Co-reporter:Thomas C. Pochapsky
PNAS 2015 Volume 112 (Issue 31 ) pp:9502-9503
Publication Date(Web):2015-08-04
DOI:10.1073/pnas.1512077112
Co-reporter:Thomas C. Pochapsky
PNAS 2014 Volume 111 (Issue 10 ) pp:3659-3660
Publication Date(Web):2014-03-11
DOI:10.1073/pnas.1401325111
Co-reporter:Eliana K. Asciutto, Matthew J. Young, Jeffry Madura, Susan Sondej Pochapsky, and Thomas C. Pochapsky
Biochemistry 2012 Volume 51(Issue 16) pp:
Publication Date(Web):April 2, 2012
DOI:10.1021/bi300007r
Removal of substrate (+)-camphor from the active site of cytochrome P450cam (CYP101A1) results in nuclear magnetic resonance-detected perturbations in multiple regions of the enzyme. The 1H–15N correlation map of substrate-free diamagnetic Fe(II) CO-bound CYP101A permits these perturbations to be mapped onto the solution structure of the enzyme. Residual dipolar couplings (RDCs) were measured for 15N–1H amide pairs in two independent alignment media for the substrate-free enzyme and used as restraints in solvated molecular dynamics (MD) simulations to generate an ensemble of best-fit structures of the substrate-free enzyme in solution. Nuclear magnetic resonance-detected chemical shift perturbations reflect changes in the electronic environment of the NH pairs, such as hydrogen bonding and ring current shifts, and are observed for residues in the active site as well as in hinge regions between secondary structural features. RDCs provide information about relative orientations of secondary structures, and RDC-restrained MD simulations indicate that portions of a β-rich region adjacent to the active site shift so as to partially occupy the vacancy left by removal of the substrate. The accessible volume of the active site is reduced in the substrate-free enzyme relative to the substrate-bound structure calculated using the same methods. Both symmetric and asymmetric broadening of multiple resonances observed upon substrate removal as well as localized increased errors in RDC fits suggest that an ensemble of enzyme conformations are present in the substrate-free form.
Co-reporter:Marina Dang, Susan Sondej Pochapsky and Thomas C. Pochapsky
Metallomics 2011 vol. 3(Issue 4) pp:339-343
Publication Date(Web):24 Dec 2010
DOI:10.1039/C0MT00065E
A hydrogen bond network has been identified that adjusts protein-substrate contacts in cytochrome P450cam (CYP101A1). Replacing the native substrate camphor with adamantanone or norcamphor causes perturbations in NMR-detected NH correlations assigned to the network, which includes portions of a β sheet and an adjacent helix that is remote from the active site. A mutation in this helix reduces enzyme efficiency and perturbs the extent of substrate-induced spin state changes at the haem iron that accompany substrate binding. In turn, the magnitude of the spin state changes induced by alternate substrate binding parallel the NMR-detected perturbations observed near the haem in the enzyme active site.
Co-reporter:Eliana K. Asciutto, Marina Dang, Susan Sondej Pochapsky, Jeffry D. Madura, and Thomas C. Pochapsky
Biochemistry 2011 Volume 50(Issue 10) pp:1664-1671
Publication Date(Web):January 25, 2011
DOI:10.1021/bi101820d
Residual dipolar couplings (RDCs) were used as restraints in fully solvated molecular dynamics simulations of reduced substrate- and carbonmonoxy-bound cytochrome P450cam (CYP101A1), a 414-residue soluble monomeric heme-containing camphor monooxygenase from the soil bacterium Pseudomonas putida. The 1DNH residual dipolar couplings used as restraints were measured in two independent alignment media. A soft annealing protocol was used to heat the starting structures while incorporating the RDC restraints. After production dynamics, structures with the lowest total violation energies for RDC restraints were extracted to identify ensembles of conformers accessible to the enzyme in solution. The simulations result in substrate orientations different from that seen in crystallographic structures and a more open and accessible enzyme active site and largely support previously reported differences between the open and closed states of CYP101A1.
Co-reporter:Wei Wang;Iva Perovic;Johnathan Chittuluru;Alice Kaganovich;Linh T. T. Nguyen;Jingling Liao;Jared R. Auclair;Derrick Johnson;Anuradha Landeru;Alana K. Simorellis;Shulin Ju;Mark R. Cookson;Francisco J. Asturias;Jeffrey N. Agar;Brian N. Webb;ChulHee Kang;Dagmar Ringe;Gregory A. Petsko;Quyen Q. Hoang
PNAS 2011 108 (43 ) pp:17797-17802
Publication Date(Web):2011-10-25
DOI:10.1073/pnas.1113260108
A heterologously expressed form of the human Parkinson disease-associated protein α-synuclein with a 10-residue N-terminal
extension is shown to form a stable tetramer in the absence of lipid bilayers or micelles. Sequential NMR assignments, intramonomer
nuclear Overhauser effects, and circular dichroism spectra are consistent with transient formation of α-helices in the first
100 N-terminal residues of the 140-residue α-synuclein sequence. Total phosphorus analysis indicates that phospholipids are
not associated with the tetramer as isolated, and chemical cross-linking experiments confirm that the tetramer is the highest-order
oligomer present at NMR sample concentrations. Image reconstruction from electron micrographs indicates that a symmetric oligomer
is present, with three- or fourfold symmetry. Thermal unfolding experiments indicate that a hydrophobic core is present in
the tetramer. A dynamic model for the tetramer structure is proposed, based on expected close association of the amphipathic
central helices observed in the previously described micelle-associated “hairpin” structure of α-synuclein.
Co-reporter:Susan Sondej Pochapsky, Marina Dang, Bo OuYang, Alana K. Simorellis and Thomas C. Pochapsky
Biochemistry 2009 Volume 48(Issue 20) pp:
Publication Date(Web):March 31, 2009
DOI:10.1021/bi900002k
Local protein backbone dynamics of the camphor hydroxylase cytochrome P450cam (CYP101) depend upon the oxidation and ligation state of the heme iron. 1H−15N correlation nuclear magnetic resonance experiments were used to compare backbone dynamics of oxidized and reduced forms of this 414-residue metalloenzyme via hydrogen−deuterium exchange kinetics (H−D exchange) and 15N relaxation measurements, and these results are compared with previously published results obtained by H−D exchange mass spectrometry. In general, the reduced enzyme exhibits lower-amplitude motions of secondary structural features than the oxidized enzyme on all of the time scales accessible to these experiments, and these differences are more pronounced in regions of the enzyme involved in substrate access to the active site (B′ helix and β3 and β5 sheets) and binding of putidaredoxin (C and L helices), the iron−sulfur protein that acts as the effector and reductant of CYP101 in vivo. These results are interpreted in terms of local structural effects of changes in the heme oxidation state, and the relevance of the observed effects to the enzyme mechanism is discussed.
Co-reporter:Yoshitomo Hamuro, Kathleen S. Molnar, Stephen J. Coales, Bo OuYang, Alana K. Simorellis, Thomas C. Pochapsky
Journal of Inorganic Biochemistry 2008 Volume 102(Issue 2) pp:364-370
Publication Date(Web):February 2008
DOI:10.1016/j.jinorgbio.2007.10.001
Backbone dynamics of the camphor monoxygenase cytochrome P450cam (CYP101) as a function of oxidation/ligation state of the heme iron were investigated via hydrogen/deuterium exchange (H/D exchange) as monitored by mass spectrometry. Main chain amide NH hydrogens can exchange readily with solvent and the rate of this exchange depends upon, among other things, dynamic fluctuations in local structural elements. A fluxional region of the polypeptide will exchange more quickly with solvent than one that is more constrained. In most regions of the enzyme, exchange rates were similar between oxidized high-spin camphor-bound and reduced camphor- and CO-bound CYP101 (CYP-S and CYP-S-CO, respectively). However, in regions of the protein that have previously been implicated in substrate access by structural and molecular dynamics investigations, the reduced enzyme shows significantly slower exchange rates than the oxidized CYP-S. This observation corresponds to increased flexibility of the oxidized enzyme relative to the reduced form. Structural features previously found to be perturbed in CYP-S-CO upon binding of the biologically relevant effector and reductant putidaredoxin (Pdx) as determined by nuclear magnetic resonance are also more protected from exchange in the reduced state. To our knowledge, this study represents the first experimental investigation of backbone dynamics within the P450 family using this methodology.
Co-reporter:Yalin Zhang, Melissa H Heinsen, Milka Kostic, Gina M Pagani, Thomas V Riera, Iva Perovic, Lizbeth Hedstrom, Barry B Snider, Thomas C Pochapsky
Bioorganic & Medicinal Chemistry 2004 Volume 12(Issue 14) pp:3847-3855
Publication Date(Web):15 July 2004
DOI:10.1016/j.bmc.2004.05.002
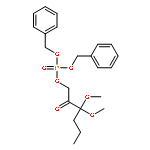
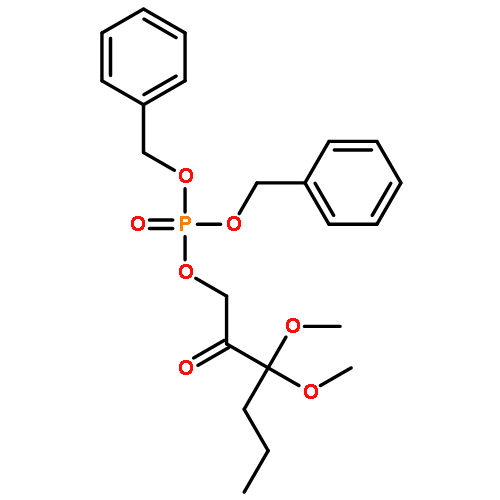
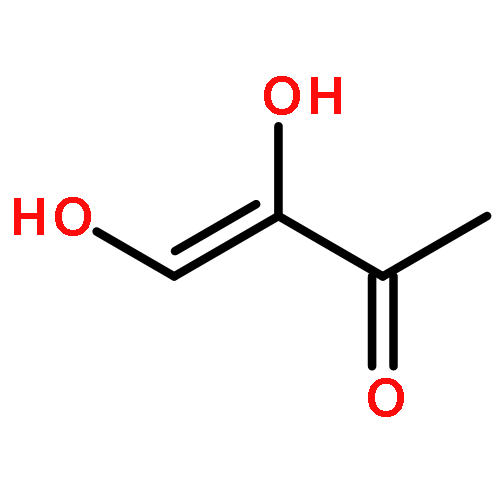
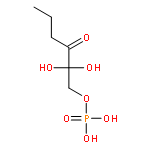
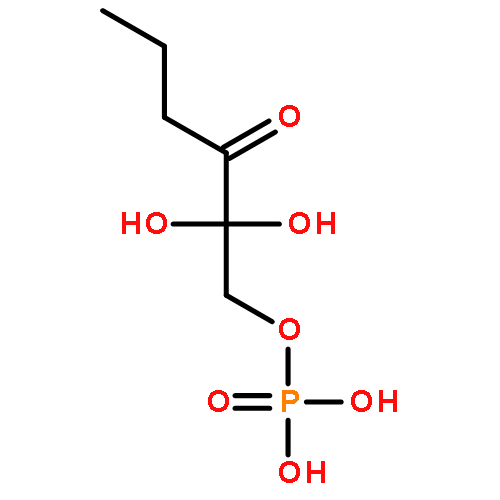
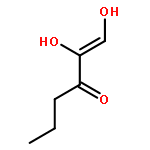
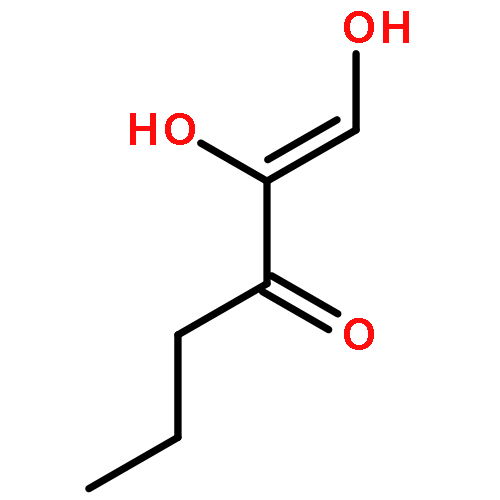
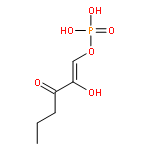
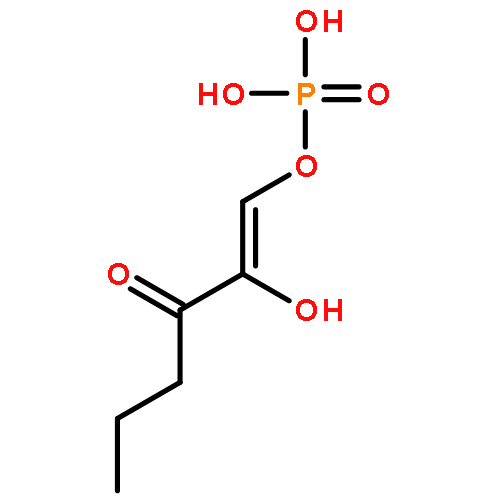
The methionine salvage pathway allows the in vivo recovery of the methylthio moiety of methionine upon the formation of methylthioadenosine (MTA) from S-adenosylmethionine (SAM). The Fe(II)-containing form of acireductone dioxygenase (ARD) catalyzes the penultimate step in the pathway in Klebsiella oxytoca, the oxidative cleavage of the acireductone 1,2-dihydroxy-3-oxo-5-(methylthio)pent-1-ene (2) by dioxygen to give formate and 2-oxo-4-(methylthio)butyrate (3). The Ni(II)-bound form (Ni–ARD) catalyzes an off-pathway shunt, forming 3-(methylthio)propionate (4), carbon monoxide, and formate. Acireductone 2 is formed by the action of another enzyme, E1 enolase/phosphatase, on precursor 1-phosphonooxy-2,2-dihydroxy-3-oxo-5-methylthiopentane (1). Simple syntheses of several analogs of 1 are described, and their activity as substrates for E1 enolase/phosphatase characterized. A new bacterial overexpression system and purification procedure for E1, a member of the haloacid dehalogenase (HAD) superfamily, is described, and further characterization of the enzyme presented.Graphic
Co-reporter:
Nature Structural and Molecular Biology 2002 9(12) pp:966 - 972
Publication Date(Web):28 October 2002
DOI:10.1038/nsb863
Co-reporter:Yoshitomo Hamuro, Kathleen S. Molnar, Stephen J. Coales, Bo OuYang, Alana K. Simorellis, Thomas C. Pochapsky
Journal of Inorganic Biochemistry (February 2008) Volume 102(Issue 2) pp:364-370
Publication Date(Web):1 February 2008
DOI:10.1016/j.jinorgbio.2007.10.001
Backbone dynamics of the camphor monoxygenase cytochrome P450cam (CYP101) as a function of oxidation/ligation state of the heme iron were investigated via hydrogen/deuterium exchange (H/D exchange) as monitored by mass spectrometry. Main chain amide NH hydrogens can exchange readily with solvent and the rate of this exchange depends upon, among other things, dynamic fluctuations in local structural elements. A fluxional region of the polypeptide will exchange more quickly with solvent than one that is more constrained. In most regions of the enzyme, exchange rates were similar between oxidized high-spin camphor-bound and reduced camphor- and CO-bound CYP101 (CYP-S and CYP-S-CO, respectively). However, in regions of the protein that have previously been implicated in substrate access by structural and molecular dynamics investigations, the reduced enzyme shows significantly slower exchange rates than the oxidized CYP-S. This observation corresponds to increased flexibility of the oxidized enzyme relative to the reduced form. Structural features previously found to be perturbed in CYP-S-CO upon binding of the biologically relevant effector and reductant putidaredoxin (Pdx) as determined by nuclear magnetic resonance are also more protected from exchange in the reduced state. To our knowledge, this study represents the first experimental investigation of backbone dynamics within the P450 family using this methodology.
Co-reporter:Eliana K. Asciutto, Jeffry D. Madura, Susan Sondej Pochapsky, Bo OuYang, Thomas C. Pochapsky
Journal of Molecular Biology (15 May 2009) Volume 388(Issue 4) pp:801-814
Publication Date(Web):15 May 2009
DOI:10.1016/j.jmb.2009.03.046
Experimental evidence has been provided for a functionally relevant cis–trans isomerization of the Ile88–Pro89 peptide bond in cytochrome P450cam (CYP101). The isomerization is proposed to be a key element of the structural reorganization leading to the catalytically competent form of CYP101 upon binding of the effector protein putidaredoxin (Pdx). A detailed comparison of the results of molecular dynamics simulations on the cis and trans conformations of substrate- and carbonmonoxy-bound ferrous CYP101 with sequence-specific Pdx-induced structural perturbations identified by nuclear magnetic resonance is presented, providing insight into the structural and dynamic consequences of the isomerization. The mechanical coupling between the Pdx binding site on the proximal face of CYP101 and the site of isomerization is described.
Co-reporter:Bo OuYang, Susan Sondej Pochapsky, Marina Dang, Thomas C. Pochapsky
Structure (11 June 2008) Volume 16(Issue 6) pp:916-923
Publication Date(Web):11 June 2008
DOI:10.1016/j.str.2008.03.011
The two-protein complex between putidaredoxin (Pdx) and cytochrome P450cam (CYP101) is the catalytically competent species for camphor hydroxylation by CYP101. We detected a conformational change in CYP101 upon binding of Pdx that reorients bound camphor appropriately for hydroxylation. Experimental evidence shows that binding of Pdx converts a single X-proline amide bond in CYP101 from trans or distorted trans to cis. Mutation of proline 89 to isoleucine yields a mixture of both bound camphor orientations, that seen in Pdx-free and that seen in Pdx-bound CYP101. A mutation in CYP101 that destabilizes the cis conformer of the Ile 88-Pro 89 amide bond results in weaker binding of Pdx. This work provides direct experimental evidence for involvement of X-proline isomerization in enzyme function.







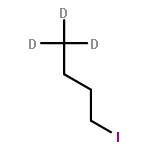
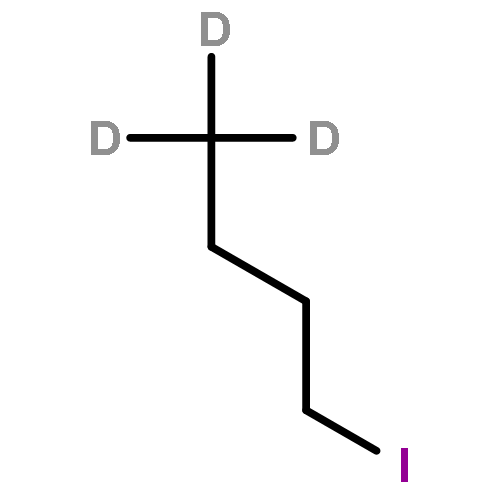
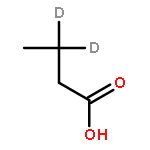
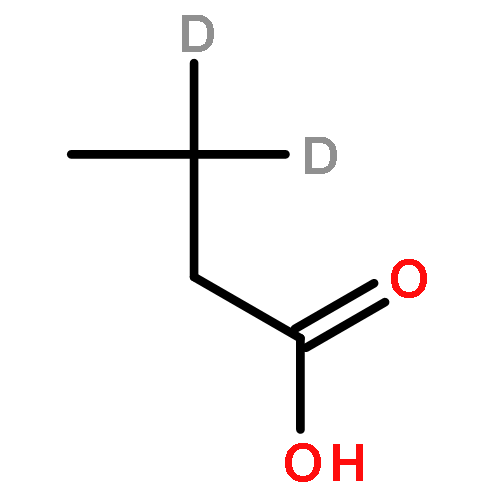
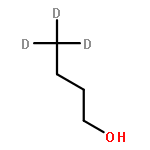
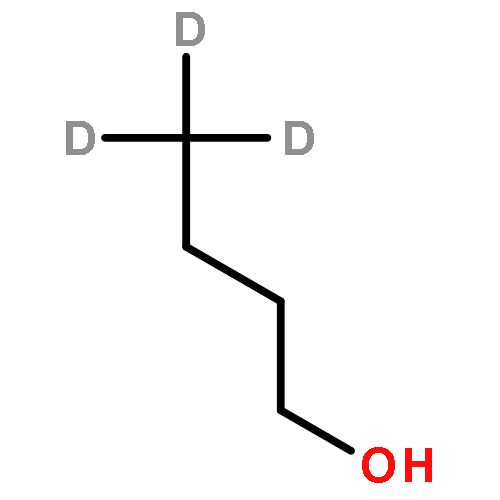
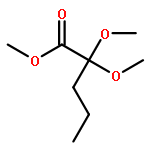
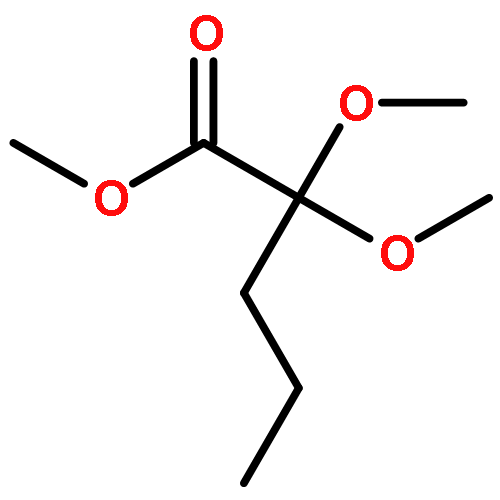
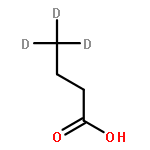
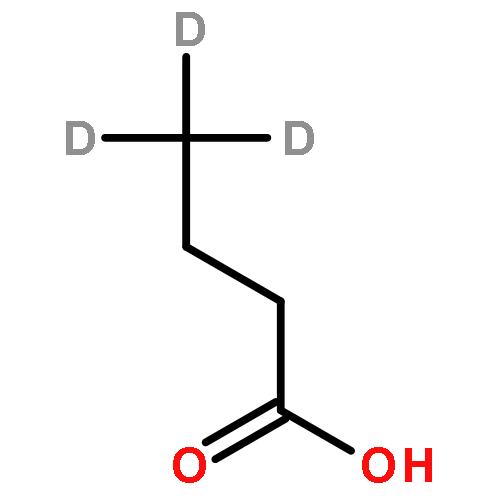
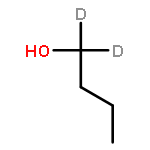



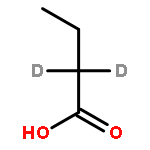
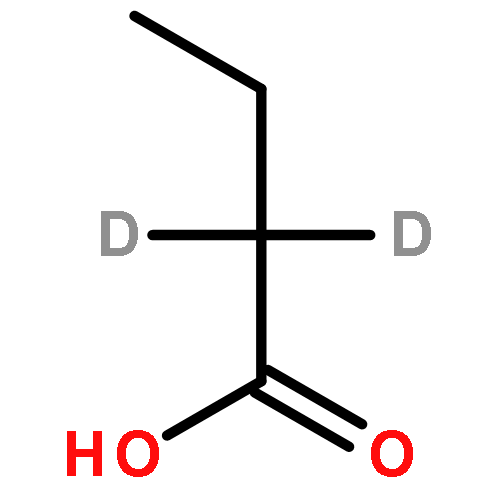
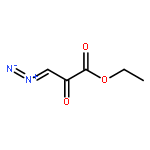

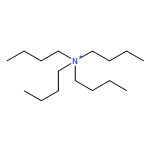



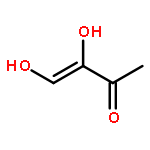




![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)