Co-reporter:D. Allan;S. C. Radzinski;M. A. Tapsak;J. J. Liggat
Silicon 2016 Volume 8( Issue 4) pp:553-562
Publication Date(Web):2016 October
DOI:10.1007/s12633-014-9247-6
The thermal degradation behaviour of novel high number average molecular mass polysilalkylenesiloxanes is reported. These have been synthesised using anionic ring-opening polymerisation of 1,1,3,3,14,14,16,16-octamethyl-2,15-dioxa-1,3,14,16-tetrasilacyclohexacosane and octamethylcyclotetrasiloxane (D4) mixtures. The thermal degradation behaviour of these materials was evaluated by a combination of thermogravimetric analysis (TGA) and thermal volatilisation analysis (TVA) and compared with a commercial sample of PDMS. The results demonstrated that the thermal degradation of the polysilalkylenesiloxanes is more complex than the PDMS, with the polysilalkylenesiloxanes exhibiting a lower degradation peak maximum temperature. The major volatile degradation products evolved from the PDMS were identified as D3 to D6 cyclic siloxane oligomers, in addition to higher molecular mass cyclic siloxane oligomers. The polysilalkylenesiloxanes, on the other hand, evolved short chain aliphatic hydrocarbons, cyclic and linear siloxane oligomers and silanes. The TVA results indicate that the polysilalkylenesiloxanes degrade mostly by random chain scission of the polymer backbone, whereas the commercial PDMS degrades by the accepted depolymerisation reaction which involves “back-biting” reactions.
Co-reporter:S. McCreath, J.P. Lewicki, J.J. Liggat, C. Lithgow, L. McCulloch, K. Miller, A. Witkowski
Polymer Degradation and Stability 2015 Volume 113() pp:144-153
Publication Date(Web):March 2015
DOI:10.1016/j.polymdegradstab.2015.01.013
Polystyrene and poly(4-methylstyrene) have very similar chemical structures with the only differences being the para methyl group of poly(4-methylstyrene). This methyl group is susceptible to oxidation at elevated temperatures. Here we demonstrate that it is possible to introduce oxidative cross-links to poly(4-methylstyrene), via the para methyl group, by thermal oxidative treatment at 230 °C, 250 °C and 270 °C in the absence of catalyst, leading to a material with markedly modified thermal degradation chemistry. Thermal gravimetric analysis and differential scanning calorimetry were used to characterise and compare untreated and post-oxidised materials and established that as the temperature of pre-treatment was increased, the subsequent thermal stability of the material increased. FTIR, NMR and microanalysis indicated that after the thermal oxidative pre-treatment ether cross-links are present alongside new oxygen containing functional groups such as aldehydes, carboxylic acids and hydroxyl groups. Finally, data obtained from pyrolysis combustion flow calorimetry confirmed that as the number of oxidative cross-links increase, a reduction in the polymer's flammability as assessed by heat release data is observed.
Co-reporter:D. Allan, J.H. Daly, J.J. Liggat
Polymer Degradation and Stability 2014 Volume 102() pp:170-179
Publication Date(Web):April 2014
DOI:10.1016/j.polymdegradstab.2014.01.016
In this paper, we report a comprehensive study on a TDI-based foam containing 13% ammonium polyphosphate (APP) in order to examine the effect that this conventional fire retardant has on the thermal and thermo-oxidative degradation behaviour of the polyurethane foam. The results from TVA and TGA analyses show that the APP foam undergoes a significantly different degradation mechanism to the standard foam that we have reported on previously. The TGA results revealed the presence of a two-stage degradation process under a non-oxidative environment. The TVA results, on the other hand, revealed that degradation of the APP foam in fact occurs in four overlapping steps compared to the two-step process which occurs for the standard foam. The additional degradation steps observed for the APP foam are proposed to correspond to degradation of the fire retardant. Evolution of volatile material was also observed to occur at a lower temperature for the APP foam and it is proposed that the primary degradation of the urethane linkages via a depolycondensation reaction is acid-catalysed by the acidic hydroxyl groups which arise from degradation of APP. The sub-ambient differential distillation trace revealed that the nature and distribution of the volatiles evolved from the APP foam were profoundly different to the standard foam, which confirms that the secondary degradation is also altered in the presence of APP.
Co-reporter:L. Turnbull, J.J. Liggat, W.A. MacDonald
Polymer Degradation and Stability 2013 Volume 98(Issue 11) pp:2244-2258
Publication Date(Web):November 2013
DOI:10.1016/j.polymdegradstab.2013.08.018
Although the fundamental degradation chemistry of poly(ethylene naphthalate), PEN, is thought to be similar to that of poly(ethylene terephthalate), PET, there is very little evidence in the literature to support this. This paper presents data on the thermal degradation of PEN, in comparison to PET, with particular reference to evolved gas analysis undertaken by thermal volatilisation analysis (TVA). Our thermal degradation studies highlight strong similarities in the degradation behaviour of PET and PEN, despite some evidence of increased thermal stability of PEN in comparison to PET. Identical primary and secondary thermal degradation mechanisms are proposed for PET and PEN, with radical degradation processes thought to dominate at high temperature.
Co-reporter:D. Allan, J. Daly, J.J. Liggat
Polymer Degradation and Stability 2013 Volume 98(Issue 2) pp:535-541
Publication Date(Web):February 2013
DOI:10.1016/j.polymdegradstab.2012.12.002
The thermal degradation behaviour of a polyurethane foam, synthesised from TDI and a polyether polyol, is reported. The thermal degradation behaviour of this material was evaluated by a combination of thermogravimetric analysis (TGA) and thermal volatilisation analysis (TVA). The results demonstrated that the thermal degradation is a complex process which consists of competing mechanisms which yield an array of degradation products. The TVA results revealed that the degradation occurs in two steps, with the initial step corresponding to degradation of the urethane linkages by two competing mechanisms. The first mechanism, proposed to be the predominant mechanism, involves simple depolymerisation of the urethane bond to yield TDI and polyol. A second, competing mechanism is proposed to occur which involves dissociation of the urethane linkages to yield DAT, CO2 and alkene-terminated polyol chains. The second degradation step has been shown to involve degradation of the polyol which was regenerated in the first degradation step. This is proposed to occur by random radical chain scission of the polyol to yield propene, formaldehyde, acetaldehyde, C3H6O isomers and high molar mass polyol chain fragments of various structures. Isothermal TVA studies have revealed that this occurs as low as 250 °C under vacuum but does not become significant until temperatures greater than 300 °C.
Co-reporter:Musarrat H. Mohammed, William M. Banks, David Hayward, John J. Liggat, Richard A. Pethrick, Barry Thomson
Polymer Degradation and Stability 2013 Volume 98(Issue 6) pp:1264-1270
Publication Date(Web):June 2013
DOI:10.1016/j.polymdegradstab.2013.02.014
The property changes occurring when poly(ether ether ketone) (PEEK) is subject to methane and carbon dioxide at high pressures (108 Pa) and high temperatures (175–200 °C) are reported. Differential scanning calorimetry, density gradient techniques, positron annihilation lifetime spectroscopy, dynamic mechanical thermal analysis and tensile tests measurements were used to monitor the changes which occur during the ageing process. Over the period of the study an overall increase in the tensile strength was noted, with little or no change in 0.2% and 2% proof stresses, whilst there was a decrease in bending modulus and glass transition temperature due to the effects of plasticization. The Young's modulus generally increases for samples exposed to a temperature of 175 °C in the presence of a mixture of 90% methane and 10% carbon dioxide, or carbon dioxide alone, but it decreases at 200 °C in the presence of carbon dioxide alone. The observed effects are consistent with the polymer undergoing initially a densification of the matrix associated with annealing-induced crystallisation, followed by plasticization as the gases permeate into the disordered regions of the matrix. When de-pressurised, the gas dissolved in the matrix attempts to leave the matrix and morphological changes are observed. The complexity of the changes in crystallinity and plasticization in the disordered phase are reflected in changes in the positron annihilation data.
Co-reporter:Alistair Apedaile;John Liggat;John Parkinson;George Nikiforidis;Leonard Berlouis;Mogon Patel
Journal of Applied Polymer Science 2012 Volume 123( Issue 5) pp:2601-2608
Publication Date(Web):
DOI:10.1002/app.34449
Abstract
Bulk condensation polymerization of (dimethylmethoxy)-m-carborane and (dichlorodimethyl)silane occurs in the presence of an Mx+Clx Lewis acid catalyst. In the literature, FeCl3 is commonly used as the catalyst of choice but little is known about the activation energy and entropy of this polymerization. By monitoring using 1H-NMR the reaction of a methoxy-terminated poly(dimethylsiloxane) and (dichlorodimethyl)silane the rate determining step in the FeCl3 catalyzed system is determined. The activation energy was calculated to be +43.6 kJ mol−1 and the entropy of the reaction was also calculated. The calculated large entropy of reaction indicates that the transition step is highly ordered. The formation of the electrophile intermediate species in the first step of the reaction has also been investigated using cyclic voltammetry. To the cyclic voltammetry data Randles-Sevcik fits have been applied to the oxidation peaks to determine the diffusion coefficients for the oxidation of Fe2+ to Fe3+. Also, the initial prediction of a reversible reaction Step 1 was shown to be incorrect as the normalized reduction peak maxima increase with scan rate, indicative of an electron transfer-chemical reaction mechanism. © 2011 Wiley Periodicals, Inc. J Appl Polym Sci, 2012
Co-reporter:L. Turnbull;J. J. Liggat;W. A. MacDonald
Journal of Applied Polymer Science 2012 Volume 124( Issue 6) pp:4517-4529
Publication Date(Web):
DOI:10.1002/app.35476
Abstract
PEN is thought to have increased thermal and hydrolytic resistance in comparison to PET. However, due to a lack of research, few studies have been published on the degradation of PEN. In our research, we report on the extent of degradation in PET and PEN after ageing under contrasting environments (dry nitrogen, dry air, wet nitrogen, and wet air) at temperatures between 140°C and 190°C. A combination of analysis techniques were employed in order to characterize and track the physical and chemical changes in the aged polyester samples, enabling the effects of temperature, water, and oxygen to be mapped onto the resultant property changes of PET and PEN. The extent of degradation has been shown to differ between both polymers and the dominant degradation mechanism in PET was shown to differ with ageing temperature. © 2011 Wiley Periodicals, Inc. J Appl Polym Sci, 2011
Co-reporter:James P. Lewicki, Krzysztof Pielichowski, Pauline Tremblot De La Croix, Bartlomiej Janowski, Deborah Todd, John J. Liggat
Polymer Degradation and Stability 2010 Volume 95(Issue 6) pp:1099-1105
Publication Date(Web):June 2010
DOI:10.1016/j.polymdegradstab.2010.02.021
Reported here is the synthesis of a series of polyurethane/POSS nanohybrid elastomers, the characterisation of their thermal stability and degradation behaviour at elevated temperatures using a combination of thermogravimetric Analysis (TGA) and thermal volatilisation analysis (TVA). A series of PU elastomer systems have been formulated incorporating varying levels of 1,2-propanediol-heptaisobutyl-POSS (PHIPOSS) as a chain extender unit, replacing butane diol. The bulk thermal stability of the nanohybrid systems has been characterised using TGA. Results indicate that covalent incorporation of POSS into the PU elastomer network increases the non-oxidative thermal stability of the systems. TVA analysis of the thermal degradation of the POSS/PU hybrid elastomers have demonstrated that the hybrid systems are indeed more thermally stable when compared to the unmodified PU matrix; evolving significantly reduced levels of volatile degradation products and exhibiting a ∼30 °C increase in onset degradation temperature. Furthermore, characterisation of the distribution of degradation products from both unmodified and hybrid systems indicate that the inclusion of POSS in the PU network is directly influencing the degradation pathways of both the soft and hard-block components of the elastomers: The POSS/PU hybrid systems show reduced levels of CO, CO2, water and increased levels of THF as products of thermal degradation.
Co-reporter:James P. Lewicki, John J. Liggat, Mogon Patel
Polymer Degradation and Stability 2009 Volume 94(Issue 9) pp:1548-1557
Publication Date(Web):September 2009
DOI:10.1016/j.polymdegradstab.2009.04.030
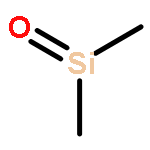
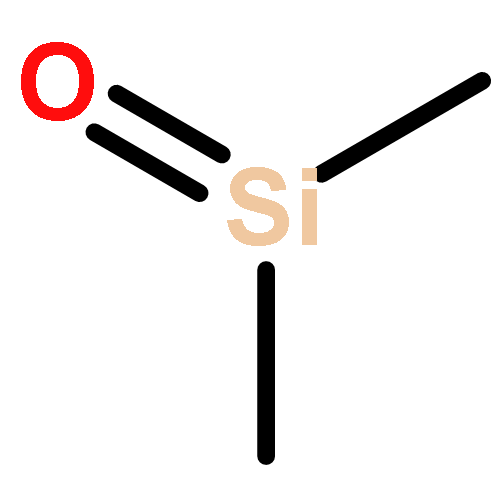
A series of novel polydimethylsiloxane/montmorillonite (PDMS/MMT) nanocomposites was prepared. The thermal degradation behaviour of these nanocomposites was studied by means of Thermal Volatilization Analysis (TVA) and Thermogravimetric Analysis (TGA). The major degradation products were identified as cyclic oligomeric siloxanes from D3 to D7, and higher oligomeric siloxane residues. Other minor degradation products include methane, bis-pentamethylcyclotrisiloxane, propene, propanal, benzene and dimethylsilanone. The results demonstrate that the nanoclay significantly alters the degradation behaviour of the PDMS network, modifying the profile of the thermal degradation and reducing the overall rate of volatiles evolution. The results also indicate that the nanoclay promotes the formation of dimethylsilanone and benzene by inducing low levels of radical chain scission.
Co-reporter:Hélène A. Lecomte, John J. Liggat
Polymer Degradation and Stability 2008 Volume 93(Issue 2) pp:498-506
Publication Date(Web):February 2008
DOI:10.1016/j.polymdegradstab.2007.11.005
Many types of fire retardants are used in poly(ethylene terephthalate), PET, formulations, and two commercial fire retardants, Ukanol® and Phosgard®, have been shown to improve significantly PET flame-retardancy when used as comonomers. Phosgard incorporates a phosphorus atom within the main chain whereas Ukanol incorporates a phosphorus atom as a pendent substituent. Despite their acknowledged effectiveness, the mode of action of these fire retardants remains unclear, and in this paper we present a comparison of the overall thermal degradation behaviour of PET and Ukanol and Phosgard fire-retarded formulations. DSC and particularly TGA data show that both Ukanol and Phosgard have some stabilising influence on PET degradation, especially under oxidative conditions. TGA and pyrolysis experiments both clearly indicate that neither of the additives acts as a char promoter. Only the Phosgard formulation shows any release of volatile phosphorus species which could act in the gas phase. On the other hand, the most striking feature of the pyrolysis experiments is the macroscopic structure of the chars produced by the fire-retarded formulations, which hints at their fire-retardancy action – an open-cell charred foam was obtained upon charring at 400 °C or 600 °C. This foaming layer between the degrading melt and the flame would lower the amount of fuel available for combustion, and would also limit the feedback of heat to the condensed phase.
Co-reporter:C. P. McAdam;N. E. Hudson;J. J. Liggat;R. A. Pethrick
Journal of Applied Polymer Science 2008 Volume 108( Issue 4) pp:2242-2251
Publication Date(Web):
DOI:10.1002/app.25599
Abstract
Nylon 6 nanocomposites were prepared by the in situ polymerization of ε-caprolactam with ultrasonically dispersed organically modified montmorillonite clay (Cloisite 30B®). Dispersions of the clay platelets with concentrations in the range 1–5 wt % in the monomer were characterized using rheological measurements. All mixtures exhibited shear-thinning, signifying that the clay particles were dispersed as platelets and forming a “house of cards” structure. Samples with Cloisite concentrations above 2 wt % showed a drop in viscosity between the initial shearing and repeated shearing, indicative of shearing breaking down the initial “house of cards” structures formed on sonication. DMTA measurements of the samples showed an increase in the β-relaxation temperature with increasing clay concentration. The bending modulus, at temperatures below Tg, showed an increase with increasing clay concentration up to 4 wt %. X-ray diffraction measurements showed that all nylon 6/Cloisite 30B samples were exfoliated apart from the 5 wt %, which showed that some intercalated material was present. The nylon crystallized into the α-crystalline phase, which is the most thermodynamically stable form. Preference for this form may be a consequence of the long time associated with the postcondensation step in the synthesis or the influence of the platelets on the nucleation step of the crystal growth. DSC measurements showed a retardation of the crystallization rate of nanocomposite samples when compared with that of pure nylon 6, due to the exfoliated clay platelets hindering chain movement. This behavior is different from that observed for the melt-mixed nylon 6/clay nanocomposites, which show an enhancement in the crystallization rate. © 2008 Wiley Periodicals, Inc. J Appl Polym Sci, 2008
Co-reporter:Sharon E Ingram;Richard A Pethrick ;John J Liggat
Polymer International 2008 Volume 57( Issue 11) pp:1206-1214
Publication Date(Web):
DOI:10.1002/pi.2452
Abstract
BACKGROUND: Cloisite 30B was added to diglycidyl ether of bisphenol F and cured with diaminodiphenylsulfone to investigate how the organoclay influenced the extent of cure.
RESULTS: A substantial increase in the extent of cure was found with the addition of Cloisite 30B, when lower cure temperatures were employed. Cloisite 30B at 2 wt% resulted in a 40 °C increase in glass transition temperature and an increase in the magnitude of the bending modulus even though a high level of intercalated material was detected.
CONCLUSIONS: It was observed that the addition of Cloisite 30B to the epoxy system increased the level of cure in the polymer, and was particularly prominent at low cure temperatures. Copyright © 2008 Society of Chemical Industry
Co-reporter:Hélène A. Lecomte, John J. Liggat
Polymer Degradation and Stability 2006 Volume 91(Issue 4) pp:681-689
Publication Date(Web):April 2006
DOI:10.1016/j.polymdegradstab.2005.05.028
Diethylene glycol (DEG) is incorporated into poly(ethylene terephthalate) (PET) during industrial synthesis in order to control crystallisation kinetics. DEG is known to be a weak point in the thermal degradation of PET, which is problematic during the recycling of the polymer.Studies on the thermal decomposition of the model polymer poly(diethylene glycol terephthalate) (PDEGT) have been performed using TG, DSC, TVA and spectroscopic techniques. They revealed a degradation behaviour with two distinct steps, where the first step initiates some 100 K below the degradation temperature of PET. The second step is similar to the behaviour of PET.Based on our observations, a new degradation mechanism specific to DEG units is proposed, where random ether groups along the backbone can back-bite and form cyclic oligomers. These cyclic species, containing ether moieties, are evolved at 245 °C and constitute the first of the two steps of degradation observed for PDEGT.
Co-reporter:Morven McAlpine;Nicholas E. Hudson;Richard A. Pethrick;David Pugh;Ian Rhoney
Journal of Applied Polymer Science 2006 Volume 99(Issue 5) pp:2614-2626
Publication Date(Web):19 DEC 2005
DOI:10.1002/app.22582
The factors that affect the dispersion of exfoliated organically modified montmorillonite in a solution of poly(methyl methacrylate) in methyl methacrylate are explored. Exfoliation of montmorillonite in the solution is achieved with the assistance of ultrasound, and rheological measurements indicate a very significant increase in the viscosity, a dramatic shear thinning behavior, and a finite yield stress, all of which are direct consequences of the exfoliated state of the clay platelets. A number of factors, including the sonication power, clay loading, use of a swelling agent, and moisture content of the modified montmorillonite, are found to influence the exfoliation process. The effect of addition of a range of titanate coupling agents (LICA-01, 12, 38, 44, and 97) on the viscosity of the nanoclay dispersions was investigated. It was found that LICA-44 had the effect of reducing the viscosity of the exfoliated montmorillonite dispersion without apparently influencing the extent of the exfoliation. Molecular modeling, UV–visible and Fourier transform infrared spectrometry were used to investigate the possible reasons for efficacy of this LICA. The LICA appears to act through a combination of steric effects and the presence of certain charges on the organic molecule. The magnitude of the negative charges on elements of the LICA appears to influence its ability to bind to the clay and also its ability to reduce the viscosity of the nanoclay. This article indicates how the apparently conflicting requirements of achieving a highly exfoliated state and also maintaining a viscosity low enough for processing can be effectively addressed. © 2005 Wiley Periodicals, Inc. J Appl Polym Sci, 2006
Co-reporter:C. Denecker;C. E. Snape;J. J. Liggat
Journal of Applied Polymer Science 2006 Volume 100(Issue 4) pp:3024-3033
Publication Date(Web):27 FEB 2006
DOI:10.1002/app.23701
In this article, we report the use of a variety of analytical methods, in particular, solid-state 1H-NMR and 13C-NMR to characterize the relationship between the condensed-phase chemistry and burning behavior as determined by a series of combustion tests for two commercially derived flexible polyurethane foams, one combustion-modified. The combustion tests showed that the foams met several regulatory requirements in terms of their fire performance, whether or not they were combustion-modified. Both foams passed the MV SS 302 and CAL 117 small-flame tests. The nonmodified foam failed the Crib 5 test, but this test had a much larger ignition source. The particular problem with the nonmodified foam was melt drip into the flame zone. This led to a steady maintenance of the fuel feed and a rapid escalation of the fire. In contrast, the combustion-modified foam showed little melt drip and self-extinguished. Thermal analysis data for the two foams showed that melamine acted in part as an endothermic heat sink. This alone did not account for the much reduced melt flow and drip of the combustion-modified foam, but the solid-state 1H-NMR data clearly showed that the molecular mobility of the combustion char from combustion-modified foam was lower than the unmodified foam char, which indicated that the flame-retardant formulation in the combustion-modified foam acted by a condensed-phase mechanism. © 2006 Wiley Periodicals, Inc. J Appl Polym Sci 100: 3024–3033, 2006
Co-reporter:Hélène A. Lecomte;Adam S. G. Curtis
Journal of Polymer Science Part A: Polymer Chemistry 2006 Volume 44(Issue 6) pp:
Publication Date(Web):26 JAN 2006
DOI:10.1002/pola.21288
Polyesters provide a good basis to work on for designing novel biodegradable materials that are also mechanically and thermally resistant. In this study, a series of aliphatic poly(ester amide)s (PEA) based on cyclohexane units was synthesized. Block-copolymers of cyclohexyl sebacate and cyclohexyl sebacamide were produced by controlling the length of the ester block and the amount of amide during a two-step melt/interfacial polycondensation reaction. Films produced from these materials could retain their shape above 373 K due to the physical network of amide hydrogen-bonding. Thermal properties were also evaluated, with various melting and softening points obtained depending on the PEA composition. The determining factor for mechanical properties was the amount of amide introduced, with films containing up to 10 mol % amide showing the best handleability and flexibility. Tensile properties typical of an amorphous viscoelastic material were observed, but with much superior elongation to break achievable (∼1700%). These materials were also shown to be hydrolyzable, noncytotoxic, and favorable for cell attachment: they may therefore have a promising future in the area of medical devices or packaging, especially as their properties can be tuned by changing their composition. © 2006 Wiley Periodicals, Inc. J Polym Sci Part A: Polym Chem 44: 1785–1795, 2006
Co-reporter:John J. Liggat;Allan R. Mackintosh
Journal of Applied Polymer Science 2004 Volume 92(Issue 5) pp:2791-2796
Publication Date(Web):17 MAR 2004
DOI:10.1002/app.20290
The fiber properties of PTT have been the subject of several reports, although very few reports describe the properties of molded specimens. In this work, the dynamic mechanical relaxation behavior of compression-molded PTT films has been investigated. The added flexibility of the PTT was found to lower the temperature of the β- and α-transitions relative to the PET and PEN. The results suggest that the β-transition is at least two relaxations for PET and PTT due to the increase in the breadth of the relaxation. The results seem to support the hypothesized mechanism of others, in that the β-transition involves the relaxation of the carbonyl entity and the aromatic C1–C4 ring flips for PTT and PET, and the relaxation of the carbonyl for PEN. The β*- and α-transitions for all three polymers seem to be cooperative in nature. © 2004 Wiley Periodicals, Inc. J Appl Polym Sci 92: 2791–2796, 2004
Co-reporter:Caroline M Dick, John J Liggat, Colin E Snape
Polymer Degradation and Stability 2001 Volume 74(Issue 3) pp:397-405
Publication Date(Web):2001
DOI:10.1016/S0141-3910(01)00174-4
Polychloroprene, also known as neoprene, is an elastomer commonly utilised in the electrical and automobile industries. Its degradation is known to occur predominantly in a two stage process: HCl is lost in the initial step, whilst the second step involves the production of volatile hydrocarbons through chain scission. In this paper we describe the use of solid state 13C NMR as a probe for structural changes in the condensed phase during these degradative steps. Cross polarisation-magic angle spinning (CP-MAS) analysis of virgin polychloroprene and a series of samples degraded at temperatures between 275 and 550 °C reveals that as degradation becomes more advanced there is a steady loss of sp3 carbon with a commensurate growth in sp2 carbon. The bulk of the chlorine loss occurs by 350 °C with the aliphatic carbon lost by 550 °C, by which temperature the residue is essentially aromatic carbon. Dipolar dephasing experiments show that this residue is essentially a network of, on average, tri-substituted phenyl rings.
Co-reporter:S.C. Martin, J.J. Liggat, C.E. Snape
Polymer Degradation and Stability 2001 Volume 74(Issue 3) pp:407-412
Publication Date(Web):2001
DOI:10.1016/S0141-3910(01)00173-2
In situ broadline 1H NMR has been used to assess the low temperature degradation of polyacrylonitrile (PAN) under a variety of atmospheres and temperatures. In combination with conventional analytical techniques (solid state 13C NMR and FTIR), the structural and conformational changes produced in the network have been correlated with its thermal behaviour. Degradation has also been shown to be both temperature and time dependent, irrespective of reaction environment. Moreover, it has been demonstrated why air is the preferred medium for stabilisation in the production of fibre precursors. Enhanced stability and resistance to char formation is observed in the air-modified polymer, which also becomes resistant to resoftening during low temperature carbonisation.



![Poly[oxy(dimethylsilylene)-1,10-decanediyl(dimethylsilylene)]](http://img.cochemist.com/ccimg/194200/194151-01-4.png)
![Poly[oxy(dimethylsilylene)-1,10-decanediyl(dimethylsilylene)]](http://img.cochemist.com/ccimg/194200/194151-01-4_b.png)
![Bis(2-hydroxyethyl) 2-[(6-oxobenzo[c][2,1]benzoxaphosphinin-6-yl)methyl]butanedioate](http://img.cochemist.com/ccimg/63600/63562-34-5.png)
![Bis(2-hydroxyethyl) 2-[(6-oxobenzo[c][2,1]benzoxaphosphinin-6-yl)methyl]butanedioate](http://img.cochemist.com/ccimg/63600/63562-34-5_b.png)










![Titanium,tris[2-[(2-aminoethyl)amino]ethanolato-kO][2,2-bis[(2-propen-1-yloxy-kO)methyl]-1-butanolato-kO]-, (OC-6-22)-](http://img.cochemist.com/ccimg/107600/107541-22-0.png)
![Titanium,tris[2-[(2-aminoethyl)amino]ethanolato-kO][2,2-bis[(2-propen-1-yloxy-kO)methyl]-1-butanolato-kO]-, (OC-6-22)-](http://img.cochemist.com/ccimg/107600/107541-22-0_b.png)
![Titanium,tris(3-aminophenolato-kO)[2,2-bis[(2-propen-1-yloxy-kO)methyl]-1-butanolato-kO]-, (OC-6-22)-](http://img.cochemist.com/ccimg/107600/107525-86-0.png)
![Titanium,tris(3-aminophenolato-kO)[2,2-bis[(2-propen-1-yloxy-kO)methyl]-1-butanolato-kO]-, (OC-6-22)-](http://img.cochemist.com/ccimg/107600/107525-86-0_b.png)
![Potassium, [(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/53200/53127-82-5.png)
![Potassium, [(trimethylsilyl)methyl]-](http://img.cochemist.com/ccimg/53200/53127-82-5_b.png)








![Titanate(3-),[P,P-bis(2-ethylhexyl) diphosphato(2-)-kO'']bis[P,P-bis(2-ethylhexyl) diphosphato(2-)-kO'',kO''''][2,2-bis[(2-propen-1-yloxy)methyl]-1-butanolato-kO]-, hydrogen (1:3)](http://img.cochemist.com/ccimg/103500/103432-54-8.png)
![Titanate(3-),[P,P-bis(2-ethylhexyl) diphosphato(2-)-kO'']bis[P,P-bis(2-ethylhexyl) diphosphato(2-)-kO'',kO''''][2,2-bis[(2-propen-1-yloxy)methyl]-1-butanolato-kO]-, hydrogen (1:3)](http://img.cochemist.com/ccimg/103500/103432-54-8_b.png)



