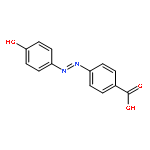
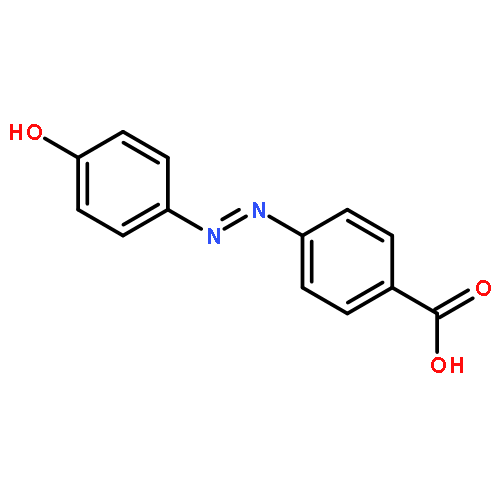
Co-reporter:Huiling Zhao, Xin Song, Hüsnü Aslan, Bo Liu, Jianguo Wang, Li Wang, Flemming Besenbacher and Mingdong Dong
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 21) pp:14168-14171
Publication Date(Web):12 May 2016
DOI:10.1039/C6CP00112B
Self-assembly provides an effective approach for the fabrication of supramolecular complexes or heterojunction materials, which have unique properties and potential applications in many fields. In this study, the self-assembled structures of stearic acid (SA) and nucleic acid base, guanine (G), are formed at the liquid–solid interface. Two main configurations, namely SA–G–SA and SA–G–G–SA, are observed and the intermolecular recognition mechanism between G and SA is proposed from the hydrogen-bonding point of view.
Co-reporter:Dong Chen, Zhichao Wei, Yuheng Yao, Bo Liu
Carbohydrate Research 2015 Volume 401() pp:51-57
Publication Date(Web):12 January 2015
DOI:10.1016/j.carres.2014.10.024
•Building trees of cellobiose and lactose are established.•The IR vibration signatures are agreeable to the experimental results.•Two conformers are first predicted here.•A tree-step computational approach can simplify the conformational determination.Great theoretical attentions have been paid on the conformational preference of individual molecular building blocks of carbohydrates because it is helpful for assignments of the experimental signals and explorations of the biological implications. A tree-step approach is applied here to simplify the conformational determination of phenyl β-cellobioside and benzyl β-lactoside, for which 35 and 23 initial structures are built, respectively. After the high-level calculations, low-energy conformers are determined and then compared with previous experimental and theoretical results. The low-energy conformers are reconstructed in our work for both cellobiose and lactose and the results show a quantitative agreement between the experimental signature and the predicted IR vibration assignment. In addition, two low-energy conformers, which are predicted in our work, have not been reported by the previous work using the traditional method. The tree-step computational approach provides an alternative timesaving and accurate method to focus on determining the preferred conformations of disaccharides.
Co-reporter:Dong Chen;Zhichao Wei
Journal of Molecular Modeling 2015 Volume 21( Issue 9) pp:
Publication Date(Web):2015 September
DOI:10.1007/s00894-015-2781-3
The hydration structure of sodium glycinate (Na+GL−) is probed by the Monte-Carlo multiple minimum (MCMM) method combined with quantum mechanical (QM) calculations at the MP2/6-311++G(d,p) level. In the gas phase, the energy of [Na+GL−]β is more than 30 kJ mol−1 higher than [Na+GL−]α. With higher degrees of hydration, our results indicate that the most stable conformers of [Na+GL−]∙(H2O)8 were derived from [Na+GL−]β instead of [Na+GL−]α. The stable conformers determined by the conductor-like polarizable continuum model (CPCM) also show that [Na+GL−]β is more stable than [Na+GL−]α in the liquid phase. By analyzing the hydration process, water…water hydrogen bonding interaction will be more preferable than ion…water interaction as the number of water molecules increases. According to the electronic density at the bond critical point on the Na-X bonds (X = O1, O2, N) in the low-energy conformers, Na+GL− will be dissociated as Na+ and GL− in the bulk water, which is not predicted by the CPCM model. The structure features and the charge redistribution of Na+GL− will provide a physical explanation for the weakening Na-O1 interaction.
Co-reporter:Yongzhi Li;Xiuhua Liu;Dong Chen;Zhichao Wei
Journal of Molecular Modeling 2013 Volume 19( Issue 9) pp:3619-3626
Publication Date(Web):2013 September
DOI:10.1007/s00894-013-1894-9
A tree-step computational approach has been applied to determine the lowest-energy conformers of luteolin-4′-O-β-D-glucoside (L4′G). Fifty-seven starting structures of the L4′G have been built, and then by performing with density functional theory (DFT) optimizations and second-order Møller-Plesset (MP2) calculations, the preferred conformations of L4′G are predicted. In order to test the accuracy of the computational approach, a hybrid Monte-Carlo multiple minimum (MCMM)/quantum mechanical (QM) approach is applied to determine the favorable conformers of L4′G. The alternative classification is employed to put similar conformations into the same catalogue according to the dihedral angles among the luteolin rings, glycosidic dihedral angles, and the orientations of hydroxyl and hydroxymethyl groups. The low-energy conformations are located after the optimizations at the HF/6-31G(d) and B3LYP/6-311+G(d) levels. Compared with the hybrid MCMM/QM approach, the tree-step computational approach not only remains accurate but also saves a lot of computing resources.
Co-reporter:S. Wang, Y.L. Li, H.L. Zhao, H. Liang, B. Liu, S. Pan
Applied Surface Science 2012 Volume 261() pp:31-36
Publication Date(Web):15 November 2012
DOI:10.1016/j.apsusc.2012.07.041
Abstract
Porous materials have drawn attention from scientists in many fields such as life sciences, catalysis and photonics since they can be used to induce some materials growth as expected. Especially, porous Langmuir–Blodgett (LB) film is an ideal material with controlled thickness and flat surface. In this paper, stearic acid (SA), which has been extensively explored in LB film technique, is chosen as the template material with known parameters to prepare the LB film, and then the porous SA monolayer film is obtained by means of etching in salt solution. The main etching mechanism is suggested that the cations in the solution block the electrostatic interaction between the polar carboxyl group of SA and the electronegative mica surface. The influencing factors (such as concentration of salt solution, valence of cation and surface pressure) of the porous SA film are systematically studied in this work. The novel method proposed in this paper makes it convenient to prepare porous monolayer film for designed material growth or cell culture.
Co-reporter:Yinli Li, Shuai Zhang, Lijun Guo, Mingdong Dong, Bo Liu, Wael Mamdouh
Colloids and Surfaces B: Biointerfaces 2012 Volume 95() pp:10-15
Publication Date(Web):15 June 2012
DOI:10.1016/j.colsurfb.2012.01.009
The extracellular matrix (ECM) plays a key role in cell culture in various physiological and pathological processes in the field of tissue engineering. Recently, the type I collagen ECM has been widely utilized in vitro model systems for the attachment of many different cell lines since it has multi-functions in human tissues. For example it accounts for 6% of the weight of strong, tendinous muscles. In this paper, we reported a new material by coating tantalum (Ta), one highly biocompatible metal, with type I collagen fibrils. The morphology of the new material was studied by high resolution atomic force microscope. It was shown that the adhesion force between type I collagen fibrils network and Ta was strong enough to overcome surface defects. A possible way to explain the phenomenon is that the longitudinal periodicity of collagen fibrils matches the grain size of the Ta domains, which results in increase of the physical adsorption contact area, thereby inducing the dramatic adhesion enhancement between collagen fibrils and Ta. The obtained material was then employed as a template for cell proliferation. Although the surface of this template is more hydrophobic by comparison with the bare Ta surface, the cells on this material were successfully incubated, indicating that the collagen coated Ta might be used as the buffer layer for proliferating cells in hydrophobic biomaterials.Graphical abstractHighlights► We prepare a new ECM by coating collagen on Ta surface and the new ECM becomes from hydrophilic to hydrophobic. ► We still successfully culture cells on the new ECM even though it is hydrophobic. ► The physical adhesion force between collagen and Ta is strong to overcome surface defects. ► The cultured cell is tightly bound to the ECM.
Co-reporter:Yuheng Yao, Dong Chen, Shuai Zhang, Yinli Li, Pinghui Tu, Bo Liu, and Mingdong Dong
The Journal of Physical Chemistry B 2011 Volume 115(Issue 19) pp:6213-6221
Publication Date(Web):April 21, 2011
DOI:10.1021/jp1117097
The first hydration shell of the deprotonated glycine is built up by the discrete hydration model. The potential energy surfaces (PESs) of the deprotonated glycine and its hydration complexes with different number of water molecules have been scanned by the Monte Carlo multiple minimum (MCMM) conformational search analysis with the MMFFs force field. Then the energy-minimized structures are predicted using the high-level ab initio calculations/MP2/6-311++G(d,p). The results of the structural parameters and the infrared spectra indicate that the first-shell water molecules around the anion of deprotonated glycine play a more important role in determining the hydration process of deprotonated glycine. The competition between the hydrate site I and the hydrate site II represents a dynamic process of hydrated complexes. The vibrational properties of C═O and N−H are determined to characterize the structure of deprotonated glycine in solution by the discrete hydration model and the conductor-like polarizable continuum model (CPCM) in the gas phase, respectively.
Co-reporter:Yinli Li, Mingdong Dong, Daniel E Otzen, Yuheng Yao, Bo Liu, Flemming Besenbacher and Wael Mamdouh
Langmuir 2009 Volume 25(Issue 23) pp:13432-13437
Publication Date(Web):June 5, 2009
DOI:10.1021/la900640f
The self-assembly of guanosine (G) molecules on solid surfaces is investigated by tapping-mode atomic force microscopy (AFM) upon controlling and introducing external factors (stimuli) to the G stock solution such as incubation time, presence/absence of metal cations, and mechanical shaking. Surprisingly, at different stages of incubation time at room temperature and in the absence of any metal cations in the G stock solution, which are known to be one of the governing factors in forming G-nanostructures, two assembly pathways resulting into two distinct supramolecular nanostructures were revealed. Astonishingly, by introducing a mechanical shaking of the tube containing the G stock solution, one-dimensional (1D) wires of G molecules are observed by AFM, and very interestingly, novel “branched” supramolecular nanostructures are formed. We have also observed that the later branched G nanostructures can grow further into a two-dimensional (2D) thin film by increasing the incubation time of the G stock solution at room temperature after it is exposed to the external mechanical stimuli. The self-assembled nanostructures of G molecules are changed significantly by tuning the assembly conditions, which show that it is indeed possible to grow complex 2D nanostructures from simple nucleoside molecules.
Co-reporter:Ping-hui Tu, Yu-heng Yao, Yin-li Li, Bo Liu
Journal of Molecular Structure: THEOCHEM 2009 Volume 894(1–3) pp:9-13
Publication Date(Web):30 January 2009
DOI:10.1016/j.theochem.2008.09.034
A scan of the potential-energy surface (PES) for phycocyanobilin has been performed by Monte-Carlo Multiple Minimum (MCMM) searching method using the force field of MMFFs. The resulting most stable/populated structures are then reoptimized with ab initio methods and density functional theory (DFT) using HF/6-31G and B3LYP/6-31G levels of theory, and all possible conformers of phycocyanobilin are investigated with these methods. Results indicate that the most stable structure is all-syn, all-Z conformation. Other local minima are also obtained, such as ZZZass, EZZass and ZZZssa, which are stabilized by intramolecular hydrogen bonds and favorable geometric structures minimizing steric interactions.
Co-reporter:Yanqing Li, Yinli Li, Yuhen Yao, Bo Liu, Menglin Chen, Xiangzhi Song, Mingdong Dong
Colloids and Surfaces B: Biointerfaces 2009 Volume 74(Issue 1) pp:136-139
Publication Date(Web):1 November 2009
DOI:10.1016/j.colsurfb.2009.07.007
The influences of temperature on xanthan biopolymer assemblies on a two-dimensional surface have been thoroughly studied. High resolution atomic force microscope images show that the xanthan nanofibrils can be used to build up well-dispersed 2D scaffold layer after 1 day annealing at 35 °C. By increasing annealing temperatures (60 °C, 90 °C) of xanthan solutions, the well-dispersed layers can be produced rapidly (6 h, 0.5 h) with micro-sized pore structures. The xanthan scaffold with pore structures potentially allows accommodating micro-sized cells for tissue engineering.
Co-reporter:Virgile Bernigaud;Kasper Drenck
Journal of The American Society for Mass Spectrometry 2008 Volume 19( Issue 6) pp:809-813
Publication Date(Web):2008 June
DOI:10.1016/j.jasms.2008.01.002
Electron-capture induced dissociation of protoporphyrin cations and anions has been studied. The cations captured two electrons in two successive collisions and were converted to the corresponding even-electron anions. About one fifth of the ions lost a hydrogen atom to become radical anions but otherwise very little fragmentation was observed. The anions captured an electron to become dianions. No hydrogen loss occurred, and the only fragmentation channel observed was loss of CO2H, to give a doubly charged carbanion. Our results indicate that protoporphyrin ions are very efficient in accommodating one or even two electrons in the lowest unoccupied molecular orbital of the porphyrin macrocycle, and that electron capture induces only limited dissociation.
Co-reporter:Huiling Zhao, Xin Song, Hüsnü Aslan, Bo Liu, Jianguo Wang, Li Wang, Flemming Besenbacher and Mingdong Dong
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 21) pp:NaN14171-14171
Publication Date(Web):2016/05/12
DOI:10.1039/C6CP00112B
Self-assembly provides an effective approach for the fabrication of supramolecular complexes or heterojunction materials, which have unique properties and potential applications in many fields. In this study, the self-assembled structures of stearic acid (SA) and nucleic acid base, guanine (G), are formed at the liquid–solid interface. Two main configurations, namely SA–G–SA and SA–G–G–SA, are observed and the intermolecular recognition mechanism between G and SA is proposed from the hydrogen-bonding point of view.

![3,5,9-Trioxa-4-phosphaheneicosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxododecyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/18200/18194-25-7.png)
![3,5,9-Trioxa-4-phosphaheneicosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxododecyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/18200/18194-25-7_b.png)