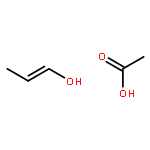
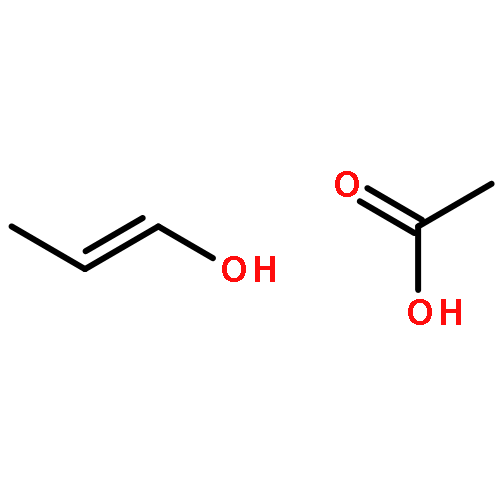
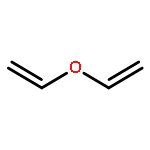
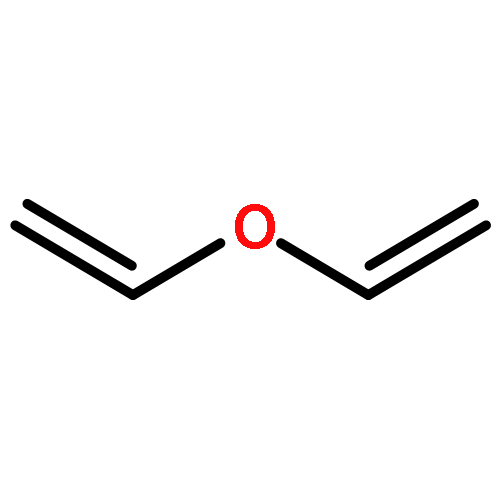
Co-reporter:Shiqing Zhang;Jianfei Sun;Haijie Cao;Qingan Qiao
Structural Chemistry 2017 Volume 28( Issue 6) pp:1831-1842
Publication Date(Web):30 May 2017
DOI:10.1007/s11224-017-0967-2
The comprehensive and reasonable mechanisms of Cl-initiated oxidation of ethyl acrylate (EA) have been proposed by computing at the M06-2X/6-311++G(3df, 2p)//M06-2X/6-31+G(d, p) level of theory. The primary reaction includes eight channels: two Cl additions and six H abstractions. Comparing all calculated results, the reactions of Cl addition are easier to occur than those of H abstraction. However, the hydrogen abstraction from the -CH2 and -CH3 groups cannot be ignored. Based on the Rice–Ramsperger–Kassel–Marcus (RRKM) theory, the rate constants are determined employing the MESMER program. The calculated total rate constant (at 298 K and 760 Torr) is 1.80 × 10−10 cm3 molecule−1 s−1 and shows negative dependence on temperature in the range of 198–648 K. The rate constants for Cl atoms of methyl acrylate (MA), EA, methyl methacrylate (MMA), and allyl acetate (AAC) are kMMA(Cl) > kEA(Cl) > kMA(Cl) > kAAC(Cl). The atmospheric lifetime of EA is 154.3 h for Cl-initiated oxidation which is compared with that of the reaction of other oxidants (OH radicals, O3 molecules, and NO3 radicals) with EA.
Co-reporter:Juan Dang and Maoxia He
RSC Advances 2016 vol. 6(Issue 21) pp:17345-17353
Publication Date(Web):27 Jan 2016
DOI:10.1039/C5RA25959B
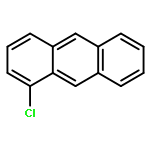
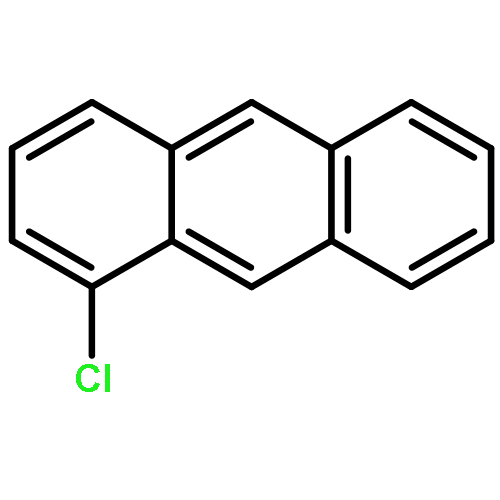
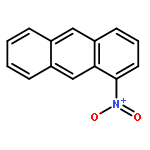
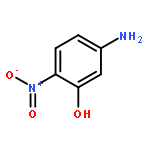
Due to their global dispersion and toxicity, polycyclic aromatic hydrocarbons (PAHs) in the atmosphere have become a serious environmental concern. Atmospheric reactions of PAHs with Cl atoms are of critical importance in specific areas such as the marine boundary layer and coastal regions. In this study, the mechanisms of the Cl radical-initiated atmospheric oxidation of anthracene (Ant) and pyrene (Pyr) were investigated using quantum chemistry calculations. The rate constants for the crucial elementary reactions were estimated. The oxidation products of the gas-phase reactions of Ant and Pyr with Cl atoms are chloro-Ants, chloro-Pyrs, 2-chloroanthracen-1-one, 1-chloropyren-2-one, 1-chloropyren-3-one, 4-chloropyren-5-one, 1-chloro-2-hydroperoxyanthracene, 2-chloro-1-hydroperoxyanthracene, 1-chloro-2-hydroperoxypyrene, 4-chloro-5-hydroperoxypyrene, epoxides, dialdehydes, 9-nitroanthracene, 1-nitroanthracene and nitropyrenes. 9-Nitroanthracene can be more easily produced by the gas-phase reaction of Ant with Cl atoms than that of Ant with OH radicals. Water plays a vital role in the formation of 9-nitroanthracene, resulting from the reactions with NO2. This comprehensive mechanistic study is the first one reported for the Cl radical-initiated atmospheric oxidation of PAHs. The calculated overall rate constants for the Cl addition reactions of Ant and Pyr are 5.87 × 10−12 and 2.81 × 10−12 cm3 per molecule per s, respectively, at 298 K and 1 atm.
Co-reporter:Xin Li, Haijie Cao, Dandan Han, Shiqing Zhang, Maoxia He
Computational and Theoretical Chemistry 2016 Volume 1087() pp:48-56
Publication Date(Web):1 July 2016
DOI:10.1016/j.comptc.2016.04.023
•Mechanism and kinetics of Cl-initiated oxidation of AAC were investigated.•Major products of AAC react with Cl in the presence of O2/NO are indicated.•ktot = 1.39 × 10−10 cm3 molecule−1 s−1 (at 298 K and 760 Torr).•Comparing with hydroxylation Cl-initiated reaction of AAC can be competitive in coastal regions.Computational investigation on Cl-initiated oxidation of allyl acetate (AAC) including reaction mechanisms and kinetics are performed using quantum chemical method. Compared with experimental results, a more comprehensive and reasonable mechanism is proposed. For the primary reaction, seven channels (two Cl-additions and five H-abstractions) are discussed. The calculated results show that the two additional channels dominate the reaction of AAC with Cl. Further investigations of two Cl-adducts are performed in the presence of O2 and NO. The rate constants are calculated using RRKM theory by employing the MESMER program. The total rate constant (1.39 × 10−10 cm3 molecule−1 s−1) is well consistent with experimental data at 298 K and 760 Torr, and shows negative temperature dependence in the range of 200–500 K. Tropospheric half-life of AAC (τ1/2 = 4.6 h for Cl-initiated oxidation) is estimated to evaluate the atmospheric implications.
Co-reporter:Shiqing Zhang, Haijie Cao, Xin Li, Jianfei Sun, Maoxia He
Computational and Theoretical Chemistry 2016 Volume 1091() pp:99-106
Publication Date(Web):1 September 2016
DOI:10.1016/j.comptc.2016.07.008
•Detailed reaction mechanisms and products of MA with Cl atom are investigated.•The Gibbs free energies, the Enthalpy changes and Entropy changes are considered.•In 198–648 K interval, the rate constants of primary reactions are obtained.•Rate constants for MA with Cl as function of temperature at 760 Torr are obtained.•The atmospheric lifetime of MA with Cl atoms is calculated.The atmospheric oxidation mechanism and kinetics of Cl with methyl acrylate are investigated in this study. The mechanisms of MA with Cl atoms have been obtained in detail at the M06-2X/6-311++G(3df, 2p)//M06-2X/6-31+G(d, p) level of theory. The reaction initiates with two kinds of reaction pathways: Cl-addition and H-abstraction. In general, Cl atoms attacking to terminal carbon (the pathway producing IM1) is more favorable. Hydrogen abstraction from the CH3 group also plays an important role. Besides, the kinetic results are determined by MESMER program on the basis of Rice–Ramsperger–Kassel–Marcus (RRKM) theory. The calculated results show that the addition reactions predominate the initial reaction under atmospheric condition and the calculated total rate constant is 1.66 × 10−10 cm3 molecule−1 s−1 at 298 K and 760 Torr.
Co-reporter:Haijie Cao, Dandan Han, Mingyue Li, Xin Li, Shiqing Zhang, Yunqiao Ding, Maoxia He, Wenxing Wang
Computational and Theoretical Chemistry 2015 Volume 1072() pp:72-78
Publication Date(Web):15 November 2015
DOI:10.1016/j.comptc.2015.09.001
•NO3 radical prefers to add to Cβ atom of MVE.•Total rate coefficient of MVE + NO3 exhibits negative dependence on temperature.•Oxirane is dominant product at low pressure.•Over 90% of nitrooxyl radical intermediates undergo collisional stabilization.This study investigates the nitrate radical oxidation of methyl vinyl ether (CH2CHOCH3, MVE) by quantum chemical methods. The oxidation initiates with the formation of open-cyclic NO3-adducts, followed by a series of unimolecular decomposition or bimolecular reactions. Kinetic data suggest that pressure and temperature have strong influence on the environmental fate of MVE. Under atmospheric condition, methyl formate, formaldehyde and NO2 are the main products by association with O2/NO. At low pressure (<50 Torr) 2-methoxyoxirane and NO2 are main products by decomposition. Total rate coefficient of the bimolecular reaction exhibits negative dependence on temperature (200–300 K) and positive dependence on pressure (1–7600 Torr). The calculated rate coefficient (7.09 × 10−13 cm3 molecule−1 s−1) is in well agreement with experimental data at 293 K and 760 Torr.
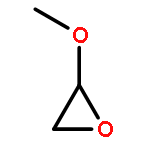
Co-reporter:Dandan Han, Haijie Cao, Mingyue Li, Xin Li, Shiqing Zhang, Maoxia He, and Jingtian Hu
The Journal of Physical Chemistry A 2015 Volume 119(Issue 4) pp:719-727
Publication Date(Web):January 7, 2015
DOI:10.1021/jp5112839
The Cl-initiated oxidation reactions of methyl vinyl ether (MVE) are analyzed by using the high-level composite method CBS-QB3. Detailed chemistry for the reactions of MVE with chlorine atoms is proposed according to the calculated thermodynamic data. The primary eight channels, including two Cl-addition reactions and six H-abstraction reactions, are discussed. In accordance with the further investigation of the two dominant additional routes, formyl chloride and formaldehyde are the major products. Over the temperature range of 200–400 K and the pressure range of 100–2000 Torr, the rate constants of primary reactions are calculated by employing the MESMER program. H-abstraction channels are negligible according to the value of rate constants. During the studied temperature range, the Arrhenius equation is obtained as ktot = 5.64 × 10–11 exp(215.1/T). The total rate coefficient is ktot = 1.25 × 10–10 cm3 molecule–1 s–1 at 298 K and 760 Torr. Finally, the atmospheric lifetime of MVE with respect to Cl is estimated to be 2.23 h.
Co-reporter:Haijie Cao, Dandan Han, Mingyue Li, Xin Li, Maoxia He, and Wenxing Wang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 24) pp:6404-6411
Publication Date(Web):May 26, 2015
DOI:10.1021/acs.jpca.5b04022
This study investigates the decomposition of 2,2′,4,4′,5-pentabrominated diphenyl ether (BDE99), a commonly detected pollutant in the environment. Debromination channels yielding tetrabrominated diphenyl ethers and hydrogen abstracting aromatic bromine atom formations play significant roles in the reaction of BDE99 + H, in which the former absolutely predominates bimolecular reactions. Polybrominated dibenzo-p-dioxins (PBDDs) and polybrominated dibenzofurans (PBDFs) can be produced during BDE99 pyrolysis, especially for PBDFs under inert conditions. The expected dominant pathways in a closed system are debromination products and PBDF formations. The bimolecular reaction with hydroxyl radical mainly leads to hydroxylated BDE99s rather than hydroxylated tetrabrominated diphenyl ethers. PBDDs are then generated from ortho-hydroxylated PBDEs. HO2 radical reactions rarely proceed. The total rate constants for the BDE99 reaction with hydrogen atoms and hydroxyl radicals exhibit positive dependence on temperature with values of 1.86 × 10–14 and 5.24 × 10–14 cm3 molecule–1 s–1 at 298.15 K, respectively.
Co-reporter:Dandan Han;Haijie Cao;Jing Li;Mingyue Li;Xin Li
Structural Chemistry 2015 Volume 26( Issue 3) pp:713-729
Publication Date(Web):2015 June
DOI:10.1007/s11224-014-0517-0
The reaction mechanisms for the atmospheric hydroxylation of n-butyl vinyl ether (n-BVE), iso-butyl vinyl ether (i-BVE), and tert-butyl vinyl ether (t-BVE) were investigated by using quantum chemical method in this paper. The geometry optimizations and frequency calculations were carried out at the MPWB1K/6-31+G(d,p) level, and the accurate energetic parameters were obtained by the MPWB1K/6-311++g(3df,2p) method. The reaction mechanisms for the title reactions have been presented. Ten possible reaction channels were discussed for the primary hydroxylation of n-BVE and t-BVE, while fourteen pathways for i-BVE. Three favorable reaction pathways were chosen for each isomer to undergo further investigation. Major products are n-butyl formate, iso-butyl formate, tert-butyl formate, and HCHO. The rate constants of the primary reactions were calculated over the temperature range of 200–400 K and the pressure range of 100–2,000 Torr by employing MESMER program. At 298 K and 760 Torr, the whole rate constants of n-BVE + OH, i-BVE + OH, and t-BVE + OH are 12.3 × 10−11, 9.32 × 10−11 and 5.75 × 10−11 cm3 molecule−1 s−1, respectively. Additionally, the lifetimes of the three isomers with respect to OH radical are \(\tau\)(n-BVE) = 1.13 h, \(\tau\)(i-BVE) = 1.49 h, and \(\tau\)(t-BVE) = 2.41 h.
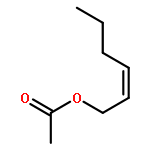
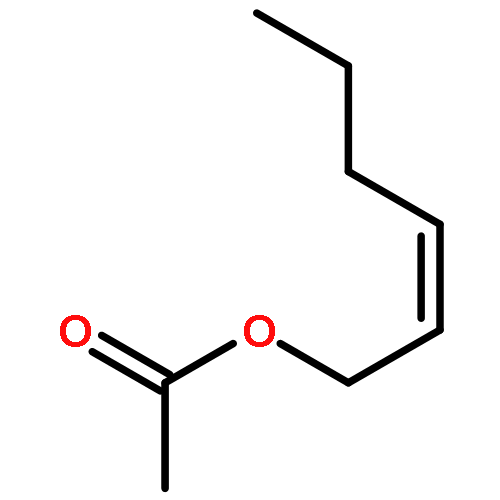
Co-reporter:Jing Li;Yanhui Sun;Haijie Cao;Dandan Han
Structural Chemistry 2014 Volume 25( Issue 1) pp:71-83
Publication Date(Web):2014 February
DOI:10.1007/s11224-013-0226-0
Gas-phase reaction mechanisms of ozone with cis/trans-3-hexenyl acetate and cis/trans-2-hexenyl acetate are performed using density functional theory. The reactions are initiated by the formation of the primary ozonides which are followed by the reactions of biradicals with H2O or NO. The formation of the secondary ozonide (SOZ) is also studied. On the basis of the above DFT calculations, the modified multichannel RRKM theory is used to evaluate the rate constants. At 298 K and 101 kPa, the calculated total rate constants are 9.84 × 10−17, 1.39 × 10−17, 2.50 × 10−17, and 7.37 × 10−17 cm3 mol−1 s−1 for cis-3-hexenyl acetate, trans-2-hexenyl acetate, cis-2-hexenyl acetate, and trans-3-hexenyl acetate, respectively. Our results are in good agreement with experimental values. The total rate coefficients are almost pressure-independent in the range of 0.01–10,000 Torr, but show temperature dependence over the whole study range (200–2,000 K). In addition, branching ratios of the favorable reaction channels are obtained.
Co-reporter:Haijie Cao, Maoxia He, Dandan Han, Jing Li, Mingyue Li, Wenxing Wang, and Side Yao
Environmental Science & Technology 2013 Volume 47(Issue 15) pp:8238-8247
Publication Date(Web):June 26, 2013
DOI:10.1021/es400088v
2,4,4′-Tribromodiphenyl ether (BDE-28) was selected as a typical congener of polybrominated diphenyl ethers (PBDEs) to examine its fate both in the atmosphere and in water solution. All the calculations were obtained at the ground state. The mechanism result shows that the oxidations between BDE-28 and OH radicals are highly feasible especially at the less-brominated phenyl ring. Hydroxylated dibrominated diphenyl ethers (OH-PBDEs) are formed through direct bromine-substitution reactions (P1∼P3) or secondary reactions of OH-adducts (P4∼P8). Polybrominated dibenzo-p-dioxins (PBDDs) resulting from o-OH-PBDEs are favored products compared with polybrominated dibenzofurans (PBDFs) generated by bromophenols and their radicals. The complete degradation of OH adducts in the presence of O2/NO, which generates unsaturated ketones and aldehydes, is less feasible compared with the H-abstraction pathways by O2. Aqueous solution reduces the feasibility between BDE-28 and the OH radical. The rate constant of BDE-28 and the OH radical is determined to be 1.79 × 10–12 cm3 molecule–1 s–1 with an atmospheric lifetime of 6.7 days.
Co-reporter:Yanhui Sun, Haijie Cao, Dandan Han, Jing Li, Maoxia He, Chen Wang
Chemical Physics 2012 Volume 402() pp:6-13
Publication Date(Web):19 June 2012
DOI:10.1016/j.chemphys.2012.03.015
The reaction mechanism for the ozonolysis of ethyl acrylate (EA) has been investigated at the CCSD(T)/6-31G(d)+CF//B3LYP/6-31+G(d,p) level of theory. The profile of the potential energy surface (PES) is constructed. Ozone adds to EA via a cyclic transition state to produce a highly unstable primary ozonide which can decompose readily. Over the temperature range of 200–2000 K, the total and individual rate constants are obtained by employing multichannel Rice–Ramsperger–Kassel–Marcus (RRKM) theory. The calculated rate constants are 1.37 × 10−18 cm3 molecule−1 s−1 at 294 K and 1.65 × 10−18 cm3 molecule−1 s−1 at 298 K under the pressure of 760 Torr. The main products of the reactions are ethyl glyoxylate and formaldehyde. These results are in good agreement with the previous experimental data. Several experimental uncertain products are identified. The branching ratios of main reaction paths are also discussed at different temperatures and pressures.Graphical abstractHighlights► The detailed reaction mechanism of EA with O3 are proposed for the first time. ► Reaction mechanism and rate constants are investigated by theoretical calculation method. ► The results show that ethyl glyoxylate and formaldehyde are the major products of the reaction. ► The rate constant is 1.37 × 10−18 cm3 molecule−1 s−1 at 294 K and the lifetime of EA is determined.
Co-reporter:Haijie Cao, Maoxia He, Dandan Han, Yanhui Sun, Sufang Zhao, Hongjuan Ma, Side Yao
Computational and Theoretical Chemistry 2012 Volume 983() pp:31-37
Publication Date(Web):1 March 2012
DOI:10.1016/j.comptc.2011.12.017
The mechanism and kinetic properties of OH-initiated gas-phase reaction of 2;4-dibrominated diphenyl ether (BDE-7) were studied at the MPWB1K/6-311+G(3df;2p)//MPWB1K/6-31+G(d;p) level of theory. Two types of reactions;hydroxyl addition and hydrogen abstraction;were investigated. The calculation results indicate that addition reactions;except for the bromo-substituted one;have lower barriers than hydrogen abstraction reactions. Moreover;hydroxyl radicals are likely to react with phenyl ring without bromine atoms. Rate constants were deduced over 200–1000 K using canonical variational transition state theory with small curvature tunneling contribution. At 298 K the calculated rate constant for the title reaction is 3.76 × 10−12 cm−3 molecule−1 s−1.Graphical abstractHighlights► Quantum chemical study on the structures and detailed reactions of BDE-7 with OH. ► Hydroxyl addition reactions are the major pathways. ► Phenyl ring without bromines is more active than the other one. ► Reaction rate constants of each elementary reaction have been obtained. ► The rate constants of BDE-7 is larger than BDE-47 while lower than BDE-15.
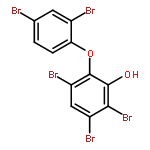
Co-reporter:Haijie Cao, Maoxia He, Yanhui Sun, and Dandan Han
The Journal of Physical Chemistry A 2011 Volume 115(Issue 46) pp:13489-13497
Publication Date(Web):October 20, 2011
DOI:10.1021/jp2059497
Hydroxylated polybrominated diphenyl ethers (OH-PBDEs),which may be generated from PBDEs, are more toxic than their matrix and have been detected in organisms. In this article, we have focused on the gas phase formation of polyhalogenated dibenzo-p-dioxins from several OH-PBDEs and their chlorinated derivatives. All of the geometries and frequencies are calculated at the MPWB1K/6-31+G(d,p) level of theory. The single point energy is obtained at the MPWB1K/6-311+G(3df,2p) level. Rate constants of each step have been calculated over a wide range of 200–2000 K using the canonical variational transition state (CVT) theory with small curvature tunneling (SCT) contribution. The rate equations are shown through Arrhenius formulas. The presence of chlorine atoms increases the reaction barrier for the formation of major products.