie 2009 Volume 121( Issue 26) pp:4926-4929
Publication Date(Web):
DOI:10.1002/ange.200901788
Co-reporter:RichardS. Grainger Dr.;Bhaven Patel;BensonM. Kariuki Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 26) pp:4832-4835
Publication Date(Web):
DOI:10.1002/anie.200901788
Co-reporter:Paolo Innocenti
Heteroatom Chemistry 2007 Volume 18(Issue 5) pp:568-571
Publication Date(Web):10 JUL 2007
DOI:10.1002/hc.20336
The application of dithiocarbamates in group transfer radical cyclization reactions is described. Carbamoyl diethyldithiocarbamates are synthesized in two high-yielding steps from secondary amines and act as sources of carbamoyl radicals through chemical or photochemical initiation. Group transfer radical cyclization reactions lead to dithiocarbamate-functionalized lactams. © 2007 Wiley Periodicals, Inc. Heteroatom Chem 18:568–571, 2007; Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/hc.20336
Co-reporter:Richard S. Grainger Dr.;Emma J. Welsh
Angewandte Chemie 2007 Volume 119(Issue 28) pp:
Publication Date(Web):6 JUN 2007
DOI:10.1002/ange.200701055
Zeit zum Lampenwechsel: Ein Alkyldithiocarbamat, selbst das Produkt der lichtinduzierten Gruppentransfercyclisierung eines Carbamoylradikals, geht einen zweiten lichtvermittelten Radikalprozess ein, der durch eine andere Lichtquelle ausgelöst wird. Diese Reaktionen, die über das gleiche Cyclohexenylradikal verlaufen, sind Schlüsselschritte in einer neuen asymmetrischen Synthese des Alkaloids Aphanorphin. TEMPO = 2,2,6,6-Tetramethyl-1-piperidinoxylradikal.
Co-reporter:Richard S. Grainger Dr.;Emma J. Welsh
Angewandte Chemie International Edition 2007 Volume 46(Issue 28) pp:
Publication Date(Web):6 JUN 2007
DOI:10.1002/anie.200701055
Time to change the light bulb: An alkyl dithiocarbamate, itself formed through a photoinitiated group-transfer cyclization of a carbamoyl radical, undergoes a second photomediated radical process initiated with a different light source. These two reactions, which proceed through the same cyclohexenyl radical intermediate, are key steps in a new asymmetric synthesis of the alkaloid aphanorphine. TEMPO=2,2,6,6-tetramethyl-1-piperidinoxyl radical.
Co-reporter:Thomas B. Parsons, Cédric Ghellamallah, Louise Male, Neil Spencer and Richard S. Grainger
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 14) pp:NaN5023-5023
Publication Date(Web):2011/05/13
DOI:10.1039/C1OB05522D
Treatment of methyl indole-3-carboxylate with bromine in acetic acid gives methyl 5,6-dibromoindole-3-carboxylate regioselectively, from which the parent 5,6-dibromoindole can be accessed via a one-pot, microwave-mediated ester hydrolysis and decarboxylation. Application of these building blocks in syntheses of natural and non-natural 5,6-dibromoindole derivatives, including meridianin F and 5,6-dibromo-2′-demethylaplysinopsin, is reported.
Co-reporter:Thomas B. Parsons, Neil Spencer, Chi W. Tsang and Richard S. Grainger
Chemical Communications 2013 - vol. 49(Issue 23) pp:NaN2298-2298
Publication Date(Web):2013/02/01
DOI:10.1039/C3CC39062D
The first synthesis of kottamide E, a marine natural product containing a 5,6-dibromoindole linked via a (Z)-enamide to an unusual 1,2-dithiolane-containing amino acid, is reported.
Co-reporter:Bhaven Patel, Julie Carlisle, Steven E. Bottle, Graeme R. Hanson, Benson M. Kariuki, Louise Male, John C. McMurtrie, Neil Spencer and Richard S. Grainger
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 7) pp:NaN2344-2344
Publication Date(Web):2011/02/14
DOI:10.1039/C0OB00976H
Acyclic bissulfonylnitroxides have never been isolated, and degrade through fragmentation. In an approach to stabilising a bissulfonylnitroxide radical, the cyclic, peri-substituted N,N-bissulfonylhydroxylamine, 2-hydroxynaphtho[1,8-de][1,3,2]dithiazine 1,1,3,3-tetraoxide (1), has been prepared by formal nitrogen insertion into the sulfur–sulfur bond of a sulfinylsulfone, naphtho[1,8-cd][1,2]dithiole 1,1,2-trioxide. The heterocyclic ring of 1 is shown to adopt a sofa conformation by X-ray crystallography, with a pseudo-axial hydroxyl group. N,N-Bissulfonylhydroxylamine 1 displays high thermal, photochemical and hydrolytic stability compared to acyclic systems. EPR analysis reveals formation of the corresponding bissulfonylnitroxide 2 upon oxidation of 1 with the Ce(IV) salts CAN and CTAN. Although 2 does not undergo fragmentation, it cannot be isolated, since hydrogen atom abstraction to reform 1 occurs in situ. The stability and reactivity of 1 and 2 are compared with the known cyclic benzo-fused N,N-bissulfonylhydroxylamine, N-hydroxy-O-benzenedisulfonimide (6), for which the X-ray data, and EPR of the corresponding nitroxide 10, are also reported for the first time.
Co-reporter:Claire McMaster, Robert N. Bream and Richard S. Grainger
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 24) pp:NaN4758-4758
Publication Date(Web):2012/05/15
DOI:10.1039/C2OB25434D
Reductive desulfurisation of dithiocarbamates is conveniently achieved using H3PO2–Et3N–ACCN in refluxing dioxane. Fused and spirocyclic β-lactams, prepared through 4-exo trig carbamoyl radical cyclisation–dithiocarbamate group transfer reactions, are reduced without fragmentation of the strained 4-membered ring. Diethyl tetraacetyl-D-glucopyranosyl dithiocarbamate is selectively reduced with or without acyloxy group migration depending on reaction conditions and choice of reductant. Deuterium incorporation from D3PO2–Et3N is observed for a system involving a nucleophilic radical intermediate, but not in the case of the electrophilic radical obtained through acyloxy group migration on a glucose derivative.
Co-reporter:Kevin R. Munro, Louise Male, Neil Spencer and Richard S. Grainger
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 39) pp:NaN6862-6862
Publication Date(Web):2013/08/30
DOI:10.1039/C3OB41390J
α-Hydroxyalkylidene carbenes, generated from thermolysis of α,β-epoxy-N-aziridinylimines, undergo diastereotopic group selective 1,5 C–H insertion reactions on 2,4-dimethyl-8-oxabicyclo[3.2.1]oct-6-ene ring systems. Protection of a tertiary alcohol at C-3 of the bridged oxabicycle as a trimethylsilyl ether reverses the sense of diastereoselectivity. 1,5 C–H insertion into a methine adjacent to an OBn group, 1,5 O–R insertion into a tertiary alcohol (R = H) or silylether (R = TMS) at C-3 to form spirocyclic dihydrofurans, 1,2-rearrangement to an alkyne and fragmentation to a ketone are competing major pathways for 2-benzyloxy-substituted 8-oxabicyclo[3.2.1]oct-6-ene systems. Dihydrofuran formation is shown to be a result of substitution on the oxabicyclic ring system through comparison with other methods of alkylidene carbene formation.
Co-reporter:Matthew J. Barrett, Paul W. Davies and Richard S. Grainger
Organic & Biomolecular Chemistry 2015 - vol. 13(Issue 32) pp:NaN8686-8686
Publication Date(Web):2015/07/07
DOI:10.1039/C5OB01241D
A protocol has been developed for direct Csp3–Csp2 bond formation at the 4- and 6-positions of dibenzothiophenes using a gold(I) catalyst with terminal alkynes and dibenzothiophene-S-oxides. The sulfoxide acts as a traceless directing group to avoid the need to prefunctionalise at carbon. The iterative use of this protocol is possible and has been employed in the preparation of novel macrocyclic structures. In addition, a cascade process shows how oxyarylations can be combined with other processes resulting in complex, highly efficient transformations.
Co-reporter:Benedicte Defaut, Thomas B. Parsons, Neil Spencer, Louise Male, Benson M. Kariuki and Richard S. Grainger
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 25) pp:NaN4932-4932
Publication Date(Web):2012/05/21
DOI:10.1039/C2OB25384D
A concise, stereoselective synthesis of the trans-hydrindane core of the marine natural product dictyoxetane is reported, starting from a Robinson annelation derived bicyclic enone. A phosphorane-mediated, pinacol-like rearrangement of a cis-diol, via a formal 1,2-hydride shift, is used to establish the requisite trans ring junction. 31P NMR supports the formation of the intermediate phosphorane, generated in situ from the reaction of a diol with Ph3PCl2.
Co-reporter:Matthew J. Barrett, Ghulam F. Khan, Paul W. Davies and Richard S. Grainger
Chemical Communications 2017 - vol. 53(Issue 42) pp:NaN5736-5736
Publication Date(Web):2017/05/04
DOI:10.1039/C7CC02244A
Alkynyl sulfoxides are shown to act as α-sulfinyl metallocarbene synthons under oxidative gold catalysis, enabling reactions that are not available from diazo-precursors. This strategy is exemplified in the synthesis of fused α-sulfinyl cyclopropanes.

![Naphtho[1,8-cd]-1,2-dithiole, 5-nitro-](http://img.cochemist.com/ccimg/63200/63187-36-0.png)
![Naphtho[1,8-cd]-1,2-dithiole, 5-nitro-](http://img.cochemist.com/ccimg/63200/63187-36-0_b.png)
![Naphtho[1,8-cd]-1,2-dithiole, 3-nitro-](http://img.cochemist.com/ccimg/63200/63187-35-9.png)
![Naphtho[1,8-cd]-1,2-dithiole, 3-nitro-](http://img.cochemist.com/ccimg/63200/63187-35-9_b.png)

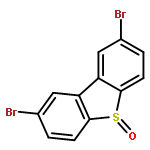


![Naphtho[1,8-cd]-1,2-dithiole, 1,1,2-trioxide](http://img.cochemist.com/ccimg/57900/57821-65-5.png)
![Naphtho[1,8-cd]-1,2-dithiole, 1,1,2-trioxide](http://img.cochemist.com/ccimg/57900/57821-65-5_b.png)
![Naphtho[1,8-de]-1,2,3-trithiin, 2-oxide](http://img.cochemist.com/ccimg/380500/380451-79-6.png)
![Naphtho[1,8-de]-1,2,3-trithiin, 2-oxide](http://img.cochemist.com/ccimg/380500/380451-79-6_b.png)
![8-Oxabicyclo[3.2.1]oct-6-en-3-one, 2-(phenylmethoxy)-, (1R,2S,5R)-rel-](http://img.cochemist.com/ccimg/238800/238757-83-0.png)
![8-Oxabicyclo[3.2.1]oct-6-en-3-one, 2-(phenylmethoxy)-, (1R,2S,5R)-rel-](http://img.cochemist.com/ccimg/238800/238757-83-0_b.png)
![8-Oxabicyclo[3.2.1]oct-6-en-3-one, 2,4-dimethyl-, (1R,2R,4R,5S)-rel-](http://img.cochemist.com/ccimg/37100/37081-60-0.png)
![8-Oxabicyclo[3.2.1]oct-6-en-3-one, 2,4-dimethyl-, (1R,2R,4R,5S)-rel-](http://img.cochemist.com/ccimg/37100/37081-60-0_b.png)
![Naphtho[1,8-cd]-1,2-dithiole, 3,8-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/350300/350253-68-8.png)
![Naphtho[1,8-cd]-1,2-dithiole, 3,8-bis(1,1-dimethylethyl)-](http://img.cochemist.com/ccimg/350300/350253-68-8_b.png)



![NAPHTHO[1,8-CD]-1,2-DITHIOLE, 1,1-DIOXIDE](http://img.cochemist.com/ccimg/40300/40227-43-8.png)
![NAPHTHO[1,8-CD]-1,2-DITHIOLE, 1,1-DIOXIDE](http://img.cochemist.com/ccimg/40300/40227-43-8_b.png)



![Naphtho[1,8-cd]-1,2-dithiole-3-carboxaldehyde](http://img.cochemist.com/ccimg/935600/935552-37-7.png)
![Naphtho[1,8-cd]-1,2-dithiole-3-carboxaldehyde](http://img.cochemist.com/ccimg/935600/935552-37-7_b.png)



![Naphtho[1,8-cd]-1,2-thiaselenole](http://img.cochemist.com/ccimg/64900/64869-35-8.png)
![Naphtho[1,8-cd]-1,2-thiaselenole](http://img.cochemist.com/ccimg/64900/64869-35-8_b.png)


![6-methyl-6-Azabicyclo[3.2.2]non-2-en-7-one](http://img.cochemist.com/ccimg/1069200/1069111-89-2.png)
![6-methyl-6-Azabicyclo[3.2.2]non-2-en-7-one](http://img.cochemist.com/ccimg/1069200/1069111-89-2_b.png)



























![2-[(4-METHYL-2-NITROPHENYL)DIAZENYL]-N-(2-METHYLPHENYL)-3-OXOBUTA<WBR />NAMIDE](http://img.cochemist.com/ccimg/36300/36291-48-2.png)
![2-[(4-METHYL-2-NITROPHENYL)DIAZENYL]-N-(2-METHYLPHENYL)-3-OXOBUTA<WBR />NAMIDE](http://img.cochemist.com/ccimg/36300/36291-48-2_b.png)
![[(2r,3r,4s,5r,6s)-3,4,5-triacetyloxy-6-ethoxycarbothioylsulfanyloxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/13700/13639-54-8.png)
![[(2r,3r,4s,5r,6s)-3,4,5-triacetyloxy-6-ethoxycarbothioylsulfanyloxan-2-yl]methyl Acetate](http://img.cochemist.com/ccimg/13700/13639-54-8_b.png)







![Naphtho[1,8-cd]-1,2-dithiole,1-oxide](http://img.cochemist.com/ccimg/49900/49833-12-7.png)
![Naphtho[1,8-cd]-1,2-dithiole,1-oxide](http://img.cochemist.com/ccimg/49900/49833-12-7_b.png)



![Benzene,1,1'-[1,2-bis(methylene)-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/2600/2548-47-2.png)
![Benzene,1,1'-[1,2-bis(methylene)-1,2-ethanediyl]bis-](http://img.cochemist.com/ccimg/2600/2548-47-2_b.png)


![Benzene, [3-(2-propynyloxy)-1-propenyl]-](http://img.cochemist.com/ccimg/111000/110966-13-7.png)
![Benzene, [3-(2-propynyloxy)-1-propenyl]-](http://img.cochemist.com/ccimg/111000/110966-13-7_b.png)
![Silane,(1,1-dimethylethyl)[[(2E)-3-iodo-2-methyl-2-propenyl]oxy]dimethyl-](http://img.cochemist.com/ccimg/180600/180590-91-4.png)
![Silane,(1,1-dimethylethyl)[[(2E)-3-iodo-2-methyl-2-propenyl]oxy]dimethyl-](http://img.cochemist.com/ccimg/180600/180590-91-4_b.png)


![Naphtho[1,8-cd]-1,2-dithiole](http://img.cochemist.com/ccimg/300/209-22-3.png)
![Naphtho[1,8-cd]-1,2-dithiole](http://img.cochemist.com/ccimg/300/209-22-3_b.png)