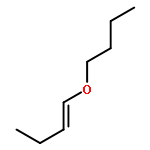
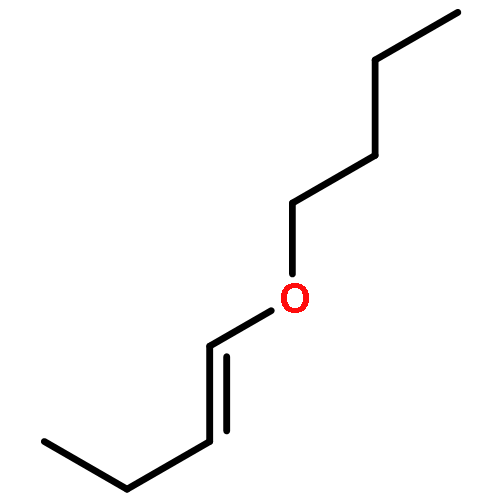
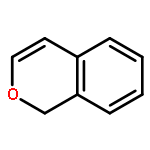
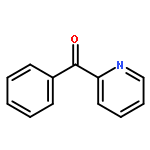
Co-reporter:Zhou Chen, Mark D. Leatherman, Olafs Daugulis, and Maurice Brookhart
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:16013-16013
Publication Date(Web):October 30, 2017
DOI:10.1021/jacs.7b10281
Copolymerizations of ethylene with vinyltrialkoxysilanes using cationic (α-diimine)Ni(Me)(CH3CN)+ complexes 4a,b/B(C6F5)3 yield high molecular weight copolymers exhibiting highly branched to nearly linear backbones depending on reaction conditions and catalyst choice. Polymerizations are first-order in ethylene pressure and inverse-order in silane concentration. Microstructural analysis of the copolymers reveals both in-chain and chain-end incorporation of −Si(OR)3 groups whose ratios depend on temperature and ethylene pressure. Detailed low-temperature NMR spectroscopic investigations show that well-defined complex 3b (α-diimine)Ni(Me)(OEt2)+ reacts rapidly at −60 °C with vinyltrialkoxysilanes via both 2,1 and 1,2 insertion pathways to yield 4- and 5-membered chelates, respectively. Such chelates are the major catalyst resting states but are in rapid equilibrium with ethylene-opened chelates, (α-diimine)Ni(R)(C2H4)+ complexes, the species responsible for chain growth. Chelate rearrangement via β-silyl elimination accounts for formation of chain-end −Si(OR)3 groups and constitutes a chain-transfer mechanism. Chelate formation and coordination of the Ni center to the ether moiety, R–O–Si, of the vinylsilane somewhat decreases the turnover frequency (TOF) relative to ethylene homopolymerization, but still remarkably high TOFs of up to 4.5 × 105 h–1 and overall productivities can be achieved. Activation of readily available (α-diimine)NiBr2 complexes 2 with a combination of AlMe3/B(C6F5)3/[Ph3C][B(C6F5)4] yields a highly active and productive catalyst system for the convenient synthesis of the copolymer, a cross-linkable PE. For example, copolymers containing 0.23 mol % silane can be generated at 60 °C, 600 psig ethylene over 4 h with a productivity of 560 kg copolymer/g Ni. This method offers an alternative route to these materials, normally prepared via radical routes, which are precursors to the commercial cross-linked polyethylene, PEX-b.
Co-reporter:Zhou Chen, Weijun Liu, Olafs Daugulis, and Maurice Brookhart
Journal of the American Chemical Society 2016 Volume 138(Issue 49) pp:16120-16129
Publication Date(Web):December 1, 2016
DOI:10.1021/jacs.6b10462
Co-reporter:Andrew D. Bolig, Thomas W. Lyons, Darren T. DiSalvo, Maurice Brookhart
Polyhedron 2016 Volume 103(Part A) pp:51-57
Publication Date(Web):8 January 2016
DOI:10.1016/j.poly.2015.07.076
The mechanism of intramolecular transfer dehydrogenation catalyzed by [Cp∗M(VTMS)2] (1, M = Rh, 2, M = Co, Cp∗ = C5Me5, VTMS = vinyltrimethylsilane) complexes has been studied using vinyl silane protected alcohols as substrates. Deuterium-labeled substrates have been synthesized and the regioselectivity of H/D transfers investigated using 1H and 2H NMR spectroscopy. The labeling studies establish a regioselective pathway consisting of alkene directed α C–H activation, 2,1 alkene insertion, and finally β-hydride elimination to give silyl enol ether products.The mechanism of intramolecular transfer dehydrogenation catalyzed by [Cp∗M(VTMS)2] (M = Rh, Co, Cp∗ = C5Me5) complexes has been studied using vinyl silane protected alcohols as substrates. Using deuterium-labeled substrates, the regioselectivity of H/D transfers was investigated using NMR spectroscopy. These studies establish a regioselective pathway involving alkene directed α-C–H activation, 2,1 alkene insertion, and finally β-hydride elimination to give silyl enol ether products.
Co-reporter:Wubing Yao, Xiangqing Jia, Xuebing Leng, Alan S. Goldman, Maurice Brookhart, Zheng Huang
Polyhedron 2016 Volume 116() pp:12-19
Publication Date(Web):25 September 2016
DOI:10.1016/j.poly.2016.02.044
A series of new (tBu2PSCOPR2)IrHCl iridium complexes with ‘hybrid’ phosphinothious-phosphinite PSCOP ligands ([tBu2PSCOPR2 = 1-(SPtBu2)-3-(OPR2)-C6H4], R = tBu, 4a, R = Cy, 4b, R = iPr, 4c, and R = Et, 4d) have been synthesized and characterized. Treatment of complexes 4a–d with sodium tert-butoxide generates the active species for catalytic transfer-dehydrogenation of cyclooctane (COA) or n-octane using tert-butylethylene (TBE) as hydrogen acceptor to form cyclooctene (COE) or octenes, respectively. The catalytic activity of these complexes and the product selectivity in alkane dehydrogenation is greatly influenced by the steric properties of the pincer ligand. In general, the less sterically bulky complex exhibits higher catalytic activity than the more hindered complex. Among the new (PSCOP)Ir-type complexes, the least crowded complex (tBu2PSCOPEt2)IrHCl 4d is most active for n-octane/TBE transfer-dehydrogenation. The relatively crowded, less active, complexes (tBu2PSCOPtBu2)IrHCl (4a) and (tBu2PSCOPCy2)IrHCl (4b) exhibit high regioselectivity for α-olefin formation at the early stages of the reaction.New (tBu2PSCOPR2)IrHCl iridium complexes ligated by hybrid phosphinothious-phosphinite PSCOP ligands have been synthesized and characterized. The steric properties of the pincer ligands prove to have a marked impact on catalytic activities of these complexes in transfer-dehydrogenation of cyclic and linear alkanes.
Co-reporter:Zhou Chen, Kate E. Allen, Peter S. White, Olafs Daugulis, and Maurice Brookhart
Organometallics 2016 Volume 35(Issue 11) pp:1756-1760
Publication Date(Web):May 6, 2016
DOI:10.1021/acs.organomet.6b00165
Traditional cationic Pd(II) and Ni(II) ethylene polymerization catalysts are supported by ortho-disubstituted aryl diimine ligands. These catalysts are capable of producing high-molecular-weight polyethylene due to positioning of bulk in the two axial sites of the square coordination plane which retards chain transfer. Similar pyridine-imine complexes bearing a single ortho-disubstituted aryl imine moiety were reported to yield very low Mn polyethylene. In earlier studies, “sandwich” diimine nickel catalysts incorporating two 8-arylnaphthylimino groups which provide exceptional shielding of the two axial sites were shown to yield ultrahigh-molecular-weight polyethylene. Here we demonstrate that 8-arylnaphthyl groups incorporated into pyridine-imine nickel catalysts that block only a single axial site are highly effective in retarding chain transfer. These catalysts produce branched polyethylene (ca. 30–90 branches per 1000 carbons) with Mn values up to 2.6 × 104 g/mol. Effects on the catalyst lifetimes and polymerization behavior as a function of substituent variations at the imine carbon and the aryl group are reported.
Co-reporter:Zhou Chen, Milad Mesgar, Peter S. White, Olafs Daugulis, and Maurice Brookhart
ACS Catalysis 2015 Volume 5(Issue 2) pp:631
Publication Date(Web):December 11, 2014
DOI:10.1021/cs501948d
Neutral nickel methyl complexes incorporating 2,8-diarylnaphthyl groups have been synthesized and characterized. Salicylaldiminato nickel systems 1a,b are exceptionally active neutral nickel single component catalysts for the polymerization of ethylene capable of producing lightly branched ultrahigh-molecular-weight polyethylene (UHMWPE). In addition, complex 1a shows a “quasi-living” polymerization behavior.Keywords: branched polyethylene; nickel catalysts; polymerization; salicyaldimine; UHMWPE
Co-reporter:Kate E. Allen, Jesús Campos, Olafs Daugulis, and Maurice Brookhart
ACS Catalysis 2015 Volume 5(Issue 1) pp:456
Publication Date(Web):December 2, 2014
DOI:10.1021/cs5016029
Cationic Pd(II) catalysts incorporating bulky 8-p-tolylnaphthyl substituted diimine ligands have been synthesized and investigated for ethylene polymerization and ethylene/methyl acrylate copolymerization. Homopolymerization of ethylene at room temperature resulted in branched polyethylene with narrow Mw/Mn values (ca. 1.1), indicative of a living polymerization. A mechanistic study revealed that the catalyst resting state was an alkyl olefin complex and that the turnover-limiting step was migratory insertion, thus the turnover frequency is independent of ethylene concentration. Copolymerization of ethylene and methyl acrylate (MA) was also achieved. MA incorporation was found to increase linearly with MA concentration, and copolymers with up to 14 mol % MA were prepared. Mechanistic studies revealed that acrylate insertion into a Pd–CH3 bond occurs at −70 °C to yield a four-membered chelate, which isomerizes first to a five-membered chelate and then to a six-membered chelate. Barriers to migratory insertion of both the (diimine)PdCH3(C2H4)+ (19.2 kcal/mol) and (diimine)PdCH3(η2-C2H3CO2Me)+ (15.2 kcal/mol) were measured by low-temperature NMR kinetics.Keywords: diimine; ethylene; living polymerization; methyl acrylate; palladium
Co-reporter:Thomas W. Lyons, David Bézier, and Maurice Brookhart
Organometallics 2015 Volume 34(Issue 16) pp:4058-4062
Publication Date(Web):August 7, 2015
DOI:10.1021/acs.organomet.5b00501
We describe efficient methods to dehydrogenate ethers by using iridium pincer complexes (iPr4Anthraphos)-Ir(H)(Cl), 4, iPr4PC(sp3)P–Ir(H)(Cl), 5, and (iPr4PCP)-Ir(H)(Cl), 6. At 120 °C, cyclic ethers were dehydrogenated with tert-butylethylene as the hydrogen acceptor with high turnover numbers (over 400 in many cases). Acyclic ethers such as diethyl ether can also be dehydrogenated catalytically with TONs up to 90. The efficient dehydrogenation of cyclic and acyclic ethers using ethylene as a more practical hydrogen acceptor has been demonstrated for the first time.
Co-reporter:Peng Kang;Zuofeng Chen;Thomas J. Meyer
Topics in Catalysis 2015 Volume 58( Issue 1) pp:30-45
Publication Date(Web):2015 February
DOI:10.1007/s11244-014-0344-y
This review summarizes the development of electrochemical CO2 reduction catalysts in the UNC Energy Frontier Research Center for Solar Fuels. Two strategies for converting CO2 to CO or formate have been explored. In one, polypyridyl complexes of Ru(II) have been used to reduce CO2 to CO in acetonitrile and in acetonitrile/water mixtures. In the absence of CO2 water is reduced to H2 by these complexes. With added weak acids in acetonitrile with added water and CO2, reduction to syngas mixtures of CO and H2 is observed. A single polypyridyl complex of Ru(II) has been shown to be both a catalyst for water oxidation and CO2 reduction in an electrochemical cell for CO2 splitting into CO and O2. In parallel, Ir pincer catalysts have been shown to act as selective electrocatalysts for reducing CO2 to formate in acetonitrile with added water and in pure water without competition from electrocatalytic H2 production. Details of the catalytic mechanisms of each have also been investigated.
Co-reporter:Chen Cheng ; Bong Gon Kim ; Damien Guironnet ; Maurice Brookhart ; Changjian Guan ; David Y. Wang ; Karsten Krogh-Jespersen ;Alan S. Goldman
Journal of the American Chemical Society 2014 Volume 136(Issue 18) pp:6672-6683
Publication Date(Web):April 18, 2014
DOI:10.1021/ja501572g
New carbazolide-based iridium pincer complexes (carbPNP)Ir(C2H4), 3a, and (carbPNP)Ir(H)2, 3b, have been prepared and characterized. The dihydride, 3b, reacts with ethylene to yield the cis-dihydride ethylene complex cis-(carbPNP)Ir(C2H4)(H)2. Under ethylene this complex reacts slowly at 70 °C to yield ethane and the ethylene complex, 3a. Kinetic analysis establishes that the reaction rate is dependent on ethylene concentration and labeling studies show reversible migratory insertion to form an ethyl hydride complex prior to formation of 3a. Exposure of cis-(carbPNP)Ir(C2H4)(H)2 to hydrogen results in very rapid formation of ethane and dihydride, 3b. DFT analysis suggests that ethane elimination from the ethyl hydride complex is assisted by ethylene through formation of (carbPNP)Ir(H)(Et)(C2H4) and by H2 through formation of (carbPNP)Ir(H)(Et)(H2). Elimination of ethane from Ir(III) complex (carbPNP)Ir(H)(Et)(H2) is calculated to proceed through an Ir(V) complex (carbPNP)Ir(H)3(Et) which reductively eliminates ethane with a very low barrier to return to the Ir(III) dihydride, 3b. Under catalytic hydrogenation conditions (C2H4/H2), cis-(carbPNP)Ir(C2H4)(H)2 is the catalyst resting state, and the catalysis proceeds via an Ir(III)/Ir(V)/Ir(III) cycle. This is in sharp contrast to isoelectronic (PCP)Ir systems in which hydrogenation proceeds through an Ir(III)/Ir(I)/Ir(III) cycle. The basis for this remarkable difference is discussed.
Co-reporter:David Bézier and Maurice Brookhart
ACS Catalysis 2014 Volume 4(Issue 10) pp:3411
Publication Date(Web):August 21, 2014
DOI:10.1021/cs500892p
Iridium ethylene complexes based on the PC(sp3)P pincer-type triptycene ligand have been synthesized. Complexes bearing various substituents on the phosphines have been investigated as catalysts in transfer dehydrogenation of alkanes. The complex 8a, which bears isopropyl groups, has demonstrated high stability and activity when used as a catalyst in the disproportionation of 1-hexene at 180 °C and in the transfer dehydrogenation of linear and cyclic alkanes with tert-butylethylene as a hydrogen acceptor at 200 °C. A similar complex bearing a CH2NMe2 group, 33, allowed support of the catalyst on γ-alumina for operation in a heterogeneous mode.Keywords: alkanes; C−H activation; dehydrogenation; iridium; pincer complexes; supported catalyst
Co-reporter:Dr. Peng Kang;Dr. Sheng Zhang; Thomas J. Meyer; Maurice Brookhart
Angewandte Chemie International Edition 2014 Volume 53( Issue 33) pp:8709-8713
Publication Date(Web):
DOI:10.1002/anie.201310722
Abstract
An iridium pincer dihydride catalyst was immobilized on carbon nanotube-coated gas diffusion electrodes (GDEs) by using a non-covalent binding strategy. The as-prepared GDEs are efficient, selective, durable, gas permeable electrodes for electrocatalytic reduction of CO2 to formate. High turnover numbers (ca. 54 000) and turnover frequencies (ca. 15 s−1) were enabled by the novel electrode architecture in aqueous solutions saturated in CO2 with added HCO3−.
Co-reporter:Marc D. Walter ; Peter S. White ; Cynthia K. Schauer
Journal of the American Chemical Society 2013 Volume 135(Issue 42) pp:15933-15947
Publication Date(Web):September 20, 2013
DOI:10.1021/ja4079539
Iridium(I) and rhodium(I) ethyl complexes, (PONOP)M(C2H5) (M = Ir (1-Et), Rh (2-Et)) and the iridium(I) propyl complex (PONOP)Ir(C3H7) (1-Pr), where PONOP is 2,6-(tBu2PO)2C5H3N, have been prepared. Low-temperature protonation of the Ir complexes yields the alkyl hydrides, (PONOP)Ir(H)(R) (1-(H)(Et)+ and 1-(H)(Pr)+), respectively. Dynamic 1H NMR characterization of 1-(H)(Et)+ establishes site exchange between the Ir–H and Ir–CH2 protons (ΔGexH‡(−110 °C) = 7.2(1) kcal/mol), pointing to a σ-ethane intermediate. By dynamic 13C NMR spectroscopy, the exchange barrier between the α and β carbons (“chain-walking”) was measured (ΔGexC‡(−110 °C) = 8.1(1) kcal/mol). The barrier for ethane loss is 17.4(1) kcal/mol (−40 °C), to be compared with the reported barrier to methane loss in 1-(H)(Me)+ of 22.4 kcal/mol (22 °C). A rhodium σ-ethane complex, (PONOP)Rh(EtH) (2-(EtH)+), was prepared by protonation of 2-Et at −150 °C. The barrier for ethane loss (ΔGdec‡(−132 °C) = 10.9(2) kcal/mol) is lower than for the methane complex, 2-(MeH)+, (ΔGdec‡(−87 °C) = 14.5(4) kcal/mol). Full spectroscopic characterization of 2-(EtH)+ is reported, a key feature of which is the upfield signal at −31.2 ppm for the coordinated CH3 group in the 13C NMR spectrum. The exchange barrier of the hydrogens of the coordinated methyl group is too low to be measured, but the chain-walking barrier of 7.2(1) kcal/mol (−132 °C) is observable by 13C NMR. The coordination mode of the alkane ligand and the exchange pathways for the Rh and Ir complexes are evaluated by DFT studies. On the basis of the computational studies, it is proposed that chain-walking occurs by different mechanisms: for Rh, the lowest energy path involves a η2-ethane transition state, while for Ir, the lowest energy exchange pathway proceeds through the symmetrical ethylene dihydride complex.
Co-reporter:Jesús Campos ; Sabuj Kundu ; Dale R. Pahls ; Maurice Brookhart ; Ernesto Carmona ;Thomas R. Cundari
Journal of the American Chemical Society 2013 Volume 135(Issue 4) pp:1217-1220
Publication Date(Web):January 11, 2013
DOI:10.1021/ja310982v
Evidence for key σ-complex intermediates in the hydrogenolysis of the iridium–methyl bond of (PONOP)Ir(H)(Me)+ (1) [PONOP = 2,6-bis(di-tert-butylphosphinito)pyridine] has been obtained. The initially formed η2-H2 complex, 2, was directly observed upon treatment of 1 with H2, and evidence for reversible formation of a σ-methane complex, 5, was obtained through deuterium scrambling from η2-D2 in 2-d2 into the methyl group of 2 prior to methane loss. This sequence of reactions was modeled by density functional theory calculations. The transition state for formation of 5 from 2 showed significant shortening of the Ir–H bond for the hydrogen being transferred; no true Ir(V) trihydride intermediate could be located. Barriers to methane loss from 2 were compared to those of 1 and the six-coordinate species (PONOP)Ir(H)(Me)(CO)+ and (PONOP)Ir(H)(Me)(Cl).
Co-reporter:Sabuj Kundu, Thomas W. Lyons, and Maurice Brookhart
ACS Catalysis 2013 Volume 3(Issue 8) pp:1768
Publication Date(Web):June 26, 2013
DOI:10.1021/cs400398w
The highly thermally stable anthraphos-based iridium pincer complex (iPr4Anthraphos)Ir(C2H4) 3, was shown to catalyze transfer dehydrogenation of pentene and pentane using various olefins as acceptors in the temperature range of 160–250 °C. Using pentene itself as an acceptor, disproportionation of pentene to pentane and pentadiene was observed, but yields of (E)- and (Z)-1,3-pentadienes (piperylenes) were limited to ∼25% as a result of a self-Diels–Alder reaction of the 1,3-dienes to produce isomeric mixtures of the C10 dimers. Using propylene as the acceptor, higher yields of piperylenes were obtained (∼40%), but self-Diels–Alder adducts were again observed along with low fractions of propylene/pentadiene Diels–Alder adducts. Using ethylene as the acceptor, the pentadienes produced via hydrogen transfer undergo an in situ Diels–Alder reaction with ethylene to produce 3-methylcyclohexene (along with toluene from further dehydrogenation) in good yields (65%). 3-Methylcylohexene was quantitatively dehydrogenated to toluene over a heterogeneous Pd/C catalyst.Keywords: catalysis; dehydrogenation; iridium pincer; piperylene; toluene
Co-reporter:Peng Kang, Thomas J. Meyer and Maurice Brookhart
Chemical Science 2013 vol. 4(Issue 9) pp:3497-3502
Publication Date(Web):20 Jun 2013
DOI:10.1039/C3SC51339D
A water-soluble Ir PCP-type pincer catalyst was developed to reduce CO2 to formate electrocatalytically in water with high efficiency and selectivity. Formate is the only reduced carbon product, formed in 93% Faradaic yield with no formation of CO. A small fraction of “background” H2 (ca. 7%) is directly produced at the electrode by solvent reduction. Detailed kinetic information relevant to the catalysis was obtained. The high selectivity for formate production over H2 originates from the aqueous stability of Ir dihydride species, the active species for hydride reduction of CO2. Under neutral pH, the Ir pincer complex does not catalyze the reduction of protons to H2 making water a viable solvent for use with this catalyst system. Addition of small amounts (ca. 1%) of acetonitrile reduces the over-potential and renders the catalysis sustainable. Mechanistic studies suggest that acetonitrile is a key ancillary ligand that ionizes formate effectively preventing catalyst deactivation.
Co-reporter:David Bézier, Sehoon Park, and Maurice Brookhart
Organic Letters 2013 Volume 15(Issue 3) pp:496-499
Publication Date(Web):January 14, 2013
DOI:10.1021/ol303296a
B(C6F5)3 efficiently catalyzes hydrosilylation of aliphatic and aromatic carboxylic acids to produce disilyl acetals under mild conditions. Catalyst loadings can be as low as 0.05 mol %, and bulky tertiary silanes are favored to give selectively the acetals. Acidic workup of the disilyl acetals results in the formation of aldehydes in good to excellent yields.
Co-reporter:Dr. Thomas W. Lyons ; Maurice Brookhart
Chemistry - A European Journal 2013 Volume 19( Issue 31) pp:10124-10127
Publication Date(Web):
DOI:10.1002/chem.201301502
Co-reporter:Danfeng Zhang, Enrico T. Nadres, Maurice Brookhart, and Olafs Daugulis
Organometallics 2013 Volume 32(Issue 18) pp:5136-5143
Publication Date(Web):September 10, 2013
DOI:10.1021/om400704h
Nickel(II) α-diimine dibromide complexes incorporating 8-p-tolylnaphthylimino groups have been prepared and used for ethylene polymerization by activation with modified methylalumoxane (MMAO). These catalysts possess increased axial bulk relative to standard diimine-nickel complexes, resulting in lower rates of chain transfer relative to chain propagation rates and thus higher polymer molecular weights and narrower PDIs. They yield the most highly branched PE produced by Ni catalysts seen to date. The easy synthesis of 8-aryl-1-naphthylamines provides ready access to a new class of α-diimine-based nickel catalysts in which the 8-substituent is ideally positioned to provide steric bulk at the axial sites.
Co-reporter:Michael C. Haibach, Sabuj Kundu, Maurice Brookhart, and Alan S. Goldman
Accounts of Chemical Research 2012 Volume 45(Issue 6) pp:947
Publication Date(Web):May 15, 2012
DOI:10.1021/ar3000713
Methods for the conversion of both renewable and non-petroleum fossil carbon sources to transportation fuels that are both efficient and economically viable could greatly enhance global security and prosperity. Currently, the major route to convert natural gas and coal to liquids is Fischer–Tropsch catalysis, which is potentially applicable to any source of synthesis gas including biomass and nonconventional fossil carbon sources. The major desired products of Fischer–Tropsch catalysis are n-alkanes that contain 9–19 carbons; they comprise a clean-burning and high combustion quality diesel, jet, and marine fuel. However, Fischer–Tropsch catalysis also results in significant yields of the much less valuable C3 to C8n-alkanes; these are also present in large quantities in oil and gas reserves (natural gas liquids) and can be produced from the direct reduction of carbohydrates. Therefore, methods that could disproportionate medium-weight (C3–C8) n-alkanes into heavy and light n-alkanes offer great potential value as global demand for fuel increases and petroleum reserves decrease.This Account describes systems that we have developed for alkane metathesis based on the tandem operation of catalysts for alkane dehydrogenation and olefin metathesis. As dehydrogenation catalysts, we used pincer-ligated iridium complexes, and we initially investigated Schrock-type Mo or W alkylidene complexes as olefin metathesis catalysts. The interoperability of the catalysts typically represents a major challenge in tandem catalysis. In our systems, the rate of alkane dehydrogenation generally limits the overall reaction rate, whereas the lifetime of the alkylidene complexes at the relatively high temperatures required to obtain practical dehydrogenation rates (ca. 125 −200 °C) limits the total turnover numbers. Accordingly, we have focused on the development and use of more active dehydrogenation catalysts and more stable olefin-metathesis catalysts. We have used thermally stable solid metal oxides as the olefin-metathesis catalysts. Both the pincer complexes and the alkylidene complexes have been supported on alumina via adsorption through basic para-substituents. This process does not significantly affect catalyst activity, and in some cases it increases both the catalyst lifetime and the compatibility of the co-catalysts.These molecular catalysts are the first systems that effect alkane metathesis with molecular-weight selectivity, particularly for the conversion of Cnn-alkanes to C2n–2n-alkanes plus ethane. This molecular-weight selectivity offers a critical advantage over the few previously reported alkane metathesis systems. We have studied the factors that determine molecular-weight selectivity in depth, including the isomerization of the olefinic intermediates and the regioselectivity of the pincer-iridium catalyst for dehydrogenation at the terminal position of the n-alkane.Our continuing work centers on the development of co-catalysts with improved interoperability, particularly olefin-metathesis catalysts that are more robust at high temperature and dehydrogenation catalysts that are more active at low temperature. We are also designing dehydrogenation catalysts based on metals other than iridium. Our ongoing mechanistic studies are focused on the apparently complex combination of factors that determine molecular-weight selectivity.
Co-reporter:Peng Kang ; Chen Cheng ; Zuofeng Chen ; Cynthia K. Schauer ; Thomas J. Meyer
Journal of the American Chemical Society 2012 Volume 134(Issue 12) pp:5500-5503
Publication Date(Web):March 5, 2012
DOI:10.1021/ja300543s
Iridium dihydride complexes supported by PCP-type pincer ligands rapidly insert CO2 to yield κ2-formate monohydride products in THF. In acetonitrile/water mixtures, these complexes become efficient and selective catalysts for electrocatalytic reduction of CO2 to formate. Electrochemical and NMR spectroscopic studies have provided mechanistic details and structures of key intermediates.
Co-reporter:Chen Cheng
Journal of the American Chemical Society 2012 Volume 134(Issue 28) pp:11304-11307
Publication Date(Web):July 6, 2012
DOI:10.1021/ja304547s
Catalytic reduction of secondary amides to imines and secondary amines has been achieved using readily available iridium catalysts such as [Ir(COE)2Cl]2 with diethylsilane as reductant. The stepwise reduction to secondary amine proceeds through an imine intermediate that can be isolated when only 2 equiv of silane is used. This system requires low catalyst loading and shows high efficiency (up to 1000 turnovers at room temperature with 99% conversion have been attained) and an appreciable level of functional group tolerance.
Co-reporter:Sehoon Park ; David Bézier
Journal of the American Chemical Society 2012 Volume 134(Issue 28) pp:11404-11407
Publication Date(Web):July 5, 2012
DOI:10.1021/ja305318c
Cationic silane complexes of general structure (POCOP)Ir(H)(HSiR3) {POCOP = 2,6-[OP(tBu)2]2C6H3} catalyze hydrosilylations of CO2. Using bulky silanes results in formation of bis(silyl)acetals and methyl silyl ethers as well as siloxanes and CH4. Using less bulky silanes such as Me2EtSiH or Me2PhSiH results in rapid formation of CH4 and siloxane with no detection of bis(silyl)acetal and methyl silyl ether intermediates. The catalyst system is long-lived, and 8300 turnovers can be achieved using Me2PhSiH with a 0.0077 mol % loading of iridium. The proposed mechanism for the conversion of CO2 to CH4 involves initial formation of the unobserved HCOOSiR3. This formate ester is then reduced sequentially to R3SiOCH2OSiR3, then R3SiOCH3, and finally to R3SiOSiR3 and CH4.
Co-reporter:Thomas W. Lyons ; Damien Guironnet ; Michael Findlater
Journal of the American Chemical Society 2012 Volume 134(Issue 38) pp:15708-15711
Publication Date(Web):August 30, 2012
DOI:10.1021/ja307612b
As oil supplies dwindle, there is a growing need to develop new routes to chemical intermediates that utilize alternative feedstocks. We report here a synthesis of para-xylene, one of the highest volume chemicals derived from petroleum, using only ethylene as a feedstock. Ethylene is an attractive alternative feedstock, as it can be derived from renewable biomass resources or harnessed from large domestic shale gas deposits. The synthesis relies on the conversion of hexene (from trimerization of ethylene) to 2,4-hexadiene followed by a Diels–Alder reaction with ethylene to form 3,6-dimethylcyclohexene. This monoene is readily dehydrogenated to para-xylene uncontaminated by the ortho and meta isomers. We report here a selective synthesis of para-xylene, uncontaminated by the ortho or meta isomers, using ethylene as the sole feedstock.
Co-reporter:Sehoon Park, Bong Gon Kim, Inigo Göttker-Schnetmann, and Maurice Brookhart
ACS Catalysis 2012 Volume 2(Issue 2) pp:307
Publication Date(Web):January 10, 2012
DOI:10.1021/cs200629t
[Ir(COE)2Cl]2 reacts with Et3SiH at 23 °C to form a binuclear iridium complex (Et3Si)2(H)2Ir(μ-Cl)2Ir(H)2(SiEt3)2, 1. Complex 1 reacts further with Et3SiH at 60 °C to form a second binuclear iridium complex, Et3Si(H)2Ir(μ-SiEt2)2Ir(H)2SiEt3, 2, containing bridging Et2Si groups. Activation of 2 with H2 produces trace quantities of a very highly reactive but unobservable species which rapidly and efficiently catalyzes alkyl redistribution reactions of silanes, RR′R″SiH. D2 and silane exchange experiments establish reactivity features of both 2 and the reactive intermediate. The intermediate cannot be observed, but it is likely a monomeric iridium silyl silylene complex that catalyzes alkyl scrambling via silane exchanges coupled with 1,3-alkyl migrations between silicon centers. DFT calculations support such a mechanism.Keywords: 1,3-alkyl migration; bridged silylene complexes; H2-triggered catalysis; iridium; low-T NMR spectroscopy; redistribution of silanes;
Co-reporter:Michael Findlater ; Katherine M. Schultz ; Wesley H. Bernskoetter ; Alison Cartwright-Sykes ; D. Michael Heinekey
Inorganic Chemistry 2012 Volume 51(Issue 8) pp:4672-4678
Publication Date(Web):February 24, 2012
DOI:10.1021/ic202630x
A series of iridium and rhodium pincer complexes have been synthesized and characterized: [(POCOP)Ir(H)(H2)] [BArf4] (1-H3), (POCOP)Rh(H2) (2-H2), [(PONOP)Ir(C2H4)] [BArf4] (3-C2H4), [(PONOP)Ir(H)2)] [BArf4] (3-H2), [(PONOP)Rh(C2H4)] [BArf4] (4-C2H4) and [(PONOP)Rh(H2)] [BArf4] (4-H2) (POCOP = κ3-C6H3-2,6-[OP(tBu)2]2; PONOP = 2,6-(tBu2PO)2C5H3N; BArf4 = tetrakis(3,5-trifluoromethylphenyl)borate). The nature of the dihydrogen–metal interaction was probed using NMR spectroscopic studies. Complexes 1-H3, 2-H2, and 4-H2 retain the H–H bond and are classified as η2-dihydrogen adducts. In contrast, complex 3-H2 is best described as a classical dihydride system. The presence of bound dihydrogen was determined using both T1 and 1JHD coupling values: T1 = 14 ms, 1JHD = 33 Hz for the dihydrogen ligand in 1-H3, T1(min) = 23 ms, 1JHD = 32 Hz for 2-H2, T1(min) = 873 ms for 3-H2, T1(min) = 33 ms, 1JHD = 30.1 Hz for 4-H2.
Co-reporter:Chen Cheng ;Dr. Maurice Brookhart
Angewandte Chemie 2012 Volume 124( Issue 37) pp:9556-9558
Publication Date(Web):
DOI:10.1002/ange.201205154
Co-reporter:Chen Cheng ;Dr. Maurice Brookhart
Angewandte Chemie International Edition 2012 Volume 51( Issue 37) pp:9422-9424
Publication Date(Web):
DOI:10.1002/anie.201205154
Co-reporter:Jongwook Choi, Amy H. Roy MacArthur, Maurice Brookhart, and Alan S. Goldman
Chemical Reviews 2011 Volume 111(Issue 3) pp:1761
Publication Date(Web):March 9, 2011
DOI:10.1021/cr1003503
Co-reporter:Michael Findlater ; Alison Cartwright-Sykes ; Peter S. White ; Cynthia K. Schauer
Journal of the American Chemical Society 2011 Volume 133(Issue 31) pp:12274-12284
Publication Date(Web):June 24, 2011
DOI:10.1021/ja204851x
Syntheses of the olefin hydride complexes [(POCOP)M(H)(olefin)][BArf4] (6a-M, M = Ir or Rh, olefin = C2H4; 6b-M, M = Ir or Rh, olefin = C3H6; POCOP = 2,6-bis(di-tert-butylphosphinito)benzene; BArf = tetrakis(3,5-trifluoromethylphenyl)borate) are reported. A single-crystal X-ray structure determination of 6b-Ir shows a square-pyramidal coordination geometry for Ir, with the hydride ligand occupying the apical position. Dynamic NMR techniques were used to characterize these complexes. The rates of site exchange between the hydride and the olefinic hydrogens yielded ΔG⧧ = 15.6 (6a-Ir), 16.8 (6b-Ir), 12.0 (6a-Rh), and 13.7 (6b-Rh) kcal/mol. The NMR exchange data also established that hydride migration in the propylene complexes yields exclusively the primary alkyl intermediate arising from 1,2-insertion. Unexpectedly, no averaging of the top and bottom faces of the square-pyramidal complexes is observed in the NMR spectra at high temperatures, indicating that the barrier for facial equilibration is >20 kcal/mol for both the Ir and Rh complexes. A DFT computational study was used to characterize the free energy surface for the hydride migration reactions. The classical terminal hydride complexes, [M(POCOP)(olefin)H]+, are calculated to be the global minima for both Rh and Ir, in accord with experimental results. In both the Rh ethylene and propylene complexes, the transition state for hydride migration (TS1) to form the agostic species is higher on the energy surface than the transition state for in-place rotation of the coordinated C–H bond (TS2), while for Ir, TS2 is the high point on the energy surface. Therefore, only for the case of the Rh complexes is the NMR exchange rate a direct measure of the hydride migration barrier. The trends in the experimental barriers as a function of M and olefin are in good agreement with the trends in the calculated exchange barriers. The calculated barriers for the hydride migration reaction in the Rh complexes are ∼2 kcal/mol higher than for the Ir complexes, despite the fact that the energy difference between the olefin hydride ground state and the agostic alkyl structure is ∼4 kcal/mol larger for Ir than for Rh. This feature, together with the high barrier for interchange of the top and bottom faces of the complexes, is proposed to arise from the unique coordination geometry of the agostic complexes and the strong preference for a cis-divacant octahedral geometry in four-coordinate intermediates.
Co-reporter:Sehoon Park
Journal of the American Chemical Society 2011 Volume 134(Issue 1) pp:640-653
Publication Date(Web):November 17, 2011
DOI:10.1021/ja209567m
The cationic Ir(III) acetone complex (POCOP)Ir(H)2(acetone)+ (POCOP = 2,6-bis(di-tert-butylphosphinito)phenyl) was shown to catalyze the reduction of a variety of tertiary amides to amines using diethylsilane as reductant. Mechanistic studies established that a minor species generated in the reaction, the neutral silyl trihydride Ir(V) complex (POCOP)IrH3(SiEt2H), was the catalytically active species. High concentrations of this species could be conveniently generated by treatment of readily available (POCOP)IrHCl with tert-butoxide in the presence of Et2SiH2 under H2. Thus, using this mixture in the presence of a trialkylammonium salt, a wide array of tertiary amides, including extremely bulky substrates, are rapidly and quantitatively reduced to tertiary amines under mild conditions with low catalyst loading. A detailed mechanistic study has been carried out and intermediates identified. In brief, (POCOP)IrH3(SiEt2H) reduces the amide to the hemiaminal silyl ether that, in the presence of a trialkylammonium salt, is ionized to the iminium ion, which is then reduced to the tertiary amine by Et2SiH2. Good functional group compatibility is demonstrated, and a high catalyst stability has provided turnover numbers as high as 10 000.
Co-reporter:Sehoon Park and Maurice Brookhart
Chemical Communications 2011 vol. 47(Issue 12) pp:3643-3645
Publication Date(Web):15 Feb 2011
DOI:10.1039/C0CC05714B
Cationic silane complex 2, catalyzes the hydrosilylation of epoxides and cyclic ethers to give the silyl-protected alcohols, regioselectively. A mechanistic study shows that the epoxide undergoes isomerization to the ketone, followed by hydrosilylation.
Co-reporter:Marc D. Walter, Peter S. White, Cynthia K. Schauer and Maurice Brookhart
New Journal of Chemistry 2011 vol. 35(Issue 12) pp:2884-2893
Publication Date(Web):18 Oct 2011
DOI:10.1039/C1NJ20602H
A series of cationic late transition metal pincer complexes with tridentate, neutral pincer ligands and their corresponding metal methyl complexes have been investigated by density functional theory (DFT). The key calculated quantities of interest for each metal–ligand pair were the energy of the metal methyl hydride relative to the metal σ-methane complex and the methane dissociation enthalpy and free energy. A few promising pincer ligand frameworks emerged as candidates for the syntheses of σ-methane complexes with enhanced thermal stability. The calculational predictions have been tested experimentally, and new iridium and rhodium complexes of a tridentate pincer ligand, 2,6-bis(di-tert-butylphosphinito)-3,5-diphenylpyrazine (N-PONOP) have been prepared as well as a cationic palladium methyl complex with 2,6-bis(di-tert-butylphosphinito)pyridine (PONOP) and subjected to several protonation experiments. Protonation of the (N-PONOP)Ir methyl complex yielded the corresponding five-coordinate iridium(III) methyl hydride cation. Kinetic studies of the C–H bond coupling and reductive elimination have been carried out. Line broadening NMR spectroscopic techniques have been established a barrier of 7.9(1) kcal mol−1 for H–Calkyl bond coupling in the iridium(III) methyl hydride (−100 °C). A protonation of the iridium pincer complexes at the uncoordinated pyrazine-N atom was not achieved.
Co-reporter:Stephanie A. Urbin, Tomislav Pintauer, Peter White, Maurice Brookhart
Inorganica Chimica Acta 2011 Volume 369(Issue 1) pp:150-158
Publication Date(Web):15 April 2011
DOI:10.1016/j.ica.2010.12.046
Synthesis of a series of cationic “wrap-around” complexes, η3-, η2- (CH2–CH–CHR–CH2–CH2–CHCHX) Pd(II)L+ (R = H, CH3; X = H, Cl, CO2Me; L = PPh3, P(C4H4N)3), is described. These chelate complexes were prepared by exposure of π-allyl chloride dimers, (η3-(CH2-CX-CH2)PdCl)2, to either 1,3-butadiene or isoprene to yield π-allyl chloride dimers of the type (η3-CH2CHCRCH2CH2CH = CH(X)PdCl)2 which result from insertion of the diene into each π-allyl unit. Abstraction of chloride with either AgSbF6 or NaB(ArF)4 in the presence of L gives the cationic wrap-around complexes in high yields. Single crystal X-ray diffraction studies of 8a (R = –CH3, X = –Cl, L = PPh3) and 9a (R = –H, X = –Cl, L = PPh3) show that Pd(II) adopts essentially a square planar geometry and the chelate arm occupies a syn orientation with respect to the allyl unit. Exposure of these wrap-around complexes to nitriles of differing basicities displaces the chelated alkene to varying extents and allows assessment of the relative strengths of chelation as a function of substituents, X and R. Initial rapid displacement of the chelated alkene yields a syn-π-allyl isomer which equilibrates with the anti-π-allyl isomer which cannot close to form a chelate. Treatment of 8b with 1,3-butadiene gives not polybutadiene but 2-chloro-4-methyl-1,E-4,6-heptatriene and 2-chloro-4-methyl-1,Z-4,6-heptatriene. Formation of these trienes is first-order in butadiene. This reaction serves as a model for chain-transfer in the polymerization of butadiene.Graphical abstractA series of “wrap-around” complexes of type A bearing different substituents X and R has been prepared. The relative binding strengths of the olefinic chelate arms in these derivatives was assessed by measuring the values of Keq1 as function of the basicities of a series of nitrile trapping ligands.Research highlights► Synthesis of (π-allyl)palladium(II) “wrap-around”complexes containing chelating olefinic arms. ► X-ray and NMR characterizations of the Pd(II) wrap-around complexes. ► Evaluation of the strength of chelation through titration with nitriles of varying basicities. ► Insertion of 1,3-dienes into the allyl units of the wrap-around complexes. ► Displacement of insertion products by butadiene as models for chain transfer reactions in diene polymerizations.
Co-reporter:Zheng Huang;Eleanor Rolfe;EmilyC. Carson;AlanS. Goldman;SaharH. El-Khalafy;AmyH. Roy MacArthur
Advanced Synthesis & Catalysis 2010 Volume 352( Issue 1) pp:125-135
Publication Date(Web):
DOI:10.1002/adsc.200900539
Abstract
A fully heterogeneous and highly efficient dual catalyst system for alkane metathesis (AM) has been developed. The system is comprised of an alumina-supported iridium pincer catalyst for alkane dehydrogenation/olefin hydrogenation and a second heterogeneous olefin metathesis catalyst. The iridium catalysts bear basic functional groups on the aromatic backbone of the pincer ligand and are strongly adsorbed on Lewis acid sites on alumina. The heterogeneous systems exhibit higher lifetimes and productivities relative to the corresponding homogeneous systems as catalyst/catalyst interactions and bimolecular decomposition reactions are inhibited. Additionally, using a “two-pot” device, the supported Ir catalysts and metathesis catalysts can be physically separated and run at different temperatures. This system with isolated catalysts shows very high turnover numbers and is selective for the formation of high molecular weight alkanes.
Co-reporter:Sehoon Park and Maurice Brookhart
Organometallics 2010 Volume 29(Issue 22) pp:6057-6064
Publication Date(Web):October 18, 2010
DOI:10.1021/om100818y
Hydrosilylation of a variety of ketones and aldehydes using the cationic iridium catalyst (POCOP)Ir(H)(acetone)+, 1 (POCOP = 2,6-bis(di-tert-butylphosphinito)phenyl), is reported. With triethyl silane, all but exceptionally bulky ketones undergo quantitative reactions employing 0.5 mol % catalyst in 20−30 min at 25 °C. Hydrosilylation of esters and amides results in over-reduction and cleavage of C−O and C−N bonds, respectively. The diastereoselectivity of hydrosilylation of 4-tert-butyl cyclohexanone has been examined using numerous silanes and is highly temperature dependent. Using EtMe2SiH, analysis of the ratio of cis:trans hydrosilylation products as a function of temperature yields values for ΔΔH‡ (ΔH‡(trans) − ΔH‡(cis)) and ΔΔS‡ (ΔS‡(trans) − ΔS‡(cis)) of −2.5 kcal/mol and −6.9 eu, respectively. Mechanistic studies show that the ketone complex (POCOP)Ir(H)(ketone)+ is the catalyst resting state and is in equilibrium with low concentration of the silane complex (POCOP)Ir(H)(HSiR3)+. The silane complex transfers R3Si+ to ketone, forming the oxocarbenium ion R3SiOCR′2+, which is reduced by the resulting neutral dihydride 3, (POCOP)Ir(H)2, to yield product R3SiOCHR′2 and (POCOP)IrH+, which closes the catalytic cycle.
Co-reporter:Abby R. O’Connor, Peter S. White, and Maurice Brookhart
Organometallics 2010 Volume 29(Issue 21) pp:5382-5389
Publication Date(Web):July 15, 2010
DOI:10.1021/om100391v
The synthesis and characterization of the complexes [(cyclohexenyl)Ni(NCMe)2][B(ArF)4] (1) and [(cyclohexenyl)Ni(mesitylene)][B(ArF)4] (2) are reported. Abstraction of the nitrile ligands in 1 with B(C6F5)3 yields a “ligand-free” [(cyclohexenyl)NiII]+ species which coordinates 1,3-dienes, including butadiene (BD), isoprene (IP), 1,3-cyclohexadiene (1,3-CHD), 1,4-cyclohexadiene (1,4-CHD), and 2,3-dimethyl-1,3-butadiene (DMBD) at low temperatures. Diene insertion is observed at temperatures above ca. −20 °C. An s-trans diene complex is formed upon coordination of DMBD. Hydrogen transfer is observed when complex 2 is exposed to 1-hexene or cyclopentadiene to generate new allyl nickel complexes and 1 equiv of cyclohexene. Polymerization of 1,3-CHD and DMBD with 2 is described, and a coupling product formed between DMBD and 2 is observed and characterized.
Co-reporter:Abby R. O'Connor
Journal of Polymer Science Part A: Polymer Chemistry 2010 Volume 48( Issue 9) pp:1901-1912
Publication Date(Web):
DOI:10.1002/pola.23955
Abstract
Polymerizations of 1,3-dienes using in situ generated catalyst [(2-methallyl)Ni][B(ArF)4], 6, (ArF = 3,5-bis(trifluoromethyl)phenyl) as well as [(2-methallyl)Ni(mes)][B(ArF)4], 14, (mes = mesitylene) are reported. Highly sensitive complex 6 polymerizes butadiene (BD) at –30 °C to yield polybutadiene with a Mn of ca. 10 K and 94% cis-1,4-enchainment while less reactive isoprene (IP) was polymerized at 23 °C to yield polyisoprene with Mn ca. 7 K. Complex 6 was also shown to polymerize a functionalized diene, 2,3-bis(4-trifluoroethoxy-4-oxobutyl)-1,3-BD, to polymer with Mn = 113 K. The stable and readily isolated arene complex 14 initiates BD and IP polymerizations at somewhat higher temperatures relative to 6 and delivers polymers with higher molecular weights. Complex [(allyl)Ni(mes)][B(ArF)4], 13, catalyzes polymerization of styrene to yield polystyrene with high conversion, Mn's = ca. 6 K and MWD = 2. The π-benzyl complex [(η3-1-methylbenzyl)Ni(mes)] [B(ArF)4], 19, was detected as an intermediate following chain transfer by in situ NMR studies. © 2010 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 48: 1901–1912, 2010
Co-reporter:Wesley H. Bernskoetter;Cynthia K. Schauer;Karen I. Goldberg
Science 2009 Vol 326(5952) pp:553-556
Publication Date(Web):23 Oct 2009
DOI:10.1126/science.1177485
Co-reporter:Marc D. Walter ; Rebecca A. Moorhouse ; Stephanie A. Urbin ; Peter S. White
Journal of the American Chemical Society 2009 Volume 131(Issue 25) pp:9055-9069
Publication Date(Web):May 27, 2009
DOI:10.1021/ja9025598
Several cationic (allyl)Pd(II) complexes were synthesized and shown to be highly active for (2,3)-vinyl addition polymerization of norbornene (NB) to yield polymers with low molecular weight distributions (MWDs) ranging from 1.2−1.4. Despite the low MWDs, slow initiation was followed by rapid propagation preventing molecular weight control of the poly(norbornene). Several intermediates in these polymerizations initiated with [(2-R-allyl)Pd(mesitylene)]+ complexes were fully characterized (NMR and X-ray diffraction). Consistent with previous observations the allyl and NB units couple in cis-exo fashion to yield a σ,π-complex capped by mesitylene. Mesitylene is readily displaced by NB to form an agostic intermediate in which NB acts as a bidentate ligand and binds to the cationic Pd center via the π-system and a γ-agostic interaction with the syn hydrogen at C7. The identity of this complex was established by NMR spectroscopy and single-crystal X-ray diffraction. It is significant since it suggests bidentate binding of NB in the propagating species, which cannot be observed by NMR spectroscopy. The NMR studies suggest that the second insertion, i.e., insertion of NB in the agostic intermediate, is the slow initiation step and the subsequent insertions are extremely fast. Therefore, slow chelate opening is the major limitation preventing a living polymerization. This hypothesis was explored using a series of cationic substituted π-allyl complexes; significantly increased reactivity was observed when electron-withdrawing groups were introduced into the allyl moiety. However, despite these modifications initiation remained slow relative to chain propagation.
Co-reporter:Wesley H. Bernskoetter ; Susan Kloek Hanson ; Sara K. Buzak ; Zoe Davis ; Peter S. White ; Rodney Swartz ; Karen I. Goldberg
Journal of the American Chemical Society 2009 Volume 131(Issue 24) pp:8603-8613
Publication Date(Web):June 2, 2009
DOI:10.1021/ja901706b
New iridium complexes of a tridentate pincer ligand, 2,6-bis(di-tert-butylphosphinito)pyridine (PONOP), have been prepared and used in the study of hydrocarbon C−H bond activation. Intermolecular oxidative addition of a benzene C−H bond was directly observed with [(PONOP)IrI(cyclooctene)][PF6] at ambient temperature, resulting in a cationic five-coordinate iridium(III) phenyl hydride product. Protonation of the (PONOP)IrI methyl complex yielded the corresponding iridium(III) methyl hydride cation, a rare five-coordinate, 16-valence electron transition metal alkyl hydride species which was characterized by X-ray diffraction. Kinetic studies of C−H bond coupling and reductive elimination reactions from the five-coordinate complexes have been carried out. Exchange NMR spectroscopy measurements established a barrier of 17.8(4) kcal/mol (22 °C) for H−Caryl bond coupling in the iridium(III) phenyl hydride cation and of 9.3(4) kcal/mol (−105 °C) for the analogous H−Calkyl coupling in the iridium(III) methyl hydride cation. The origin of the higher barrier of H−Caryl relative to H−Calkyl bond coupling is proposed to be influenced by a hindered rotation about the Ir−Caryl bond, a result of the sterically demanding PONOP ligand.
Co-reporter:Marc D. Walter, Peter S. White and Maurice Brookhart
Chemical Communications 2009 (Issue 42) pp:6361-6363
Publication Date(Web):28 Sep 2009
DOI:10.1039/B916457J
Cationic [(tmeda)Pd(OEt2)(Me)][B(ArF)4] was synthesized by protonation of (tmeda)PdMe2 and allowed for the first time the nature of the propagating species in the vinyl addition polymerization of norbornene to be discerned. In this system γ-agostic interactions stabilize the propagating species and permit stepwise monomer insertions to be observed by 1H NMR spectroscopy. This and earlier studies provide a new proposal for the propagating species in ligand-less late transition metal-catalyzed norbornene polymerizations.
Co-reporter:Jian Yang
Advanced Synthesis & Catalysis 2009 Volume 351( Issue 1-2) pp:175-187
Publication Date(Web):
DOI:10.1002/adsc.200800528
Abstract
A highly efficient procedure for the reduction of a broad range of alkyl halides by triethylsilane based on a cationic iridium bis(phosphinite) pincer catalyst has been discovered and developed. This reduction chemistry is chemoselective and has unique selectivities compared with conventional radical-based processes and the aluminum trichloride/triethylsilane (AlCl3/Et3SiH) and triphenylmethyl tetrakis[pentafluorophenyl]borate/triethylsilane {[Ph3C] [B(C6F5)4]/Et3SiH} systems. Reductions use three equivalents of triethylsilane relative to the halide and can be carried out with very low catalyst loadings and in a solvent-free manner, which may provide an environmentally attractive and safe alternative to many currently practiced methods for reduction of alkyl halides. Mechanistic studies reveal a unique catalytic cycle. The cationic iridium hydride 2,6-bis[di-(tert-butyl)phosphinyloxy)phenyl(hydrido)iridium, (POCOP)IrH+ {POCOP= 2,6-[OP(t-Bu)2]2C6H3} binds and activates the silane. This complex serves as a potent silylating reagent to generate silyl halonium ions, Et3SiXR+, which are reduced by the neutral iridium dihydride to yield alkane product and regenerate the cationic (POCOP)IrH+, thus closing the catalytic cycle. All key intermediates have been identified by in situ NMR monitoring and kinetic studies have been completed. An application of this reduction system to the catalytic hydrodehalogenation of a metal chloride complex is also described.
Co-reporter:Francis C. Rix, Michael J. Rachita, Mark I. Wagner, Maurice Brookhart, Barbara Milani and James C. Barborak
Dalton Transactions 2009 (Issue 41) pp:8977-8992
Publication Date(Web):03 Sep 2009
DOI:10.1039/B911392D
A mechanistic interpretation of the [(1,10-phenanthroline)Pd(CH3)(CH3CN)]+[BArF]− (1a) and [(2,2′-bipyridine)Pd(CH3)(CH3CN)]+[BArF]− (1b) (BArF = 3,5-(CF3)2-C6H3) catalyzed perfectly alternating copolymerization of styrenes with CO is reported. The copolymerization in CH2Cl2 or chlorobenzene has been found to be first order in styrene and inverse first order in CO concentrations. The microscopic steps involved in the catalytic cycle have been studied via low temperature NMR techniques. Palladium alkyl chelate complex [(2,2′-bipyridine)Pd(CHArCH2C(O)CH3]+[BArF]− (5bσ) and [(2,2′-bipyridine)Pd(η3-CH(CH2C(O)CH3)Ar)]+[BArF]− (5bπ), existing in equilibrium, were prepared. Treatment of 5σ,π with 13CO followed by 4-tert-butylstyrene at −78 oC allowed for 13C NMR monitoring of the alternating chain growth of a series of palladium acyl carbonyl complexes. The acyl carbonyl species, representing the catalyst resting state, is in equilibrium with a palladium acyl styrene complex. The equilibrium constant, K4, measured between [(phen)Pd(CO)(C(O)CH3]+[BArF]− (3a) and [(phen)Pd(C(O)CH3)-(C6H5CCH2)]+[BArF]− (8a), was determined to be 2.84 ± 2.8 × 10−7 at −66 °C. The barrier to migratory insertion in 8a was determined (ΔG‡ (−66 °C) = 15.6 ± 0.1 kcal mol−1). From the experimentally determined kinetic and thermodynamic data for the copolymerization of styrene with CO a mechanistic model has been constructed. The ability of this model to predict catalyst turnover frequency (TOF) was used as a test of its validity. A series of para-substituted styrenes, p-XC6H4CHCH2 (X = –OCH3, –CH3, –H, –Cl), were copolymerized with CO. A Hammett treatment of TOF for the series showed that electron-donating groups increase the rate of copolymerization (ρp = −0.8). The ratio of chain transfer to chain propagation was found to increase with styrene concentration and decrease with CO concentration. Polymer end group analysis showed the presence of α, β-enone end groups. The reactivity of model systems, coupled with a study of the effect of added acetonitrile, support a chain transfer mechanism involving β-hydrogen transfer to monomer from a palladium alkyl styrene intermediate.
Co-reporter:Marc D. Walter;Rebecca A. Moorhouse;Peter S. White
Journal of Polymer Science Part A: Polymer Chemistry 2009 Volume 47( Issue 10) pp:2560-2573
Publication Date(Web):
DOI:10.1002/pola.23340
Abstract
Several cationic (allyl)Ni(II) complexes were synthesized and shown to be highly active for (2,3)-vinyl addition polymerization of norbornene to yield polymers with low molecular weight distributions (MWDs) ranging from 1.4–1.9. In all cases slow initiation was followed by rapid propagation which prevents molecular weight control of the poly(norbornene). One of the intermediates in the polymerization process has been identified and characterized by NMR spectroscopy as the first insertion product resulting from the insertion of norbornene into the NiC allyl bond in cis-exo fashion. This insertion product was synthesized independently and NMR studies showed that the first insertion of norbornene into the NiC allyl bond is a reversible process. © 2009 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 47: 2560–2573, 2009
Co-reporter:Abby R. O’Connor, Stephanie A. Urbin, Rebecca A. Moorhouse, Peter S. White and Maurice Brookhart
Organometallics 2009 Volume 28(Issue 8) pp:2372-2384
Publication Date(Web):April 2, 2009
DOI:10.1021/om801103p
[(2-R-allyl)M(arene)]+ complexes (M = Pd, Ni; R = H, CH3, Cl; arene = mesitylene, hexamethylbenzene) have been synthesized via halide abstraction from the corresponding allyl halide dimers, [(allyl)MX]2, using either AgSbF6 in the case of M = Pd or NaB(Arf)4 (Arf = 3,5-(CF3)2C6H3) in the case of M = Ni. The [(allyl)Ni(mesitylene)]+ and [(2-methallyl)Ni(hexamethylbenzene)]+ salts have been characterized by single-crystal X-ray diffraction. The arene ligands in the Pd species are highly labile. The mesitylene ligand in the [(2-R-allyl)Pd(mesitylene)]+ complexes is rapidly displaced at temperatures as low as −120 °C by olefins and alkynes (ethylene, tert-butylethylene, cyclopentene, cyclohexene, cyclooctene, 2-butyne) to yield the bis-olefin or bis-alkyne complexes, which have been characterized by NMR spectroscopy. [(allyl)Pd(mesitylene)]+ undergoes rapid degenerate exchange with free mesitylene at low temperatures (ΔG⧧ = 10.2 kcal/mol). The arene ligand of the Ni complexes is less labile. Displacement of mesitylene from [(allyl)Ni(mesitylene)]+ by excess diethyl ether at 25 °C yields [(allyl)Ni(Et2O)2]+. Reaction of the [(2-R-allyl)Ni(mesitylene)]+ complexes (R = H, CH3) with α-olefins at 25 °C yields new allyl complexes plus propene (when R = H) or isobutylene (when R = CH3). A mechanism involving intramolecular hydrogen migration is proposed to account for these transformations.
Co-reporter:Jian Yang ; Peter S. White
Journal of the American Chemical Society 2008 Volume 130(Issue 51) pp:17509-17518
Publication Date(Web):November 20, 2008
DOI:10.1021/ja806419h
The cationic iridium pincer complex [(POCOP)Ir(H)(acetone)]+[B(C6F5)4]− {1, POCOP = 2,6-[OP(tBu)2]2C6H3} was found to be a highly active catalyst for the room-temperature cleavage and reduction of a wide variety of unactivated alkyl ethers including primary, secondary, and tertiary alkyl ethers as well as aryl alkyl ethers by triethylsilane. Mechanistic studies have revealed the full details of the catalytic cycle with the catalyst resting state(s) depending on the basicity of the alkyl ether. During the catalytic reduction of diethyl ether, cationic iridium silane complex, [(POCOP)Ir(H)(η1-Et3SiH)]+[B(C6F5)4]− (3), and Et2O are in rapid equilibrium with neutral dihydride, (POCOP)Ir(H)2 (5) and diethyl(triethylsilyl)oxonium ion, [Et3SiOEt2]+[B(C6F5)4]− (7), with 5 + 7 strongly favored. Species 7 has been isolated from the reaction mixture and fully characterized. The turnover-limiting step in this cycle is the reduction of 7 by the neutral dihydride 5. The relative rates of reduction of 7 by dihydride 5 and Et3SiH were determined to be ∼30,000:1. In the cleavage of the less basic ethers anisole and EtOSiEt3, the cationic iridium silane complex, 3, was found to be the catalyst resting state. The hydride reduction of the intermediate oxonium ion EtO(SiEt3)2+, 9, occurs via attack by Et3SiH. In the case of anisole, the intermediate PhMeOSiEt3+, 10, is reduced by 5 and/or Et3SiH.
Co-reporter:Jian Yang;PeterS. White Dr.;CynthiaK. Schauer
Angewandte Chemie International Edition 2008 Volume 47( Issue 22) pp:4141-4143
Publication Date(Web):
DOI:10.1002/anie.200705359
Co-reporter:Wesley H. Bernskoetter
Organometallics 2008 Volume 27(Issue 9) pp:2036-2045
Publication Date(Web):April 1, 2008
DOI:10.1021/om701148t
A family of primary amines has been catalytically dehydrogenated to nitriles by [C6H3-2,6-(OPtBu2)2]IrH2 using tert-butylethylene as a hydrogen acceptor. The catalytic mechanism has been investigated by a series of kinetic and isotopic labeling experiments, in addition to the isolation of intermediates along the reaction pathway. The turnover frequency exhibits a first-order dependence on the concentration of amine, an inverse first-order dependence on nitrile, and a zero-order dependence on tert-butylethylene. The mechanism of amine dehydrogenation is proposed to proceed from an iridium(I) nitrile complex, the catalyst resting state, via two preturnover limiting equilibria, followed by a slow β-hydride elimination event from a transient iridium(III) amido hydride species. Measuring rate constants for the stoichiometric dehydrogenation of isobutylamine over a 41 °C temperature range established activation parameters for β-hydride elimination of ΔH⧧ 24.8(9) kcal/mol and ΔS⧧ = −10(2) eu. Dehydrogenation of a series of para-substituted benzyl amines indicated enhanced conversions to nitriles for substrates bearing electron-donating substituents.
Co-reporter:Eric L. Dias, Maurice Brookhart and Peter S. White
Chemical Communications 2001 (Issue 5) pp:423-424
Publication Date(Web):14 Feb 2001
DOI:10.1039/B007815H
The remarkably stable cationic, three-coordinate, 14-electron
rhodium complex 1 has been synthesized, isolated and used as a catalyst for
hydrosilation, Mukaiyama aldol and cyclopropanation reactions.
Co-reporter:Volker P. W. Böhm Dr. Dr.
Angewandte Chemie 2001 Volume 113(Issue 24) pp:
Publication Date(Web):18 DEC 2001
DOI:10.1002/1521-3757(20011217)113:24<4832::AID-ANGE4832>3.0.CO;2-V
Gleich zwei Vorschriften wurden für die katalytische Dehydrokupplung sekundärer Phosphane mit dem Rhodium(I)-Komplex [Cp*Rh(CH2=CHSiMe3)2] entwickelt: In Gegenwart von Olefinen findet eine Transferhydrierung zum entsprechenden Alkan und die Bildung des Diphosphans statt. Ohne Olefinzusatz und bei erhöhten Temperaturen entsteht das Diphosphan unter Freisetzung von molekularem Wasserstoff [Gl. (1)].
Co-reporter:Volker P. W. Böhm Dr. Dr.
Angewandte Chemie International Edition 2001 Volume 40(Issue 24) pp:
Publication Date(Web):18 DEC 2001
DOI:10.1002/1521-3773(20011217)40:24<4694::AID-ANIE4694>3.0.CO;2-7
Two reaction protocols have been developed for the catalytic dehydrocoupling of secondary phosphanes by the rhodium(I) complex [Cp*Rh{CH2=CH(TMS)}2]: In the presence of an olefin, transfer hydrogenation occurs to give the corresponding alkane and the diphosphane. Without the addition of an olefin, the reaction proceeds by loss of dihydrogen but more elevated reaction temperatures must be used [Eq. (1)].
Co-reporter:Maurice Brookhart ;Brian E. Grant Dr.;Christian P. Lenges Dipl.-Chem.;Marc H. Prosenc Dr.;Peter S. White Dr.
Angewandte Chemie 2000 Volume 112(Issue 9) pp:
Publication Date(Web):2 MAY 2000
DOI:10.1002/(SICI)1521-3757(20000502)112:9<1742::AID-ANGE1742>3.0.CO;2-V
Co-reporter:R. Leigh Huff;Michael J. Rachita;Jordan L. Bennett
Journal of Polymer Science Part A: Polymer Chemistry 2000 Volume 38(Issue S1) pp:4627-4640
Publication Date(Web):27 NOV 2000
DOI:10.1002/1099-0518(200012)38:1+<4627::AID-POLA20>3.0.CO;2-9
A series of neutral phosphinosulfonamide complexes of nickel(II) were synthesized that catalyzed the oligomerization of ethylene to branched oligomers with average degrees of polymerization between 10 and 35. Branching numbers varied from 17 to 80 branches per 1000 carbons, depending on the catalyst structure and reaction conditions. The catalysts were active in a variety of solvents, including toluene, CH2Cl2, tetrahydrofuran, ethyl acetate, and methanol, but showed decreasing activity at temperatures higher than 40 °C. Electron-rich phosphinosulfonamides produced the highest catalyst activities in a series of structure–reactivity studies. The mechanism of oligomer formation was investigated with 1H NMR spectroscopy, which indicated that branching arose from the isomerization of the nickel alkyl species during propagation rather than the reincorporation of α-olefin products. © 2000 John Wiley & Sons, Inc. J Polym Sci A: Polym Chem 38: 4627–4640, 2000
Co-reporter:Soumik Biswas ; Zheng Huang ; Yuriy Choliy ; David Y. Wang ; Maurice Brookhart ; Karsten Krogh-Jespersen ;Alan S. Goldman
Journal of the American Chemical Society () pp:
Publication Date(Web):July 5, 2012
DOI:10.1021/ja301464c
The isomerization of olefins by complexes of the pincer-ligated iridium species (tBuPCP)Ir (tBuPCP = κ3-C6H3-2,6-(CH2PtBu2)2) and (tBuPOCOP)Ir (tBuPOCOP = κ3-C6H3-2,6-(OPtBu2)2) has been investigated by computational and experimental methods. The corresponding dihydrides, (pincer)IrH2, are known to hydrogenate olefins via initial Ir–H addition across the double bond. Such an addition is also the initial step in the mechanism most widely proposed for olefin isomerization (the “hydride addition pathway”); however, the results of kinetics experiments and DFT calculations (using both M06 and PBE functionals) indicate that this is not the operative pathway for isomerization in this case. Instead, (pincer)Ir(η2-olefin) species undergo isomerization via the formation of (pincer)Ir(η3-allyl)(H) intermediates; one example of such a species, (tBuPOCOP)Ir(η3-propenyl)(H), was independently generated, spectroscopically characterized, and observed to convert to (tBuPOCOP)Ir(η2-propene). Surprisingly, the DFT calculations indicate that the conversion of the η2-olefin complex to the η3-allyl hydride takes place via initial dissociation of the Ir–olefin π-bond to give a σ-complex of the allylic C–H bond; this intermediate then undergoes C–H bond oxidative cleavage to give an iridium η1-allyl hydride which “closes” to give the η3-allyl hydride. Subsequently, the η3-allyl group “opens” in the opposite sense to give a new η1-allyl (thus completing what is formally a 1,3 shift of Ir), which undergoes C–H elimination and π-coordination to give a coordinated olefin that has undergone double-bond migration.
Co-reporter:Sehoon Park and Maurice Brookhart
Chemical Communications 2011 - vol. 47(Issue 12) pp:NaN3645-3645
Publication Date(Web):2011/02/15
DOI:10.1039/C0CC05714B
Cationic silane complex 2, catalyzes the hydrosilylation of epoxides and cyclic ethers to give the silyl-protected alcohols, regioselectively. A mechanistic study shows that the epoxide undergoes isomerization to the ketone, followed by hydrosilylation.
Co-reporter:Marc D. Walter;Peter S. White
Chemical Communications 2009(Issue 42) pp:NaN6363-6363
Publication Date(Web):2009/11/14
DOI:10.1039/B916457J
Cationic [(tmeda)Pd(OEt2)(Me)][B(ArF)4] was synthesized by protonation of (tmeda)PdMe2 and allowed for the first time the nature of the propagating species in the vinyl addition polymerization of norbornene to be discerned. In this system γ-agostic interactions stabilize the propagating species and permit stepwise monomer insertions to be observed by 1H NMR spectroscopy. This and earlier studies provide a new proposal for the propagating species in ligand-less late transition metal-catalyzed norbornene polymerizations.
Co-reporter:Peng Kang, Thomas J. Meyer and Maurice Brookhart
Chemical Science (2010-Present) 2013 - vol. 4(Issue 9) pp:NaN3502-3502
Publication Date(Web):2013/06/20
DOI:10.1039/C3SC51339D
A water-soluble Ir PCP-type pincer catalyst was developed to reduce CO2 to formate electrocatalytically in water with high efficiency and selectivity. Formate is the only reduced carbon product, formed in 93% Faradaic yield with no formation of CO. A small fraction of “background” H2 (ca. 7%) is directly produced at the electrode by solvent reduction. Detailed kinetic information relevant to the catalysis was obtained. The high selectivity for formate production over H2 originates from the aqueous stability of Ir dihydride species, the active species for hydride reduction of CO2. Under neutral pH, the Ir pincer complex does not catalyze the reduction of protons to H2 making water a viable solvent for use with this catalyst system. Addition of small amounts (ca. 1%) of acetonitrile reduces the over-potential and renders the catalysis sustainable. Mechanistic studies suggest that acetonitrile is a key ancillary ligand that ionizes formate effectively preventing catalyst deactivation.
Co-reporter:Francis C. Rix, Michael J. Rachita, Mark I. Wagner, Maurice Brookhart, Barbara Milani and James C. Barborak
Dalton Transactions 2009(Issue 41) pp:NaN8992-8992
Publication Date(Web):2009/09/03
DOI:10.1039/B911392D
A mechanistic interpretation of the [(1,10-phenanthroline)Pd(CH3)(CH3CN)]+[BArF]− (1a) and [(2,2′-bipyridine)Pd(CH3)(CH3CN)]+[BArF]− (1b) (BArF = 3,5-(CF3)2-C6H3) catalyzed perfectly alternating copolymerization of styrenes with CO is reported. The copolymerization in CH2Cl2 or chlorobenzene has been found to be first order in styrene and inverse first order in CO concentrations. The microscopic steps involved in the catalytic cycle have been studied via low temperature NMR techniques. Palladium alkyl chelate complex [(2,2′-bipyridine)Pd(CHArCH2C(O)CH3]+[BArF]− (5bσ) and [(2,2′-bipyridine)Pd(η3-CH(CH2C(O)CH3)Ar)]+[BArF]− (5bπ), existing in equilibrium, were prepared. Treatment of 5σ,π with 13CO followed by 4-tert-butylstyrene at −78 oC allowed for 13C NMR monitoring of the alternating chain growth of a series of palladium acyl carbonyl complexes. The acyl carbonyl species, representing the catalyst resting state, is in equilibrium with a palladium acyl styrene complex. The equilibrium constant, K4, measured between [(phen)Pd(CO)(C(O)CH3]+[BArF]− (3a) and [(phen)Pd(C(O)CH3)-(C6H5CCH2)]+[BArF]− (8a), was determined to be 2.84 ± 2.8 × 10−7 at −66 °C. The barrier to migratory insertion in 8a was determined (ΔG‡ (−66 °C) = 15.6 ± 0.1 kcal mol−1). From the experimentally determined kinetic and thermodynamic data for the copolymerization of styrene with CO a mechanistic model has been constructed. The ability of this model to predict catalyst turnover frequency (TOF) was used as a test of its validity. A series of para-substituted styrenes, p-XC6H4CHCH2 (X = –OCH3, –CH3, –H, –Cl), were copolymerized with CO. A Hammett treatment of TOF for the series showed that electron-donating groups increase the rate of copolymerization (ρp = −0.8). The ratio of chain transfer to chain propagation was found to increase with styrene concentration and decrease with CO concentration. Polymer end group analysis showed the presence of α, β-enone end groups. The reactivity of model systems, coupled with a study of the effect of added acetonitrile, support a chain transfer mechanism involving β-hydrogen transfer to monomer from a palladium alkyl styrene intermediate.
.jpg)




![Chloro(pentamethylcyclopentadienyl)[(2-pyridinyl-kN)phenyl-kC]iridum(III)](http://img.cochemist.com/ccimg/945500/945491-51-0.png)
![Chloro(pentamethylcyclopentadienyl)[(2-pyridinyl-kN)phenyl-kC]iridum(III)](http://img.cochemist.com/ccimg/945500/945491-51-0_b.png)


![Benzene, 1-[(1,1-dimethylethyl)sulfonyl]-2-iodosyl-](http://img.cochemist.com/ccimg/242500/242461-32-1.png)
![Benzene, 1-[(1,1-dimethylethyl)sulfonyl]-2-iodosyl-](http://img.cochemist.com/ccimg/242500/242461-32-1_b.png)


![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)