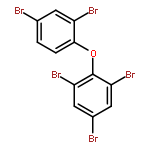
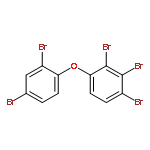
Co-reporter:Xiao-yu Jia, Di-rong Gong, Yi Han, Tai-cheng Duan and Hang-ting Chen
Analytical Methods 2012 vol. 4(Issue 2) pp:575-580
Publication Date(Web):25 Jan 2012
DOI:10.1039/C2AY05740A
An on-line anion-exchange membrane separation system has been coupled to inductively coupled plasma mass spectrometry (ICP-MS) for the total vanadium(V) and chromium (Cr) determination and V(IV) and Cr(III) species determination simultaneously. The positive charged V(IV) and Cr(III) species have been efficiently separated from V(V) and Cr(VI), and selectively determined by the on-line electrodialyzer-ICP-MS. To determine the total concentrations of V and Cr in water samples, H2SO4 and Na2S2O3 were added to convert all vanadium and chromium species into their positive species, thus, V(V) and Cr(VI) in the samples were quantified by subtraction. The cool plasma and reaction gas can also be avoided as the severe interference from chloride, carbon and nitrogen-based molecular ion has been substantially suppressed by the anion-exchange membrane. The detection limits of the analytes were 0.02 μg L−1 for V(IV) and 0.06 μg L−1 for Cr(III) with this on-line system, respectively, and the relative standard deviation (n = 10) of 10 μg L−1V(IV) and Cr(III) were 3.7% and 2.1%, respectively. The developed method has been applied for the speciation of V and Cr in three water samples, with V(V) and Cr(III) found as the main species in these samples. The accuracy and feasibility of this method have also been verified by spike tests and a certified reference material (CRM) of environmental water (GSBZ 50029-94) and a CRM of riverine water (GBW 08608), good recoveries have been obtained and the analytical results are also in good agreement with the certified values.
Co-reporter:Xiaoyu Jia, Yi Han, Chao Wei, Taicheng Duan and Hangting Chen
Journal of Analytical Atomic Spectrometry 2011 vol. 26(Issue 7) pp:1380-1386
Publication Date(Web):17 Mar 2011
DOI:10.1039/C0JA00121J
Ionic liquid based dispersive liquid–liquid microextraction (IL-DLLME) combined with high performance liquid chromatography-inductively coupled plasma mass spectrometry (HPLC-ICP-MS) for the determination of mercury species in liquid cosmetic samples is described. Firstly mercury (Hg2+), methylmercury (MeHg+) and ethylmercury (EtHg+) were complexed with ammonium pyrrolidinedithiocarbamate (APDC), and then the complexes were extracted into 1-hexyl-3-methylimidazolium hexafluorophosphate ([C6MIM][PF6]) using DLLME. Under the optimized conditions, the enrichment factors of 760, 115, 235 for Hg2+, MeHg+ and EtHg+ were obtained from only 5.00 mL sample solution. The detection limits of the analytes (as Hg) were 1.3 ng L−1 for Hg2+, 7.2 ng L−1 for MeHg+ and 5.4 ng L−1 for EtHg+, respectively. The relative standard deviation (n = 10) of 0.5 ng mL−1 Hg2+, MeHg+ and EtHg+ were 7.4%, 5.2% and 2.3%, respectively. Finally, the developed method was successfully applied for the speciation of mercury in four liquid cosmetic samples, Hg2+ was found as the main species in these samples, and a spike test was performed to verify the accuracy of the method.
Co-reporter:Yi Han, Xiaoyu Jia, Taicheng Duan and Hangting Chen
Analytical Methods 2011 vol. 3(Issue 4) pp:842-848
Publication Date(Web):17 Feb 2011
DOI:10.1039/C0AY00742K
A simple, inexpensive and reliable analytical method was developed for the determination of polybrominated diphenyl ethers (PBDEs) in whole milk by gas chromatography-mass spectrometry (GC-MS). Matrix components such as proteins and fats in the whole milk were removed by saponification procedure and the combined PBDEs were released. Then, a simple one-step liquid–liquid extraction procedure in the centrifuge tube (in-tube LLE) was performed and PBDEs were extracted into petroleum ether. After centrifugation, dehydration and nitrogen drying of petroleum ether phase, the extract was dissolved in acetonitrile. The acetonitrile solution was directly used as dispersant for the subsequent dispersive liquid–liquid microextraction (DLLME). Under the optimal conditions, the enrichment factors (EFs) of PBDEs ranged from 270 to 307. The calibration was performed by use of matrix-matched calibration standards, the correlation coefficients were better than 0.99 and the limits of detection (LODs) ranged from 0.012 to 0.29 μg L−1. The recoveries for the whole milk samples analysis spiked at 1 μg L−1 were between 78% and 120% with relative standard deviations (RSDs) ranging from 1.7% to 11% (n = 5). In this study, a simple, valid and combined sample preparation method was applied in the analysis of PBDEs in whole milk sample and the satisfactory recoveries for PBDEs were obtained.
Co-reporter:Xiaoyu Jia, Yi Han, Xinli Liu, Taicheng Duan, Hangting Chen
Spectrochimica Acta Part B: Atomic Spectroscopy 2011 Volume 66(Issue 1) pp:88-92
Publication Date(Web):January 2011
DOI:10.1016/j.sab.2010.12.003
The dispersive liquid–liquid microextraction (DLLME) combined with high performance liquid chromatography-inductively coupled plasma mass spectrometry for the speciation of mercury in water samples was described. Firstly methylmercury (MeHg+) and mercury (Hg2+) were complexed with sodium diethyldithiocarbamate, and then the complexes were extracted into carbon tetrachloride by using DLLME. Under the optimized conditions, the enrichment factors of 138 and 350 for MeHg+ and Hg2+ were obtained from only 5.00 mL sample solution. The detection limits of the analytes (as Hg) were 0.0076 ng mL−1 for MeHg+ and 0.0014 ng mL−1 for Hg2+, respectively. The relative standard deviations for ten replicate measurements of 0.5 ng mL−1 MeHg+ and Hg2+ were 6.9% and 4.4%, respectively. Standard reference material of seawater (GBW(E)080042) was analyzed to verify the accuracy of the method and the results were in good agreement with the certified values. Finally, the developed method was successfully applied for the speciation of mercury in three environmental water samples.
Co-reporter:Xinli Liu, Taicheng Duan, Yi Han, Xiaoyu Jia and Hangting Chen
Journal of Analytical Atomic Spectrometry 2010 vol. 25(Issue 2) pp:206-209
Publication Date(Web):18 Nov 2009
DOI:10.1039/B915054D
The determination of As(III) in high purity Sb2O3 was taken as example to illustrate a high selective solid phase extraction (SPE) with non-polar resins as the sorbent. The extraction was based on the fact that in concentrated HCl solutions, As(III) forming molecular covalent AsCl3 could be attracted by the hydrophobic phase of the resins, whereas the matrix Sb existing only as the ionic Sb–Cl complex could not be adsorbed. The SPE column was made from the Amberlite XAD-16 resin and was on-line coupled with hydride generation atomic fluorescence spectrometer for As(III) detection. Under the optimized conditions, the detection limit (3σ) of 0.26 μg g−1, the RSD (n = 6) of 6.2% for 50 μg L−1 of As(III) and a linear calibration curve from 2 μg L−1 to 200 μg L−1 with R = 0.9998 were obtained. The recoveries for 10 μg L−1 of As(III) spiked in 1 g L−1 of Sb(III) solution was in the range of 97–102%. The method was applied to the determination of trace As(III) in two highly pure Sb2O3 samples.
Co-reporter:Xin-Li LIU, Tai-Cheng DUAN, Yi HAN, Xiao-Yu JIA, Wei-Na ZHANG, Hang-Ting CHEN
Chinese Journal of Analytical Chemistry 2010 Volume 38(Issue 5) pp:693-696
Publication Date(Web):May 2010
DOI:10.1016/S1872-2040(09)60044-X
Slurry nebulization-inductively coupled plasma mass spectrometry (ICP-MS) was developed for the direct determination of trace boron (B) in high-purity graphite powders. After the graphite powders were ground and sifted, the particle size of graphite was collected less than 5 μm. Polyvinylpyrrolidone (PVP) was used as the dispersant in slurry preparation. The optimal mass ratio of PVP to the graphite was found to be 0.5. Well-proportioned and stable slurry solution was obtained by magnetic stirrer. 0.1 M NH4OH as the aqueous medium could provide the optimal pH of 10 for the stable slurry and eliminate the memory effect of B. ICP-MS was operated in a higher resolution mode (0.6 amu) to eliminate the interference with the matrix 12C by peak tailing. Oxygen was added into the plasma at a flow rate of 60 mL min−1 to resolve carbon deposition on the sampler and skimmer cones and accelerate particle dissociation in the plasma. External calibration with aqueous solution standards was established for quantification. Beryllium was chosen as the internal standard to evaluate the efficiency of matrix effect correction. A correlation coefficient of 0.9995 was obtained for B concentration ranging 2–200 μg L−1. The detection limit (3S) of B was 0.095 μg g−1. As a practical application, the proposed method was used for the determination of trace B in four nuclear graphite samples (claimed 99.999% purity), with the satisfactory recoveries for the spike tests in the range of 97.2%–103.1%.
Co-reporter:Xiaoyu Jia;Yi Han;Xinli Liu;Taicheng Duan;Hangting Chen
Microchimica Acta 2010 Volume 171( Issue 1-2) pp:49-56
Publication Date(Web):2010 October
DOI:10.1007/s00604-010-0402-5
Dispersive liquid–liquid microextraction (DLLME) was combined with flow injection inductively coupled plasma mass spectrometry for simultaneous determination of cadmium, lead and bismuth in water samples. The metal elements were complexed with sodium diethyldithiocarbamate, and then the complexes were extracted into carbon tetrachloride by using DLLME. Under the optimized conditions, the enrichment factors for Cd, Pb and Bi are 460, 900 and 645 in 5 mL of a spiked water sample, respectively. The calibration graphs for the three metals are linear in the range of concentrations from <10 ng L−1 to 1,000 ng L−1. The detection limits are 0.5 ng L−1, 1.6 ng L−1 and 4.7 ng L−1, respectively. The relative standard deviations for ten replicate measurements of 50 ng L−1 cadmium, lead and bismuth are 2.6%, 6.7%, and 4.9%, respectively, and the relative recoveries in various water samples at a spiking level of 50 ng L−1 range from 83.6% to 107.0%.
Co-reporter:Pengran Guo, Taicheng Duan, Xuejie Song, Hangting Chen
Talanta 2007 Volume 71(Issue 2) pp:778-783
Publication Date(Web):15 February 2007
DOI:10.1016/j.talanta.2006.05.038
Sequential extraction procedures were widely applied for speciation of radioactive elements. In this study, the sequential extraction procedure developed by Martínez-Aguirre was employed for quantification of different chemical forms of thorium in the soil. The total amount of thorium in contaminated soil was much higher by four-fold than the local background value. The soil properties affect the amount of thorium and distribution of fractions in contaminated soil. Results showed that the proportion of thorium in soils from Baotou was found as the residual fraction (F5 + F6) > absorbed fraction (F3), coprecipitated fraction (F4) > carbonates fraction (F2) and exchangeable fraction (F1) that could be available to plants. The recovery, calculated by ratio of the sum of the six fractions to the pseudo-total content of thorium, was in the range from 96% to 110%. A comparison was carried out between the sequential extraction and the single extraction to evaluate the selectivity of the extractants. It was found that the amount of thorium of absorbed fraction (F3) was higher in the single extraction than that estimated in the sequential extraction, possibly duo to transform of the labile form. While for non-residual fraction analysis, the single extraction scheme was a desirable alternative to the sequential extraction procedure. According to correlativity analysis of various fractions, it might be predicted that how the non-residual fractions of thorium were directionally transformed into interrelated fractions under the changes of conditions.
Co-reporter:Xuejie Song, Taicheng Duan, Pengran Guo, Hangting Chen
Microchemical Journal 2006 Volume 84(1–2) pp:22-25
Publication Date(Web):September 2006
DOI:10.1016/j.microc.2006.03.010
The determination of Nb and Ta in Nb–Ta minerals was accomplished by slurry nebulization inductively coupled plasma optical emission spectrometry (ICP-OES), using a clog-free V-groove ceramic nebulizer. Samples were first wet-ground to appropriate particle sizes with narrow size distribution and 90% of the particles in the slurry were smaller than 2.32 μm in diameter. Subsamples were then dispersed in pH 9 aqueous solutions, and agitated in an ultrasonic bath for 15 min prior to analysis. Due to the lack of slurry standards matching well with the samples, calibration was simply carried out using aqueous solution standards. Results were compared with those obtained from a conventional fusion decomposition procedure and acid digestion procedures and a good agreement between the measured and referred values was obtained. The technique provided a good alternative for the rapid determination of Nb and/or Ta in their corresponding minerals.
Co-reporter:Taicheng Duan, Xuejie Song, Jingwei Xu, Pengran Guo, Hangting Chen, Hongfei Li
Spectrochimica Acta Part B: Atomic Spectroscopy 2006 Volume 61(Issue 9) pp:1069-1073
Publication Date(Web):September 2006
DOI:10.1016/j.sab.2006.09.005
Using a solid phase extraction mini-column home-made from a neutral extractant Cyanex 923, inorganic Hg could be on-line preconcentrated and simultaneously separated from methyl mercury. The preconcentrated Hg (II) was then eluted with 10% HNO3 and subsequently reduced by NaBH4 to form Hg vapor before determination by cold vapor atomic absorption spectrometry (CVAAS). Optimal conditions for and interferences on the Hg preconcentration and measurement were at 1% HCl, for a 25 mL sample uptake volume and a 10 mL min− 1 sample loading rate. The detection limit was 0.2 ng L− 1 and much lower than that of conventional method (around 15.8 ng L− 1). The relative standard deviation (RSD) is 1.8% for measurements of 40 ng L− 1 of Hg and the linear working curve is from 20 to 2000 ng L− 1 (with a correlation coefficient of 0.9996). The method was applied in determination of inorganic Hg in city lake and deep well water (from Changchun, Jilin, China), and recovery test results for both samples were satisfactory.
Co-reporter:Taicheng Duan, Xuejie Song, Dan Jin, Hongfei Li, Jingwei Xu, Hangting Chen
Talanta 2005 Volume 67(Issue 5) pp:968-974
Publication Date(Web):31 October 2005
DOI:10.1016/j.talanta.2005.04.017
In this work, a method was developed for determination of ultra-trace levels of Cd in tea samples by atomic fluorescence spectrometry (AFS). A flow injection solid phase extraction (FI-SPE) separation and preconcentration technique, to on-line couple with a sequential injection hydride generation (SI-HG) technique is employed in this study. Cd was preconcentrated on the SPE column, which was made from a neutral extractant named Cyanex 923, while other matrix ions or interfering ions were completely or mostly separated off. Conditions for the SPE separation and preconcentration, as well as conditions for the HG technique, were studied. Due to the separation of interfering elements, Cd hydride generation efficiency could be greatly enhanced with the sole presence of Co2+ with a concentration of 200 μg L−1, which is much lower than those in other works previously reported. Interferences on both the Cd separation and preconcentration, and Cd hydride generation (HG) were investigated; it showed that both the separation and preconcentration system, and the HG system had a strong anti-interference ability. The SPE column could be repeatedly used at least 400 times, a R.S.D. of 0.97% was obtained for 6 measurements of Cd with 0.2 μg L−1 and a correlation coefficiency of 1.0000 was obtained for the measurement of a series of solutions with Cd concentrations from 0.1 to 2 μg L−1. The method has a low detection limit of 10.8 ng L−1 for a 25 mL solution and was successfully validated by using two tea standard reference materials (GBW08513 and GBW07605).
Co-reporter:Duan Taicheng, Chen Hangting and Zeng Xianjin
Journal of Analytical Atomic Spectrometry 2002 vol. 17(Issue 4) pp:410-413
Publication Date(Web):06 Mar 2002
DOI:10.1039/B109660P
A method was developed for the determination of trace and ultratrace amounts of REE, Cd, In, Tl, Th, Nb, Ta, Zr and Hf in soils and sediments. With NaOH–Na2O2 as the flux, Ti(OH)4–Fe(OH)3 co-precipitation as the preconcentration technique and inductively coupled plasma mass spectrometry (ICP-MS) for measurement, the whole procedure was concise and suitable for batch analysis of multi-element solutions. An investigation was carried out of the Ti(OH)4–Fe(OH)3 co-precipitation system, and the results obtained showed that the natural situation of Ti tightly coexisting with Nb, Ta, Zr and Hf in geological samples plays a very important role in the complete co-precipitation of the four elements. The accuracy of this procedure was established using six Chinese soil and sediment certified reference materials (GSS and GSD), and the relative errors
between the found and certified values were mostly below 10%.
Co-reporter:Xiaoyu Jia, Yi Han, Chao Wei, Taicheng Duan and Hangting Chen
Journal of Analytical Atomic Spectrometry 2011 - vol. 26(Issue 7) pp:NaN1386-1386
Publication Date(Web):2011/03/17
DOI:10.1039/C0JA00121J
Ionic liquid based dispersive liquid–liquid microextraction (IL-DLLME) combined with high performance liquid chromatography-inductively coupled plasma mass spectrometry (HPLC-ICP-MS) for the determination of mercury species in liquid cosmetic samples is described. Firstly mercury (Hg2+), methylmercury (MeHg+) and ethylmercury (EtHg+) were complexed with ammonium pyrrolidinedithiocarbamate (APDC), and then the complexes were extracted into 1-hexyl-3-methylimidazolium hexafluorophosphate ([C6MIM][PF6]) using DLLME. Under the optimized conditions, the enrichment factors of 760, 115, 235 for Hg2+, MeHg+ and EtHg+ were obtained from only 5.00 mL sample solution. The detection limits of the analytes (as Hg) were 1.3 ng L−1 for Hg2+, 7.2 ng L−1 for MeHg+ and 5.4 ng L−1 for EtHg+, respectively. The relative standard deviation (n = 10) of 0.5 ng mL−1 Hg2+, MeHg+ and EtHg+ were 7.4%, 5.2% and 2.3%, respectively. Finally, the developed method was successfully applied for the speciation of mercury in four liquid cosmetic samples, Hg2+ was found as the main species in these samples, and a spike test was performed to verify the accuracy of the method.
Co-reporter:Xinli Liu, Taicheng Duan, Yi Han, Xiaoyu Jia and Hangting Chen
Journal of Analytical Atomic Spectrometry 2010 - vol. 25(Issue 2) pp:NaN209-209
Publication Date(Web):2009/11/18
DOI:10.1039/B915054D
The determination of As(III) in high purity Sb2O3 was taken as example to illustrate a high selective solid phase extraction (SPE) with non-polar resins as the sorbent. The extraction was based on the fact that in concentrated HCl solutions, As(III) forming molecular covalent AsCl3 could be attracted by the hydrophobic phase of the resins, whereas the matrix Sb existing only as the ionic Sb–Cl complex could not be adsorbed. The SPE column was made from the Amberlite XAD-16 resin and was on-line coupled with hydride generation atomic fluorescence spectrometer for As(III) detection. Under the optimized conditions, the detection limit (3σ) of 0.26 μg g−1, the RSD (n = 6) of 6.2% for 50 μg L−1 of As(III) and a linear calibration curve from 2 μg L−1 to 200 μg L−1 with R = 0.9998 were obtained. The recoveries for 10 μg L−1 of As(III) spiked in 1 g L−1 of Sb(III) solution was in the range of 97–102%. The method was applied to the determination of trace As(III) in two highly pure Sb2O3 samples.
Co-reporter:
Analytical Methods (2009-Present) 2012 - vol. 4(Issue 2) pp:
Publication Date(Web):
DOI:10.1039/C2AY05740A
An on-line anion-exchange membrane separation system has been coupled to inductively coupled plasma mass spectrometry (ICP-MS) for the total vanadium(V) and chromium (Cr) determination and V(IV) and Cr(III) species determination simultaneously. The positive charged V(IV) and Cr(III) species have been efficiently separated from V(V) and Cr(VI), and selectively determined by the on-line electrodialyzer-ICP-MS. To determine the total concentrations of V and Cr in water samples, H2SO4 and Na2S2O3 were added to convert all vanadium and chromium species into their positive species, thus, V(V) and Cr(VI) in the samples were quantified by subtraction. The cool plasma and reaction gas can also be avoided as the severe interference from chloride, carbon and nitrogen-based molecular ion has been substantially suppressed by the anion-exchange membrane. The detection limits of the analytes were 0.02 μg L−1 for V(IV) and 0.06 μg L−1 for Cr(III) with this on-line system, respectively, and the relative standard deviation (n = 10) of 10 μg L−1V(IV) and Cr(III) were 3.7% and 2.1%, respectively. The developed method has been applied for the speciation of V and Cr in three water samples, with V(V) and Cr(III) found as the main species in these samples. The accuracy and feasibility of this method have also been verified by spike tests and a certified reference material (CRM) of environmental water (GSBZ 50029-94) and a CRM of riverine water (GBW 08608), good recoveries have been obtained and the analytical results are also in good agreement with the certified values.
Co-reporter:Yi Han, Xiaoyu Jia, Taicheng Duan and Hangting Chen
Analytical Methods (2009-Present) 2011 - vol. 3(Issue 4) pp:NaN848-848
Publication Date(Web):2011/02/17
DOI:10.1039/C0AY00742K
A simple, inexpensive and reliable analytical method was developed for the determination of polybrominated diphenyl ethers (PBDEs) in whole milk by gas chromatography-mass spectrometry (GC-MS). Matrix components such as proteins and fats in the whole milk were removed by saponification procedure and the combined PBDEs were released. Then, a simple one-step liquid–liquid extraction procedure in the centrifuge tube (in-tube LLE) was performed and PBDEs were extracted into petroleum ether. After centrifugation, dehydration and nitrogen drying of petroleum ether phase, the extract was dissolved in acetonitrile. The acetonitrile solution was directly used as dispersant for the subsequent dispersive liquid–liquid microextraction (DLLME). Under the optimal conditions, the enrichment factors (EFs) of PBDEs ranged from 270 to 307. The calibration was performed by use of matrix-matched calibration standards, the correlation coefficients were better than 0.99 and the limits of detection (LODs) ranged from 0.012 to 0.29 μg L−1. The recoveries for the whole milk samples analysis spiked at 1 μg L−1 were between 78% and 120% with relative standard deviations (RSDs) ranging from 1.7% to 11% (n = 5). In this study, a simple, valid and combined sample preparation method was applied in the analysis of PBDEs in whole milk sample and the satisfactory recoveries for PBDEs were obtained.
.gif)