Co-reporter:Samantha I. Johnson, Harry B. Gray, James D. Blakemore, and William A. Goddard III
Inorganic Chemistry September 18, 2017 Volume 56(Issue 18) pp:11375-11375
Publication Date(Web):September 1, 2017
DOI:10.1021/acs.inorgchem.7b01698
Recent work has shown that Cp*Rh(bpy) [Cp* = pentamethylcyclopentadienyl, bpy = 2,2′- bipyridine] undergoes endo protonation at the [Cp*] ligand in the presence of weak acid (Et3NH+; pKa = 18.8 in MeCN). Upon exposure to stronger acid (e.g., DMFH+; pKa = 6.1), hydrogen is evolved with unity yield. Here, we study the mechanisms by which this catalyst evolves dihydrogen using density functional theory (M06) with polarizable continuum solvation. The calculations show that the complex can be protonated by weak acid first at the metal center with a barrier of 3.2 kcal/mol; this proton then migrates to the ring to form the detected intermediate, a rhodium(I) compound bearing endo η4-Cp*H. Stronger acid is required to evolve hydrogen, which calculations show happens via a concerted mechanism. The acid approaches and protonates the metal, while the second proton simultaneously migrates from the ring with a barrier of ∼12 kcal/mol. Under strongly acidic conditions, we find that hydrogen evolution can proceed through a traditional metal–hydride species; protonation of the initial hydride to form an H–H bond occurs before migration of the hydride (in the form of a proton) to the [Cp*] ring (i.e., H–H bond formation is faster than hydride–proton tautomerization). This work demonstrates the role of acid strength in accessing different mechanisms of hydrogen evolution. Calculations also predict that modification of the bpy ligand by a variety of functional groups does not affect the preference for [Cp*] protonation, although the driving force for protonation changes. However, we predict that exchange of bpy for a bidentate phosphine ligand will stabilize a rhodium(III) hydride, reversing the preference for bound [Cp*H] found in all computed bpy derivatives and offering an appealing alternative ligand platform for future experimental and computational mechanistic studies of H2 evolution.
Co-reporter:Kun Sun, Tao Cheng, Lina Wu, Yongfeng Hu, Jigang Zhou, Aimee Maclennan, Zhaohua Jiang, Yunzhi Gao, William A. Goddard III, and Zhijiang Wang
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15608-15608
Publication Date(Web):October 9, 2017
DOI:10.1021/jacs.7b09251
Wide application of carbon dioxide (CO2) electrochemical energy storage requires catalysts with high mass activity. Alloy catalysts can achieve superior performance to single metals while reducing the cost by finely tuning the composition and morphology. We used in silico quantum mechanics rapid screening to identify Au–Fe as a candidate improving CO2 reduction and then synthesized and tested it experimentally. The synthesized Au–Fe alloy catalyst evolves quickly into a stable Au–Fe core–shell nanoparticle (AuFe-CSNP) after leaching out surface Fe. This AuFe-CSNP exhibits exclusive CO selectivity, long-term stability, nearly a 100-fold increase in mass activity toward CO2 reduction compared with Au NP, and 0.2 V lower in overpotential. Calculations show that surface defects due to Fe leaching contribute significantly to decrease the overpotential.
Co-reporter:Jonathan E. Mueller;Adri C. T. van Duin
The Journal of Physical Chemistry C March 25, 2010 Volume 114(Issue 11) pp:4939-4949
Publication Date(Web):Publication Date (Web): February 26, 2010
DOI:10.1021/jp9035056
To enable the study of hydrocarbon reactions catalyzed by nickel surfaces and particles using reactive molecular dynamics on thousands of atoms as a function of temperature and pressure, we have developed the ReaxFF reactive force field to describe adsorption, decomposition, reformation and desorption of hydrocarbons as they interact with the nickel surface. The ReaxFF parameters were determined by fitting to the geometries and energy surfaces from quantum mechanics (QM) calculations for a large number of reaction pathways for hydrocarbon molecules chemisorbed onto nickel (111), (100) and (110) surfaces, supplemented with QM equations of state for nickel and nickel carbides. We demonstrate the validity and accuracy of ReaxFF by applying it to study the reaction dynamics of hydrocarbons as catalyzed by nickel particles and surfaces. For the dissociation of methyl on the (111), (100), and stepped (111) surfaces of nickel, we observe the formation of chemisorbed CH plus subsurface carbide. We observe that the (111) surface is the least reactive, the (100) surface has the fastest reaction rates, and the stepped (111) surface has an intermediate reaction rate. The importance of surface defects in accelerating reaction rates is highlighted by these results.
Co-reporter:Wei Liu, Mu-Jeng Cheng, Robert J. Nielsen, William A. Goddard III, and John T. Groves
ACS Catalysis June 2, 2017 Volume 7(Issue 6) pp:4182-4182
Publication Date(Web):May 10, 2017
DOI:10.1021/acscatal.7b00655
The oxygen rebound mechanism, proposed four decades ago, is invoked in a wide range of oxygen- and heteroatom-transfer reactions. In this process, a high-valent metal-oxo species abstracts a hydrogen atom from the substrate to generate a carbon-centered radical, which immediately recombines with the hydroxometal intermediate with very fast rate constants that can be in the nanosecond to picosecond regime. In addition to catalyzing C–O bond formation, we found that manganese porphyrins can also directly catalyze C–H halogenations and pseudohalogenations, including chlorination, bromination, and fluorination as well as C–H azidation. For these cases, we showed that long-lived substrate radicals are involved, indicating that radical rebound may involve a barrier in some cases. In this study, we show that axial ligands significantly affect the oxygen rebound rate. Fluoride, hydroxide, and oxo ligands all slow down the oxygen rebound rate by factors of 10–40-fold. The oxidation of norcarane by a manganese porphyrin coordinated with fluoride or hydroxide leads to the formation of significant amounts of radical rearranged products. cis-Decalin oxidation afforded both cis- and trans-decalol. Xanthene afforded dioxygen trapped products and the radical dimer product, bixanthene, under aerobic and anaerobic conditions, respectively. DFT calculations probing the rebound step show that the rebound barrier increases significantly (by 3.3, 5.4, and 6.0 kcal/mol, respectively) with fluoride, hydroxide, and oxo as axial ligands.Keywords: DFT; heterorebound catalysis; iron porphyrin; manganese porphyrin; oxygen rebound;
Co-reporter:Ted H. Yu;Yao Sha;Boris V. Merinov
The Journal of Physical Chemistry C July 8, 2010 Volume 114(Issue 26) pp:11527-11533
Publication Date(Web):2017-2-22
DOI:10.1021/jp1024735
Based on studies on Pt3Co and Pt3Ni, we developed the hypothesis that improved alloy catalysts for the oxygen reduction reaction (ORR) at fuel cell cathodes should have a surface layer that is noble (e.g., Pt, Pd, or Rh) while the second layer should have ∼50% electropositive metal to decrease the critical barriers for ORR, and we used quantum mechanics (QM) to examine 80 binary alloys of composition Y3X, where Y = Pt, Pd, Rh, and X is any of the three rows of transition metals (columns 3−11). This study identified that for Pd3X, good segregating alloys include X = Re (best), W, Os, Mo, Ru, Ir, Tc, Rh, Co, Ta, Nb, and Ni. Of these we selected Pd3W as particularly promising since it is known experimentally to form an ordered alloy and was found to have a desirable d-band center. We then examined the critical barriers for various steps of the ORR with Pd3W and compared them to the analogous barriers for Pt, Pt3Co, and Pd. These results suggest that Pd3W will exhibit ORR properties dramatically improved over pure Pd and close to that of pure Pt. The cost of Pd3W is ∼6 times less than pure Pt, suggesting that Pd3W catalysts might lead to significant decreases in catalyst cost, while maintaining performance.
Co-reporter:Ross Fu, William A. Goddard III, Mu-Jeng Cheng, and Robert J. Nielsen
ACS Catalysis January 6, 2017 Volume 7(Issue 1) pp:356-356
Publication Date(Web):November 28, 2016
DOI:10.1021/acscatal.6b02781
We propose the vanadium bis(2-phenoxyl)phosphinite pincer complex, denoted (OPO)V, as a low-temperature water-soluble catalyst for monoxygenation of propane to isopropanol with functionalization and catalyst regeneration using molecular oxygen. We use density functional theory (DFT) study to predict that the barrier for (OPO)V to activate the secondary hydrogen of propane is ΔG⧧ = 25.2 kcal/mol at 298 K, leading to isopropanol via the new reduction-coupled oxo activation (ROA) mechanism. We then show that reoxidation by dioxygen to complete the cycle is also favorable with ΔG⧧ = 6.2 kcal/mol at 298 K. We conclude that (OPO)V represents a promising homogeneous catalyst for the monoxygenation of propane and other alkanes (including ethane), warranting experimental validation.Keywords: alkane C−H activation; alkane oxidation; density functional theory; homogeneous catalysis; pincer ligand; vanadium;
Co-reporter:Tao Cheng, Hai Xiao, and William A. Goddard
Journal of the American Chemical Society August 30, 2017 Volume 139(Issue 34) pp:11642-11642
Publication Date(Web):August 16, 2017
DOI:10.1021/jacs.7b03300
Recent experiments show that the grain boundaries (GBs) of copper nanoparticles (NPs) lead to an outstanding performance in reducing CO2 and CO to alcohol products. We report here multiscale simulations that simulate experimental synthesis conditions to predict the structure of a 10 nm Cu NP (158 555 atoms). To identify active sites, we first predict the CO binding at a large number of sites and select four exhibiting CO binding stronger than the (211) step surface. Then, we predict the formation energy of the *OCCOH intermediate as a descriptor for C–C coupling, identifying two active sites, both of which have an under-coordinated surface square site adjacent to a subsurface stacking fault. We then propose a periodic Cu surface (4 by 4 supercell) with a similar site that substantially decreases the formation energy of *OCCOH, by 0.14 eV.
Co-reporter:William A. Goddard, III;Tao Cheng;Yuanyue Liu;Hai Xiao
PNAS 2017 Volume 114 (Issue 26 ) pp:6685-6688
Publication Date(Web):2017-06-27
DOI:10.1073/pnas.1702405114
We propose and validate with quantum mechanics methods a unique catalyst for electrochemical reduction of CO2 (CO2RR) in which selectivity and activity of CO and C2 products are both enhanced at the borders of oxidized and metallic surface regions. This Cu metal embedded in oxidized matrix
(MEOM) catalyst is consistent with observations that Cu2O-based electrodes improve performance. However, we show that a fully oxidized matrix (FOM) model would not explain the experimentally
observed performance boost, and we show that the FOM is not stable under CO2 reduction conditions. This electrostatic tension between the Cu+ and Cu0 surface sites responsible for the MEOM mechanism suggests a unique strategy for designing more efficient and selective electrocatalysts
for CO2RR to valuable chemicals (HCOx), a critical need for practical environmental and energy applications.
Co-reporter:William A. Goddard, III;Junko Yano;Ethan J. Crumlin;Hai Xiao;Tao Cheng;Marco Favaro
PNAS 2017 Volume 114 (Issue 26 ) pp:6706-6711
Publication Date(Web):2017-06-27
DOI:10.1073/pnas.1701405114
A national priority is to convert CO2 into high-value chemical products such as liquid fuels. Because current electrocatalysts are not adequate, we aim to discover
new catalysts by obtaining a detailed understanding of the initial steps of CO2 electroreduction on copper surfaces, the best current catalysts. Using ambient pressure X-ray photoelectron spectroscopy
interpreted with quantum mechanical prediction of the structures and free energies, we show that the presence of a thin suboxide
structure below the copper surface is essential to bind the CO2 in the physisorbed configuration at 298 K, and we show that this suboxide is essential for converting to the chemisorbed
CO2 in the presence of water as the first step toward CO2 reduction products such as formate and CO. This optimum suboxide leads to both neutral and charged Cu surface sites, providing
fresh insights into how to design improved carbon dioxide reduction catalysts.
Co-reporter:Tao Cheng, III;Hai Xiao
PNAS 2017 Volume 114 (Issue 8 ) pp:1795-1800
Publication Date(Web):2017-02-21
DOI:10.1073/pnas.1612106114
A critical step toward the rational design of new catalysts that achieve selective and efficient reduction of CO2 to specific hydrocarbons and oxygenates is to determine the detailed reaction mechanism including kinetics and product selectivity
as a function of pH and applied potential for known systems. To accomplish this, we apply ab initio molecular metadynamics
simulations (AIMμD) for the water/Cu(100) system with five layers of the explicit solvent under a potential of −0.59 V [reversible
hydrogen electrode (RHE)] at pH 7 and compare with experiment. From these free-energy calculations, we determined the kinetics
and pathways for major products (ethylene and methane) and minor products (ethanol, glyoxal, glycolaldehyde, ethylene glycol,
acetaldehyde, ethane, and methanol). For an applied potential (U) greater than −0.6 V (RHE) ethylene, the major product, is produced via the Eley–Rideal (ER) mechanism using H2O + e–. The rate-determining step (RDS) is C–C coupling of two CO, with ΔG‡ = 0.69 eV. For an applied potential less than −0.60 V (RHE), the rate of ethylene formation decreases, mainly due to the
loss of CO surface sites, which are replaced by H*. The reappearance of C2H4 along with CH4 at U less than −0.85 V arises from *CHO formation produced via an ER process of H* with nonadsorbed CO (a unique result). This
*CHO is the common intermediate for the formation of both CH4 and C2H4. These results suggest that, to obtain hydrocarbon products selectively and efficiency at pH 7, we need to increase the CO
concentration by changing the solvent or alloying the surface.
Co-reporter:Michael S. Webster-Gardiner, Paige E. Piszel, Ross Fu, Bradley A. McKeown, ... T. Brent Gunnoe
Journal of Molecular Catalysis A: Chemical 2017 Volume 426, Part B(Volume 426, Part B) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.molcata.2016.07.045
•Rh(I) catalyzed H/D exchange.•Ligand effects.•Electrophilic aromatic substitution.A series of new rhodium (I) complexes supported by bidentate nitrogen-donor ligands with varying electronic and steric properties were synthesized in situ and evaluated for catalytic arene C–H/D activation. In trifluoroacetic acid (HTFA), these complexes are proposed to mediate H/D exchange of arene C–H/D bonds by an electrophilic aromatic substitution mechanism that involves Rh-mediated activation of HTFA (or DTFA). DFT calculations support the proposed pathway for the H/D exchange reactions.Download high-res image (58KB)Download full-size image
Co-reporter:Tao Cheng;William A Goddard, III;Qi An;Hai Xiao;Boris Merinov;Sergey Morozov
Physical Chemistry Chemical Physics 2017 vol. 19(Issue 4) pp:2666-2673
Publication Date(Web):2017/01/25
DOI:10.1039/C6CP08055C
The sluggish oxygen reduction reaction (ORR) is a major impediment to the economic use of hydrogen fuel cells in transportation. In this work, we report the full ORR reaction mechanism for Pt(111) based on Quantum Mechanics (QM) based Reactive metadynamics (RμD) simulations including explicit water to obtain free energy reaction barriers at 298 K. The lowest energy pathway for 4 e− water formation is: first, *OOH formation; second, *OOH reduction to H2O and O*; third, O* hydrolysis using surface water to produce two *OH and finally *OH hydration to water. Water formation is the rate-determining step (RDS) for potentials above 0.87 Volt, the normal operating range. Considering the Eley–Rideal (ER) mechanism involving protons from the solvent, we predict the free energy reaction barrier at 298 K for water formation to be 0.25 eV for an external potential below U = 0.87 V and 0.41 eV at U = 1.23 V, in good agreement with experimental values of 0.22 eV and 0.44 eV, respectively. With the mechanism now fully understood, we can use this now validated methodology to examine the changes upon alloying and surface modifications to increase the rate by reducing the barrier for water formation.
Co-reporter:Tao Cheng, III;Hai Xiao
PNAS 2017 Volume 114 (Issue 8 ) pp:1795-1800
Publication Date(Web):2017-02-21
DOI:10.1073/pnas.1612106114
A critical step toward the rational design of new catalysts that achieve selective and efficient reduction of CO2 to specific hydrocarbons and oxygenates is to determine the detailed reaction mechanism including kinetics and product selectivity
as a function of pH and applied potential for known systems. To accomplish this, we apply ab initio molecular metadynamics
simulations (AIMμD) for the water/Cu(100) system with five layers of the explicit solvent under a potential of −0.59 V [reversible
hydrogen electrode (RHE)] at pH 7 and compare with experiment. From these free-energy calculations, we determined the kinetics
and pathways for major products (ethylene and methane) and minor products (ethanol, glyoxal, glycolaldehyde, ethylene glycol,
acetaldehyde, ethane, and methanol). For an applied potential (U) greater than −0.6 V (RHE) ethylene, the major product, is produced via the Eley–Rideal (ER) mechanism using H2O + e–. The rate-determining step (RDS) is C–C coupling of two CO, with ΔG‡ = 0.69 eV. For an applied potential less than −0.60 V (RHE), the rate of ethylene formation decreases, mainly due to the
loss of CO surface sites, which are replaced by H*. The reappearance of C2H4 along with CH4 at U less than −0.85 V arises from *CHO formation produced via an ER process of H* with nonadsorbed CO (a unique result). This
*CHO is the common intermediate for the formation of both CH4 and C2H4. These results suggest that, to obtain hydrocarbon products selectively and efficiency at pH 7, we need to increase the CO
concentration by changing the solvent or alloying the surface.
Co-reporter:John Beatty, Tao ChengYuan Cao, M. Sky Driver, William A. Goddard III, Jeffry A. Kelber
The Journal of Physical Chemistry Letters 2017 Volume 8(Issue 1) pp:188-192
Publication Date(Web):December 14, 2016
DOI:10.1021/acs.jpclett.6b02325
We report directly grown strongly adherent graphene on Co3O4(111) by carbon molecular beam epitaxy (C MBE) at 850 K and density functional theory (DFT) findings that the first graphene layer is reconstructed to fit the Co3O4 surface, while subsequent layers retain normal graphene structure. This adherence to the Co3O4 structure results from partial bonding of half the carbons to top oxygens of the substrate. This structure is validated by X-ray photoelectron spectroscopy and low-energy electron diffraction studies, showing layer-by-layer graphene growth with ∼0.08 electrons/carbon atom transferred to the oxide from the first graphene layer, in agreement with DFT. In contrast, for Cr2O3 DFT finds no strong bonding to the surface and C MBE on Cr2O3(0001) yields only graphite formation at 700 K, with C desorption above 800 K. Thus strong graphene-to-oxide charge transfer aids nucleation of graphene on incommensurate oxide substrates and may have implications for spintronics.
Co-reporter:Qi An, William A. Goddard III, Kelvin Y. Xie, Gi-dong Sim, Kevin J. Hemker, Tyler Munhollon, M. Fatih Toksoy, and Richard A. Haber
Nano Letters 2016 Volume 16(Issue 12) pp:7573-7579
Publication Date(Web):November 7, 2016
DOI:10.1021/acs.nanolett.6b03414
The theoretical strength of a material is the minimum stress to deform or fracture the perfect single crystal material that has no defects. This theoretical strength is considered as an upper bound on the attainable strength for a real crystal. In contradiction to this expectation, we use quantum mechanics (QM) simulations to show that for the boron carbide (B4C) hard ceramic, this theoretical shear strength can be exceeded by 11% by imposing nanoscale twins. We also predict from QM that the indentation strength of nanotwinned B4C is 12% higher than that of the perfect crystal. Further, we validate this effect experimentally, showing that nanotwinned samples are harder by 2.3% than the twin-free counterpart of B4C. The origin of this strengthening mechanism is suppression of twin boundary (TB) slip within the nanotwins due to the directional nature of covalent bonds at the TB.Keywords: deformation mechanism; DFT; hardness; nanoindentation; Superhard ceramics;
Co-reporter:Yuanyue Liu, Hai Xiao, and William A. Goddard III
Nano Letters 2016 Volume 16(Issue 5) pp:3335-3340
Publication Date(Web):April 21, 2016
DOI:10.1021/acs.nanolett.6b00964
Two-dimensional (2D) halide perovskites are emerging as promising candidates for nanoelectronics and optoelectronics. To realize their full potential, it is important to understand the role of those defects that can strongly impact material properties. In contrast to other popular 2D semiconductors (e.g., transition metal dichalcogenides MX2) for which defects typically induce harmful traps, we show that the electronic activities of defects in 2D perovskites are significantly tunable. For example, even with a fixed lattice orientation one can change the synthesis conditions to convert a line defect (edge or grain boundary) from electron acceptor to inactive site without deep gap states. We show that this difference originates from the enhanced ionic bonding in these perovskites compared with MX2. The donors tend to have high formation energies and the harmful defects are difficult to form at a low halide chemical potential. Thus, we unveil unique properties of defects in 2D perovskites and suggest practical routes to improve them.
Co-reporter:Qi An, K. Madhav Reddy, Huafeng Dong, Ming-Wei Chen, Artem R. Oganov, and William A. Goddard III
Nano Letters 2016 Volume 16(Issue 7) pp:4236-4242
Publication Date(Web):June 2, 2016
DOI:10.1021/acs.nanolett.6b01204
Nanotwinned structures in superhard ceramics rhombohedral boron suboxide (R-B6O) have been examined using a combination of transmission electron microscopy (TEM) and quantum mechanics (QM). QM predicts negative relative energies to R-B6O for various twinned R-B6O (denoted as τ-B6O, 2τ-B6O, and 4τ-B6O), consistent with the recently predicted B6O structure with Cmcm space group (τ-B6O) which has an energy 1.1 meV/B6O lower than R-B6O. We report here TEM observations of this τ-B6O structure, confirming the QM predictions. QM studies under pure shear deformation and indentation conditions are used to determine the deformation mechanisms of the new τ-B6O phase which are compared to R-B6O and 2τ-B6O. The lowest stress slip system of τ-B6O is (010)/⟨001⟩ which transforms τ-B6O to R-B6O under pure shear deformation. However, under indentation conditions, the lowest stress slip system changes to (001)/⟨110⟩, leading to icosahedra disintegration and hence amorphous band formation.
Co-reporter:Tao Cheng, Hai Xiao, and William A. Goddard III
Journal of the American Chemical Society 2016 Volume 138(Issue 42) pp:13802-13805
Publication Date(Web):October 11, 2016
DOI:10.1021/jacs.6b08534
Copper is the only elemental metal that reduces a significant fraction of CO2 to hydrocarbons and alcohols, but the atomistic reaction mechanism that controls the product distributions is not known because it has not been possible to detect the reaction intermediates on the electrode surface experimentally, or to carry out Quantum Mechanics (QM) calculations with a realistic description of the electrolyte (water). Here, we carry out QM calculations with an explicit description of water on the Cu(100) surface (experimentally shown to be stable under CO2 reduction reaction conditions) to examine the initial reaction pathways to form CO and formate (HCOO–) from CO2 through free energy calculations at 298 K and pH 7. We find that CO formation proceeds from physisorbed CO2 to chemisorbed CO2 (*CO2δ−), with a free energy barrier of ΔG⧧ = 0.43 eV, the rate-determining step (RDS). The subsequent barriers of protonating *CO2δ− to form COOH* and then dissociating COOH* to form *CO are 0.37 and 0.30 eV, respectively. HCOO– formation proceeds through a very different pathway in which physisorbed CO2 reacts directly with a surface H* (along with electron transfer), leading to ΔG⧧ = 0.80 eV. Thus, the competition between CO formation and HCOO– formation occurs in the first electron-transfer step. On Cu(100), the RDS for CO formation is lower, making CO the predominant product. Thus, to alter the product distribution, we need to control this first step of CO2 binding, which might involve controlling pH, alloying, or changing the structure at the nanoscale.
Co-reporter:Yuanyue Liu, Hai Xiao, and William A. Goddard III
Journal of the American Chemical Society 2016 Volume 138(Issue 49) pp:15853-15856
Publication Date(Web):November 22, 2016
DOI:10.1021/jacs.6b10834
Two-dimensional (2D) metal carbides and nitrides, called MXenes, have attracted great interest for applications such as energy storage. We demonstrate their potential as Schottky-barrier-free metal contacts to 2D semiconductors, providing a solution to the contact-resistance problem in 2D electronics. On the basis of first-principles calculations, we find that the surface chemistry strongly affects Fermi level of MXenes: O termination always increases the work function with respect to that of bare surface, OH always decreases it, whereas F exhibits either trend depending on the specific material. This phenomenon originates from the effect of surface dipoles, which together with the weak Fermi level pinning, enable Schottky-barrier-free hole (or electron) injection into 2D semiconductors through van der Waals junctions with some of the O-terminated (or all the OH-terminated) MXenes. Furthermore, we suggest synthetic routes to control surface terminations based on calculated formation energies. This study enhances understanding of the correlation between surface chemistry and electronic/transport properties of 2D materials, and also gives predictions for improving 2D electronics.
Co-reporter:Hai XiaoTao Cheng, William A. Goddard III
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:130-136
Publication Date(Web):December 7, 2016
DOI:10.1021/jacs.6b06846
Practical environmental and energy applications of the electrochemical reduction of CO2 to chemicals and fuels require far more efficient and selective electrocatalysts beyond the only working material Cu, but the wealth of experimental data on Cu can serve to validate any proposed mechanisms. To provide design guidelines, we use quantum mechanics to predict the detailed atomistic mechanisms responsible for C1 and C2 products on Cu. Thus, we report the pH dependent routes to the major products, methane and ethylene, and identify the key intermediates where branches to methanol, ketene, ethanol, acetylene, and ethane are kinetically blocked. We discovered that surface water on Cu plays a key role in the selectivity for hydrocarbon products over the oxygen-containing alcohol products by serving as a strong proton donor for electrochemical dehydration reductions. We suggest new experiments to validate our predicted mechanisms.
Co-reporter:Yuan Ping, Robert J. Nielsen, and William A. Goddard III
Journal of the American Chemical Society 2016 Volume 139(Issue 1) pp:149-155
Publication Date(Web):December 9, 2016
DOI:10.1021/jacs.6b07557
How to efficiently oxidize H2O to O2 (oxygen evolution reaction, OER) in photoelectrochemical cells (PEC) is a great challenge due to its complex charge transfer process, high overpotential, and corrosion. So far no OER mechanism has been fully explained atomistically with both thermodynamic and kinetics. IrO2 is the only known OER catalyst with both high catalytic activity and stability in acidic conditions. This is important because PEC experiments often operate at extreme pH conditions. In this work, we performed first-principles calculations integrated with implicit solvation at constant potentials to examine the detailed atomistic reaction mechanism of OER at the IrO2 (110) surface. We determined the surface phase diagram, explored the possible reaction pathways including kinetic barriers, and computed reaction rates based on the microkinetic models. This allowed us to resolve several long-standing puzzles about the atomistic OER mechanism.
Co-reporter:Guodong Li, Saurabh Bajaj, Umut Aydemir, Shiqiang Hao, Hai Xiao, William A. Goddard III, Pengcheng Zhai, Qingjie Zhang, and G. Jeffrey Snyder
Chemistry of Materials 2016 Volume 28(Issue 7) pp:2172
Publication Date(Web):March 22, 2016
DOI:10.1021/acs.chemmater.6b00112
Skutterudite CoSb3 based thermoelectric devices have high potential for engineering applications because both n- and p-type doped CoSb3 demonstrate excellent thermoelectric performance. A crucial point concerning the application of CoSb3 is to understand and control its defect chemistry. To reveal the native conductivity behavior of nonstoichiometric CoSb3, we investigated the intrinsic point defects in CoSb3 using density functional theory. We found CoSb3 is p-type in either Co or Sb rich regions of phase stability. Interstitial Co (Coi) and interstitial Co-pair (Coi-p) are the dominant point defects in the Co rich region. However, Coi-p will be difficult to form because the formation temperature of Coi-p is much lower than the synthesis temperature of CoSb3. The unexpected acceptor nature of the Coi or Coi-p defects is explained by the breakage of multiple Sb4-rings. Co vacancy (Cov) is found to be the p-type defect in the Sb rich region. Furthermore, the solubility of excess Co in CoSb3 is expected to be larger than that of Sb because of the lower formation energy and higher carrier concentration of Coi compared with those of Cov.
Co-reporter:Qi An, Jin Qian, Robert R. Nielsen, Luca Sementa, Giovanni Barcaro, Fabio R. Negreiros, Alessandro Fortunelli and William A. Goddard III
Journal of Materials Chemistry A 2016 vol. 4(Issue 31) pp:12036-12045
Publication Date(Web):14 Jun 2016
DOI:10.1039/C6TA03669D
Experimental evidence that surface acoustic waves (SAW) can significantly enhance the rate of catalytic oxidation of CO to CO2 over the Pt(110) catalyst surface [S. Kelling et al., Faraday Disc., 1997, 107, 435–444] is examined using quantum mechanics (QM) simulations. First we determined the QM based mechanism for the O2-rich régime of the reaction, and the energy landscape of CO interacting with an O-covered reconstructed Pt(110) surface at both static and dynamic levels, but in the absence of SAW. We then utilized ab initio molecular dynamic (AIMD) simulations to determine how SAW might modify the kinetics. We focus here on the short (picosecond time scale) shock spikes induced by switching of domains in the piezoelectric driver on which the catalyst is deposited. We find that SAW-induced spikes promote dynamic changes in the diffusion and desorption, from which we estimate the influence of SAW on CO oxidation rate over Pt(110). We find good agreement with the experimentally observed catalytic enhancement by SAW. With an atomistic mechanism in place one can now consider how to use SAW to enhance other catalytic reactions.
Prediction of structures and properties of 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N) green energetic materials from DFT and ReaxFF molecular modeling
Co-reporter:Saber Naserifar, Sergey Zybin, Cai-Chao Ye and William A. Goddard III
Journal of Materials Chemistry A 2016 vol. 4(Issue 4) pp:1264-1276
Publication Date(Web):09 Nov 2015
DOI:10.1039/C5TA06426K
2,4,6-Triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N) were suggested by Klapötke et al. as candidates for green high energy density materials (HEDM), but a successful synthesis has not yet been reported. In order to predict the properties of these systems, we used quantum mechanics (PBE flavor of density functional theory) to predict the most stable conformations of MTO and MTO3N and their optimum packing into the most stable crystal structures. We found that MTO has the P21 space-group with a density of ρ = 1.92 g cm−3 while MTO3N has the P21/c space-group with a density of ρ = 2.10 g cm−3. The heats of reaction (ΔHrxn) were computed to be 1036 kcal kg−1 for MTO, 1412 kcal kg−1 for MTO3N, and 1653 kcal kg−1 for a mixture of them. These properties are comparable to those of such other useful energetic materials as RDX (ρ = 1.80 g cm−3, ΔHrxn = 1266 kcal kg−1), HMX, and PETN, making MTO and MTO3N excellent candidates for environmentally friendly HEDMs. In addition, we predicted the stability of –NH2, –NO, and –NO2 groups in water solution. We also show that the ReaxFF-lg reactive FF leads to an accurate description of the structural properties of MTO and MTO3N crystals making it practical to carry out large-scale reactive molecular dynamics simulations practical for these systems to determine the sensitivity and performance (CJ point calculation and velocity) under shear, shock, and thermal loads.
Co-reporter:Guodong Li, Qi An, William A. Goddard III, Riley Hanus, Pengcheng Zhai, Qingjie Zhang, G. Jeffrey Snyder
Acta Materialia 2016 Volume 103() pp:775-780
Publication Date(Web):15 January 2016
DOI:10.1016/j.actamat.2015.11.021
Abstract
CoSb3 based skutterudite thermoelectric material has superior thermoelectric properties, but the low fracture toughness prevents its widespread commercial application. To determine the origin of its brittle failure, we examined the response of shear deformation in CoSb3 along the most plausible slip system (010)/<100>, using large-scale molecular dynamics simulations. We find that the brittle failure of CoSb3 arises from the formation of shear bands due to the destruction of Sb4-rings and the slippage of Co-octahedraes. This leads to the breakage of Co-octahedraes and cavitation, resulting in the crack opening and mechanical failure.
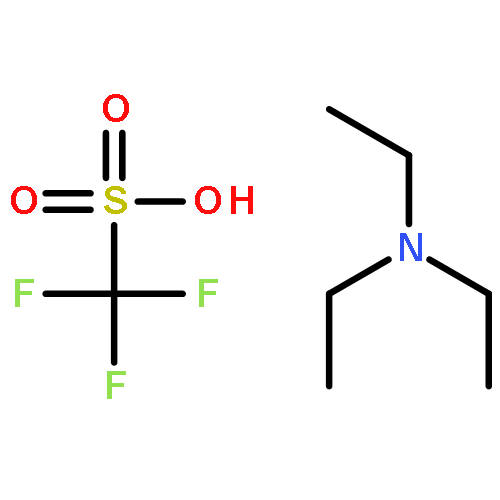
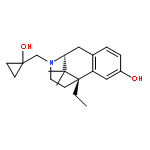
Co-reporter:Caitlin E. Scott; Kwang H. Ahn; Steven T. Graf; William A. GoddardIII; Debra A. Kendall;Ravinder Abrol
Journal of Chemical Information and Modeling 2016 Volume 56(Issue 1) pp:201-212
Publication Date(Web):December 3, 2015
DOI:10.1021/acs.jcim.5b00581
Human cannabinoid type 1 (CB1) G-protein coupled receptor is a potential therapeutic target for obesity. The previously predicted and experimentally validated ensemble of ligand-free conformations of CB1 [Scott, C. E. et al. Protein Sci. 2013, 22, 101−113; Ahn, K. H. et al. Proteins 2013, 81, 1304–1317] are used here to predict the binding sites for known CB1-selective inverse agonists including rimonabant and its seven known derivatives. This binding pocket, which differs significantly from previously published models, is used to identify 16 novel compounds expected to be CB1 inverse agonists by exploiting potential new interactions. We show experimentally that two of these compounds exhibit inverse agonist properties including inhibition of basal and agonist-induced G-protein coupling activity, as well as an enhanced level of CB1 cell surface localization. This demonstrates the utility of using the predicted binding sites for an ensemble of CB1 receptor structures for designing new CB1 inverse agonists.
Co-reporter:Dezhou Guo, Sergey V. Zybin, Qi An, William A. Goddard III and Fenglei Huang
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 3) pp:2015-2022
Publication Date(Web):17 Nov 2015
DOI:10.1039/C5CP04516A
The combustion or detonation of reacting materials at high temperature and pressure can be characterized by the Chapman–Jouguet (CJ) state that describes the chemical equilibrium of the products at the end of the reaction zone of the detonation wave for sustained detonation. This provides the critical properties and product kinetics for input to macroscale continuum simulations of energetic materials. We propose the ReaxFF Reactive Dynamics to CJ point protocol (Rx2CJ) for predicting the CJ state parameters, providing the means to predict the performance of new materials prior to synthesis and characterization, allowing the simulation based design to be done in silico. Our Rx2CJ method is based on atomistic reactive molecular dynamics (RMD) using the QM-derived ReaxFF force field. We validate this method here by predicting the CJ point and detonation products for three typical energetic materials. We find good agreement between the predicted and experimental detonation velocities, indicating that this method can reliably predict the CJ state using modest levels of computation.
Co-reporter:Marios D. Demetriou;Konrad Samwer;Qi An;Michael C. Floyd;Danielle O. Duggins;William L. Johnson, III
PNAS 2016 Volume 113 (Issue 26 ) pp:7053-7058
Publication Date(Web):2016-06-28
DOI:10.1073/pnas.1607506113
To gain insight into the large toughness variability observed between metallic glasses (MGs), we examine the origin of fracture
toughness through bending experiments and molecular dynamics (MD) simulations for two binary MGs: Pd82Si18 and Cu46Zr54. The bending experiments show that Pd82Si18 is considerably tougher than Cu46Zr54, and the higher toughness of Pd82Si18 is attributed to an ability to deform plastically in the absence of crack nucleation through cavitation. The MD simulations
study the initial stages of cavitation in both materials and extract the critical factors controlling cavitation. We find
that for the tougher Pd82Si18, cavitation is governed by chemical inhomogeneity in addition to topological structures. In contrast, no such chemical correlations
are observed in the more brittle Cu46Zr54, where topological low coordination number polyhedra are still observed around the critical cavity. As such, chemical inhomogeneity
leads to more difficult cavitation initiation in Pd82Si18 than in Cu46Zr54, leading to a higher toughness. The absence of chemical separation during cavitation initiation in Cu46Zr54 decreases the energy barrier for a cavitation event, leading to lower toughness.
Co-reporter:Shaojie Jiang;Dr. Yanan Fang;Dr. Ruipeng Li;Dr. Hai Xiao;Jason Crowley;Dr. Chenyu Wang; Timothy J. White; William A. Goddard III;Dr. Zhongwu Wang;Dr. Tom Baikie; Jiye Fang
Angewandte Chemie International Edition 2016 Volume 55( Issue 22) pp:6540-6544
Publication Date(Web):
DOI:10.1002/anie.201601788
Abstract
We report the pressure-induced crystallographic transitions and optical behavior of MAPbI3 (MA=methylammonium) using in situ synchrotron X-ray diffraction and laser-excited photoluminescence spectroscopy, supported by density functional theory (DFT) calculations using the hybrid functional B3PW91 with spin-orbit coupling. The tetragonal polymorph determined at ambient pressure transforms to a ReO3-type cubic phase at 0.3 GPa. Upon continuous compression to 2.7 GPa this cubic polymorph converts into a putative orthorhombic structure. Beyond 4.7 GPa it separates into crystalline and amorphous fractions. During decompression, this phase-mixed material undergoes distinct restoration pathways depending on the peak pressure. In situ pressure photoluminescence investigation suggests a reduction in band gap with increasing pressure up to ≈0.3 GPa and then an increase in band gap up to a pressure of 2.7 GPa, in excellent agreement with our DFT calculation prediction.
Co-reporter:Shaojie Jiang;Dr. Yanan Fang;Dr. Ruipeng Li;Dr. Hai Xiao;Jason Crowley;Dr. Chenyu Wang; Timothy J. White; William A. Goddard III;Dr. Zhongwu Wang;Dr. Tom Baikie; Jiye Fang
Angewandte Chemie 2016 Volume 128( Issue 22) pp:6650-6654
Publication Date(Web):
DOI:10.1002/ange.201601788
Abstract
We report the pressure-induced crystallographic transitions and optical behavior of MAPbI3 (MA=methylammonium) using in situ synchrotron X-ray diffraction and laser-excited photoluminescence spectroscopy, supported by density functional theory (DFT) calculations using the hybrid functional B3PW91 with spin-orbit coupling. The tetragonal polymorph determined at ambient pressure transforms to a ReO3-type cubic phase at 0.3 GPa. Upon continuous compression to 2.7 GPa this cubic polymorph converts into a putative orthorhombic structure. Beyond 4.7 GPa it separates into crystalline and amorphous fractions. During decompression, this phase-mixed material undergoes distinct restoration pathways depending on the peak pressure. In situ pressure photoluminescence investigation suggests a reduction in band gap with increasing pressure up to ≈0.3 GPa and then an increase in band gap up to a pressure of 2.7 GPa, in excellent agreement with our DFT calculation prediction.
Co-reporter:Ho-Cheng Tsai
The Journal of Physical Chemistry C 2016 Volume 120(Issue 1) pp:207-214
Publication Date(Web):December 11, 2015
DOI:10.1021/acs.jpcc.5b06847
In this study, we used quantum mechanics (QM) to investigate steam reforming of methane on Ni-alloy catalyst surfaces and to examine the effect of anode material modifications on the catalytic processes in a solid oxide fuel cell (SOFC). The conventional Ni anode suffers from coking, coarsening, and sulfur poisoning because of the decomposition of hydrocarbon fuels, Ni particle agglomeration at high operating temperature, and impurities contained in fuels. Ni-electrode surface modification, such as alloying Ni with other metals (e.g., Fe and Cu), is probably the most practical and promising way of developing SOFC anodes tolerant to coking and sulfur poisoning. According to experimental data, Ni4Fe shows a good catalytic performance and excellent long-term stability as an SOFC anode catalyst. We have performed QM calculations of segregation energy for various surface structures of five-layer Ni4M slabs (M = Co, Fe, and Mn) and found that Ni atoms show segregation preference for the surface layer and the most favorable Ni4M surface structure has two M atoms in the 2nd layer and one M atom in the 3rd and in the 4th layer (the numbering starts from the bottom layer). This structure was used for our further QM calculations of binding energies for CHx, C, and H. We find that the Ni4M(111) surfaces bind CHx species weaker (by 1–10 kcal/mol) than pure Ni, and the binding energy of C is always ∼10 kcal/mol lower for the Ni4M alloys compared to pure Ni. This is consistent with improved catalytic characteristics of certain Ni-based alloys compared to pure Ni obtained in experiment. Reaction energy barriers for methane decomposition on the Ni4M(111) catalyst surfaces were calculated as well. On the basis of these results, the rate-determining step for the methane decomposition was found to be the CH → C + H reaction. Our results predict that Ni4Fe and Ni4Mn have both better activity and better coking resistance and can be considered as candidates for an SOFC anode catalyst suitable for the CH4 fuel reforming.
Co-reporter:Matthew E. O’Reilly, Samantha I. Johnson, Robert J. Nielsen, William A. Goddard III, and T. Brent Gunnoe
Organometallics 2016 Volume 35(Issue 12) pp:2053-2056
Publication Date(Web):June 9, 2016
DOI:10.1021/acs.organomet.6b00285
Transition-metal-mediated nucleophilic aromatic substitution (SNAr) reactions prefer that a suitably strong nucleophile be in an aprotic medium. Usually, using protic nucleophile/medium requires high reaction temperatures (>180 °C) to overcome the attenuated nucleophilicity for attack on the arene π system. Surprisingly, we demonstrate herein a RhIII-mediated SNAr reaction of a fluoroarene moiety with RCO2H (R = CH3, CF3) in acid media that proceeds at moderate temperatures (<100 °C). We show both by experimental and with DFT calculations that the mechanism proceeds through an internal nucleophilic aromatic substitution (I-SNAr), where the nucleophile coordinates to the metal ion prior to substitution, thereby mitigating the acid influence.
Co-reporter:Jason M. Crowley; Jamil Tahir-KheliIII
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 7) pp:1198-1203
Publication Date(Web):March 4, 2016
DOI:10.1021/acs.jpclett.5b02870
An important property with any new material is the band gap. Standard density functional theory methods grossly underestimate band gaps. This is known as the band gap problem. Here, we show that the hybrid B3PW91 density functional returns band gaps with a mean absolute deviation (MAD) from experiment of 0.22 eV over 64 insulators with gaps spanning a factor of 500 from 0.014 to 7 eV. The MAD is 0.28 eV over 70 compounds with gaps up to 14.2 eV, with a mean error of −0.03 eV. To benchmark the quality of the hybrid method, we compared the hybrid method to the rigorous GW many-body perturbation theory method. Surprisingly, the MAD for B3PW91 is about 1.5 times smaller than the MAD for GW. Furthermore, B3PW91 is 3–4 orders of magnitude faster computationally. Hence, B3PW91 is a practical tool for predicting band gaps of materials before they are synthesized and represents a solution to the band gap prediction problem.
Co-reporter:Dr. Karel J. Hartlieb;Dr. Wei-Guang Liu;Dr. Albert C. Fahrenbach;Anthea K. Blackburn;Dr. Marco Frasconi;Dr. Nema Hafezi;Dr. Sanjeev K. Dey;Dr. Amy A. Sarjeant;Charlotte L. Stern; William A. Goddard III; J. Fraser Stoddart
Chemistry - A European Journal 2016 Volume 22( Issue 8) pp:2736-2745
Publication Date(Web):
DOI:10.1002/chem.201502157
Abstract
The promiscuous encapsulation of π-electron-rich guests by the π-electron-deficient host, cyclobis(paraquat-p-phenylene) (CBPQT4+), involves the formation of 1:1 inclusion complexes. One of the most intensely investigated charge-transfer (CT) bands, assumed to result from inclusion of a guest molecule inside the cavity of CBPQT4+, is an emerald-green band associated with the complexation of tetrathiafulvalene (TTF) and its derivatives. This interpretation was called into question recently in this journal based on theoretical gas-phase calculations that reinterpreted this CT band in terms of an intermolecular side-on interaction of TTF with one of the bipyridinium (BIPY2+) units of CBPQT4+, rather than the encapsulation of TTF inside the cavity of CBPQT4+. We carried out DFT calculations, including solvation, that reveal conclusively that the CT band emerging upon mixing TTF with CBPQT4+ arises from the formation of a 1:1 inclusion complex. In support of this conclusion, we have performed additional experiments on a [2]rotaxane in which a TTF unit, located in the middle of its short dumbbell, is prevented sterically from interacting with either one of the two BIPY2+ units of a CBPQT4+ ring residing on a separate [2]rotaxane in a side-on fashion. This [2]rotaxane has similar UV/Vis and 1H NMR spectroscopic properties with those of 1:1 inclusion complexes of TTF and its derivatives with CBPQT4+. The [2]rotaxane exists as an equimolar mixture of cis- and trans-isomers associated with the disubstituted TTF unit in its dumbbell component. Solid-state structures were obtained for both isomers, validating the conclusion that the TTF unit, which gives rise to the CT band, resides inside CBPQT4+.
Co-reporter:Jeong-hyun Kim, Soo-Kyung Kim, Jae-Hyuk Lee, Young-Joon Kim, William A. Goddard III, Yong-Chul Kim
Journal of Molecular Graphics and Modelling 2016 Volume 66() pp:115-122
Publication Date(Web):May 2016
DOI:10.1016/j.jmgm.2016.03.014
•The 3D structures of drosophila and aedes sex peptide receptors were generated by using the GEnSeMBLE method.•To investigate the binding mechanism of the new small molecule agonists, molecular docking studies were performed.•The Ser3.25, Tyr5.35 and Phe2.67 residues of DrmSPR would be important for binding of small molecule agonist compound 1.•Molecular docking studies supported the previously observed structure-activity relationships of the reference compounds.•The structural differences between DrmSPR and AedesSPR lead to the species selectivity of compound 1.The Drosophila melanogaster sex peptide receptor (DrmSPR), which is a G protein-coupled receptor (GPCR), is known as the specific receptor for sex peptide (SP). It is responsible for the reproductive behavior in the Drosophila model system; in particular, it is involved in the post-mating responses such as the increase in egg-laying ability and decrease in receptivity in females. In a previous study, we discovered a small molecule agonist of DrmSPR for the first time, which could not, however, activate Aedes aegypti SPR (AedesSPR). To investigate the binding mechanism of the small molecule agonist of DrmSPR, the ensemble structures of low-lying packing structures of DrmSPR and AedesSPR were assembled using the GEnSeMBLE (GPCR Ensemble of Structures in Membrane BiLayer Environment) method. The generated homology models exhibited the typical pattern of inter-helical interactions of the class A GPCRs. The docking experiments of the small molecule agonist suggest that Tyr5.35 and Phe2.67 residues may be involved in a hydrophobic interaction and that Ser3.25 forms a hydrogen bond with the agonist. Additionally, we found that the docking results were consistent with the experimental data of the reference compounds with variable agonistic activities. Moreover, a potential distinction of the putative binding sites in two GPCR models of DrmSPR and AedesSPR, which was determined in this study, can explain the selective action of the agonist for DrmSPR but not for AedesSPR.
Co-reporter:Mu-Jeng Cheng III
Topics in Catalysis 2016 Volume 59( Issue 17-18) pp:1506-1517
Publication Date(Web):2016 October
DOI:10.1007/s11244-016-0669-9
We report here first principles predictions (density functional theory with periodic boundary conditions) of the structures, mechanisms, and activation barriers for the catalytic activation and functionalization of propane by the M1 phase of the Mitsubishi-BP America generation of Mo–V–Nb–Te–O mixed metal oxide (MMO) catalysts. Our calculations show that the reduction-coupled oxo activation (ROA) principle, which we reported at Irsee VI to play the critical role for the selective oxidation of n-butane to maleic anhydride by vanadium phosphorous oxide, also plays the critical role for the MMO activation of propane, as speculated during Irsee VI. However for MMO, this ROA principle involves Te=O and V rather than P=O and V. The ability of the Te=O bond to activate the propane CH bond depends sensitively upon the number of V atoms that are coupled through a bridging O to the Te=O center. Based on this ROA mechanism, we suggest synthetic procedures aimed at developing a single phase MMO catalyst with dramatically improved selectivity for ammoxidation. We also suggest a modified single phase composition suitable for simultaneous oxidative dehydrogenation of ethane and propane to ethene and propene, respectively, which is becoming more important with the increase in petroleum fracking. Moreover, we also suggest some organometallic molecules that activate alkane CH bonds through the ROA principle.
Co-reporter:Tao Cheng;Zipeng Zhao;Mufan Li;Alessandro Fortunelli;Chih-Yen Chen;Qinghua Zhang;Rong Yu;Enbo Zhu;Boris V. Merinov;Jinghua Guo;Qingying Jia;Yu Huang III;Lin Gu;Xiangfeng Duan;Ted Yu;Zhaoyang Lin;Liang Zhang
Science 2016 Volume 354(Issue 6318) pp:
Publication Date(Web):
DOI:10.1126/science.aaf9050
An activity lift for platinum
Platinum is an excellent but expensive catalyst for the oxygen reduction reaction (ORR), which is critical for fuel cells. Alloying platinum with other metals can create shells of platinum on cores of less expensive metals, which increases its surface exposure, and compressive strain in the layer can also boost its activity (see the Perspective by Stephens et al.). Bu et al. produced nanoplates—platinum-lead cores covered with platinum shells—that were in tensile strain. These nanoplates had high and stable ORR activity, which theory suggests arises from the strain optimizing the platinum-oxygen bond strength. Li et al. optimized both the amount of surface-exposed platinum and the specific activity. They made nanowires with a nickel oxide core and a platinum shell, annealed them to the metal alloy, and then leached out the nickel to form a rough surface. The mass activity was about double the best reported values from previous studies.
Science, this issue p. 1410, p. 1414; see also p. 1378
Co-reporter:Yuanyue Liu;Boris V. Merinov III;
Proceedings of the National Academy of Sciences 2016 113(14) pp:3735-3739
Publication Date(Web):March 21, 2016
DOI:10.1073/pnas.1602473113
It is well known that graphite has a low capacity for Na but a high capacity for other alkali metals. The growing interest
in alternative cation batteries beyond Li makes it particularly important to elucidate the origin of this behavior, which
is not well understood. In examining this question, we find a quite general phenomenon: among the alkali and alkaline earth
metals, Na and Mg generally have the weakest chemical binding to a given substrate, compared with the other elements in the
same column of the periodic table. We demonstrate this with quantum mechanics calculations for a wide range of substrate materials
(not limited to C) covering a variety of structures and chemical compositions. The phenomenon arises from the competition
between trends in the ionization energy and the ion–substrate coupling, down the columns of the periodic table. Consequently,
the cathodic voltage for Na and Mg is expected to be lower than those for other metals in the same column. This generality
provides a basis for analyzing the binding of alkali and alkaline earth metal atoms over a broad range of systems.
Co-reporter:Yufeng Huang; Robert J. Nielsen; William A. GoddardIII;Manuel P. Soriaga
Journal of the American Chemical Society 2015 Volume 137(Issue 20) pp:6692-6698
Publication Date(Web):May 5, 2015
DOI:10.1021/jacs.5b03329
We report density functional theory (M06L) calculations including Poisson–Boltzmann solvation to determine the reaction pathways and barriers for the hydrogen evolution reaction (HER) on MoS2, using both a periodic two-dimensional slab and a Mo10S21 cluster model. We find that the HER mechanism involves protonation of the electron rich molybdenum hydride site (Volmer–Heyrovsky mechanism), leading to a calculated free energy barrier of 17.9 kcal/mol, in good agreement with the barrier of 19.9 kcal/mol estimated from the experimental turnover frequency. Hydronium protonation of the hydride on the Mo site is 21.3 kcal/mol more favorable than protonation of the hydrogen on the S site because the electrons localized on the Mo–H bond are readily transferred to form dihydrogen with hydronium. We predict the Volmer–Tafel mechanism in which hydrogen atoms bound to molybdenum and sulfur sites recombine to form H2 has a barrier of 22.6 kcal/mol. Starting with hydrogen atoms on adjacent sulfur atoms, the Volmer–Tafel mechanism goes instead through the M–H + S–H pathway. In discussions of metal chalcogenide HER catalysis, the S–H bond energy has been proposed as the critical parameter. However, we find that the sulfur–hydrogen species is not an important intermediate since the free energy of this species does not play a direct role in determining the effective activation barrier. Rather we suggest that the kinetic barrier should be used as a descriptor for reactivity, rather than the equilibrium thermodynamics. This is supported by the agreement between the calculated barrier and the experimental turnover frequency. These results suggest that to design a more reactive catalyst from edge exposed MoS2, one should focus on lowering the reaction barrier between the metal hydride and a proton from the hydronium in solution.
Co-reporter:Yuan Ping; William A. GoddardIII;Giulia A. Galli
Journal of the American Chemical Society 2015 Volume 137(Issue 16) pp:5264-5267
Publication Date(Web):April 13, 2015
DOI:10.1021/jacs.5b00798
The design of optimal interfaces between photoelectrodes and catalysts is a key challenge in building photoelectrochemical cells to split water. Iridium dioxide (IrO2) is an efficient catalyst for oxygen evolution, stable in acidic conditions, and hence a good candidate to be interfaced with photoanodes. Using first-principles quantum mechanical calculations, we investigated the structural and electronic properties of tungsten trioxide (WO3) surfaces interfaced with an IrO2 thin film. We built a microscopic model of the interface that exhibits a formation energy lower than the surface energy of the most stable IrO2 surface, in spite of a large lattice mismatch, and has no impurity states pinning the Fermi level. We found that, upon full coverage of WO3 by IrO2, the two oxides form undesirable Ohmic contacts. However, our calculations predicted that if both oxides are partially exposed to water solvent, the relative position of the absorber conduction band and the catalyst Fermi level favors charge transfer to the catalyst and hence water splitting. We propose that, for oxide photoelectrodes interfaced with IrO2, it is advantageous to form rough interfaces with the catalyst, e.g., by depositing nanoparticles, instead of sharp interfaces with thin films.
Co-reporter:Hai Xiao; Tao Cheng; William A. GoddardIII;Ravishankar Sundararaman
Journal of the American Chemical Society 2015 Volume 138(Issue 2) pp:483-486
Publication Date(Web):December 30, 2015
DOI:10.1021/jacs.5b11390
Energy and environmental concerns demand development of more efficient and selective electrodes for electrochemical reduction of CO2 to form fuels and chemicals. Since Cu is the only pure metal exhibiting reduction to form hydrocarbon chemicals, we focus here on the Cu (111) electrode. We present a methodology for density functional theory calculations to obtain accurate onset electrochemical potentials with explicit constant electrochemical potential and pH effects using implicit solvation. We predict the atomistic mechanisms underlying electrochemical reduction of CO, finding that (1) at acidic pH, the C1 pathway proceeds through COH to CHOH to form CH4 while C2 (C3) pathways are kinetically blocked; (2) at neutral pH, the C1 and C2 (C3) pathways share the COH common intermediate, where the branch to C–C coupling is realized by a novel CO–COH pathway; and (3) at high pH, early C–C coupling through adsorbed CO dimerization dominates, suppressing the C1 pathways by kinetics, thereby boosting selectivity for multi-carbon products.
Co-reporter:Mu-Jeng ChengIII
Journal of the American Chemical Society 2015 Volume 137(Issue 41) pp:13224-13227
Publication Date(Web):September 30, 2015
DOI:10.1021/jacs.5b07073
We used density functional theory quantum mechanics with periodic boundary conditions to determine the atomistic mechanism underlying catalytic activation of propane by the M1 phase of Mo-V-Nb-Te-O mixed metal oxides. We find that propane is activated by Te═O through our recently established reduction-coupled oxo activation mechanism. More importantly, we find that the C–H activation activity of Te═O is controlled by the distribution of nearby V atoms, leading to a range of activation barriers from 34 to 23 kcal/mol. On the basis of the new insight into this mechanism, we propose a synthesis strategy that we expect to form a much more selective single-phase Mo-V-Nb-Te-O catalyst.
Co-reporter:Dominik Munz, Michael Webster-Gardiner, Ross Fu, Thomas Strassner, William A. Goddard III, and T. Brent Gunnoe
ACS Catalysis 2015 Volume 5(Issue 2) pp:769
Publication Date(Web):December 30, 2014
DOI:10.1021/cs501620f
The H/D exchange of arenes in acidic media by transition-metal and main-group-metal complexes and common inorganic salts was studied. The influence of Lewis acidity, anions, charge, and ligands was evaluated. The results indicate that the determination of H/D exchange activity in acidic media is not related to the formation of metal–carbon bonds (i.e., C–H activation). The combined experimental data (regioselectivity, activation energy, kinetics, isotope effects, solvent effects) and DFT calculations point toward a proton catalysis mechanism. Thus, highly Lewis acidic metal compounds, such as aluminum(III) triflate, were extraordinarily active for the H/D exchange reactions. Indeed, the degree of H/D exchange reactivity allows for a comparative measurement of Lewis acidities.Keywords: acids; benzene; CH activation; H/D exchange; Lewis acid; toluene
Co-reporter:Alessandro Fortunelli, William A. Goddard III, Luca Sementa, Giovanni Barcaro, Fabio R. Negreiros and Andrés Jaramillo-Botero
Chemical Science 2015 vol. 6(Issue 7) pp:3915-3925
Publication Date(Web):29 Apr 2015
DOI:10.1039/C5SC00840A
Recently Debe et al. reported that Pt3Ni7 leads to extraordinary Oxygen Reduction Reaction (ORR) activity. However, several reports show that hardly any Ni remains in the layers of the catalysts close to the surface (“Pt-skin effect”). This paradox that Ni is essential to the high catalytic activity with the peak ORR activity at Pt3Ni7 while little or no Ni remains close to the surface is explained here using large-scale first-principles-based simulations. We make the radical assumption that processing Pt–Ni catalysts under ORR conditions would leach out all Ni accessible to the solvent. To simulate this process we use the ReaxFF reactive force field, starting with random alloy particles ranging from 50% Ni to 90% Ni and containing up to ∼300000 atoms, deleting the Ni atoms, and equilibrating the resulting structures. We find that the Pt3Ni7 case and a final particle radius around 7.5 nm lead to internal voids in communication with the exterior, doubling the external surface footprint, in fair agreement with experiment. Then we examine the surface character of these nanoporous systems and find that a prominent feature in the surface of the de-alloyed particles is a rhombic structure involving 4 surface atoms which is crystalline-like but under-coordinated. Using density-functional theory, we calculate the energy barriers of ORR steps on Pt nanoporous catalysts, focusing on the Oad-hydration reaction (Oad + H2Oad → OHad + OHad) but including the barriers of O2 dissociation (O2ad → Oad + Oad) and water formation (OHad + Had → H2Oad). We find that the reaction barrier for the Oad-hydration rate-determining-step is reduced significantly on the de-alloyed surface sites compared to Pt(111). Moreover we find that these active sites are prevalent on the surface of particles de-alloyed from a Pt–Ni 30:70 initial composition. These simulations explain the peak in surface reactivity at Pt3Ni7, and provide a rational guide to use for further optimization of improved catalytic and nanoporous materials.
Co-reporter:Guodong Li, Qi An, Wenjuan Li, William A. Goddard III, Pengcheng Zhai, Qingjie Zhang, and G. Jeffrey Snyder
Chemistry of Materials 2015 Volume 27(Issue 18) pp:6329
Publication Date(Web):September 4, 2015
DOI:10.1021/acs.chemmater.5b02268
Skutterudites based on CoSb3 have high thermoelectric efficiency, but the low fracture strength is a serious consideration for commercial applications. To understand the origin of the brittleness in CoSb3, we examine the response along various shear and tensile deformations using density functional theory. We find that the Co–Sb bond dominates the ideal strength. Among all the shear and tensile deformation paths, shearing along the (001)/⟨100⟩ slip system has the lowest ideal strength, indicating it is the most likely slip system to be activated under pressure. We also find that, because the Sb–Sb covalent bond is softer than the Co–Sb bond, the Sb-rings are less rigid than the Co–Sb frameworks, which leads to the Sb-rings softening before the Co–Sb frameworks. Further deformation leads to deconstruction of Sb-rings and collapse of Co–Sb frameworks, resulting in structural failure. Moreover, we find that filling of the CoSb3 void spaces with such typical fillers as Na, Ba, or Yb has little effect on the ideal strength and failure mode, which can be understood because they have little effect on the Sb-rings.
Co-reporter:Qi An and William A. Goddard III
Chemistry of Materials 2015 Volume 27(Issue 8) pp:2855
Publication Date(Web):April 6, 2015
DOI:10.1021/cm5046918
Boron suboxide (B6O), boron carbide (B4C), and related materials are superhard. However, they exhibit low fracture toughness, which limits their engineering applications. Here we show the shear deformation mechanism of B6O using density functional theory along the most plausible slip system (01̅11)/<101̅1>. We discovered an unusual phenomenon in which the highly sheared system recovers its original crystal structure, which indicates the possibility of being sheared to a large strain without failure. We also found a similar structural recovery in boron subphosphide (B12P2) for shearing along the same slip system. In contrast, for components of B4C, we found brittle failure. These novel deformation mechanisms under high shear deformation conditions suggest that a key element to designing ductile hard materials is to couple the icosahedra via one- or two-atom chains that allow the system to shear by walking the intericosahedral bonds and chain bonds alternately to accommodate large shear without fracturing the icosahedra.
Co-reporter:Ho-Cheng Tsai, Yu-Chi Hsieh, Ted H. Yu, Yi-Juei Lee, Yue-Han Wu, Boris V. Merinov, Pu-Wei Wu, San-Yuan Chen, Radoslav R. Adzic, and William A. Goddard III
ACS Catalysis 2015 Volume 5(Issue 3) pp:1568
Publication Date(Web):January 22, 2015
DOI:10.1021/cs501020a
Proton exchange membrane fuel cells (PEMFCs) have attracted much attention as an alternative source of energy with a number of advantages, including high efficiency, sustainability, and environmentally friendly operation. However, the low kinetics of the oxygen reduction reaction (ORR) restricts the performance of PEMFCs. Various types of catalysts have been developed to improve the ORR efficiency, but this problem still needs further investigations and improvements. In this paper, we propose advanced Os/Pt core–shell catalysts based on our previous study on segregation of both bare surfaces and surfaces exposed to ORR adsorbates, and we evaluate the catalytic activity of the proposed materials by density functional theory (DFT). Quantum mechanics was applied to calculate binding energies of ORR species and reaction energy barriers on Os/Pt core–shell catalysts. Our calculations predict a much better catalytic activity of the Os/Pt system than that of pure Pt. We find that the ligand effect of the Os substrate is more important than the lattice compression strain effect. To validate our DFT prediction, we demonstrate the fabrication of Os/Pt core–shell nanoparticles using the underpotential deposition (UPD) technique and succeeding galvanic displacement reaction between the Pt ions and Cu-coated Os nanoparticles. The Os/Pt/C samples were evaluated for electrocatalytic activities toward the ORR in acidic electrolytes. The samples with two consecutive UPD-displacement reaction cycles show 3.5 to 5 times better ORR activities as compared to those of commercially available Pt/C. Our results show good agreement between the computational predictions and electrochemical experimental data for the Os/Pt core–shell ORR catalysts.Keywords: core−shell catalysts; DFT; electrocatalysis; ORR; PtOs; UPD
Co-reporter:Cai-Chao Ye, Qi An, William A. Goddard III, Tao Cheng, Wei-Guang Liu, Sergey V. Zybin and Xue-Hai Ju
Journal of Materials Chemistry A 2015 vol. 3(Issue 5) pp:1972-1978
Publication Date(Web):25 Nov 2014
DOI:10.1039/C4TA05676K
Di-tetrazine-tetroxide (DTTO) was predicted in 2001 to have a density (up to 2.3 g cm−3) and heat of detonation (up to 421.0 kcal mol−1) better than other explosives, making it the “holy grail” of energetic materials (EMs), but all attempts at synthesis have failed. We report Density Functional Theory (DFT) molecular dynamics simulations (DFT-MD) on DTTO crystal for the two most stable monomers. We predict that the most stable isomer (c1) has a density of ρ = 1.96 g cm−3 with an estimated detonation velocity (Dv) of 9.70 km s−1 and a detonation pressure (Dp) of 43.0 GPa, making it comparable to RDX (ρ = 1.82 g cm−3, Dv = 8.75 km s−1, Dp = 35.0 GPa), HMX (ρ = 1.91 g cm−3, Dv = 9.10 km s−1, Dp = 39.3 GPa) and CL-20 (ρ = 2.04 g cm−3, Dv = 9.38 km s−1, Dp = 44.1 GPa). The DFT-MD studies find that the initial reaction at lower pressure is unimolecular decomposition to form two N2O molecules (barrier 45.9 kcal mol−1), while at higher pressure it is intermolecular oxygen-transfer with a barrier of 40.1 kcal mol−1. For the c2 isomer (less stable by 1.2 kcal mol−1) the initial reaction involves two DTTO molecules reacting to form a dimer which then releases N2 as a direct product (barrier 48.1 kcal mol−1), a unique initial reaction among EMs. These results suggest that DTTO may have a higher thermal stability (barrier >7.0 kcal mol−1 higher) than RDX, HMX, and CL-20.
Reaction mechanism from quantum molecular dynamics for the initial thermal decomposition of 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N), promising green energetic materials
Co-reporter:Cai-Chao Ye, Qi An, Tao Cheng, Sergey Zybin, Saber Naserifar, Xue-Hai Ju and William A. Goddard III
Journal of Materials Chemistry A 2015 vol. 3(Issue 22) pp:12044-12050
Publication Date(Web):24 Apr 2015
DOI:10.1039/C5TA02486B
Klapötke and co-workers recently designed two new materials, 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N), envisioned as candidates for green high-energy materials. However, all attempts at synthesis have failed. In order to validate the expected properties for these systems and to determine why these materials are too unstable to synthesize, we used the PBE flavor of Density Functional Theory (DFT) to predict the crystal structures for MTO and MTO3N and then we carried out DFT molecular dynamics simulations (DFT-MD) to determine the initial reaction mechanisms for decomposition. Klapötke estimated that MTO would have a density of ρ = 1.859 g cm−3 with an estimated detonation velocity (Dv) of 8.979 km s−1, making it comparable to RDX (ρ = 1.82 g cm−3, Dv = 8.75 km s−1) and β-HMX (ρ = 1.91 g cm−3, Dv = 9.10 km s−1). His estimated impact sensitivity >30 J, make it much better than HMX (7 J) and RDX (7.5 J). Our predicted crystal structure for MTO (P2(1) space group) leads to ρ = 1.859 g cm−3, in good agreement with expectations. Our DFT-MD studies find that the first step in the decomposition of MTO is intermolecular hydrogen-transfer reaction (barrier 3.0 kcal mol−1) which is followed quickly by H2O and NO release with reaction barriers of 46.5 and 35.5 kcal mol−1. In contrast for MTO3N (P2(1)/c predicted space group), we find that the first steps are a bimolecular decomposition to release NO2 (ΔH = 44.1 kcal mol−1, ΔG = 54.7 kcal mol−1) simultaneous with unimolecular NO2 cleavage (ΔH = 59.9 and ΔG = 58.2 kcal mol−1) a unique initial reaction among EMs. These results suggest that MTO3N would be significantly more thermally stabile (barrier > 6.0 kcal mol−1 higher) than RDX and HMX, making it an excellent candidate to be insensitive new green energetic materials. However we find that MTO leads to very favorable hydrogen transfer reactions that may complicate synthesis and crystallization, making MTO3N the more promising system.
Co-reporter:Dezhou Guo, Qi An, Sergey V. Zybin, William A. Goddard III, Fenglei Huang and Bin Tang
Journal of Materials Chemistry A 2015 vol. 3(Issue 10) pp:5409-5419
Publication Date(Web):26 Jan 2015
DOI:10.1039/C4TA06858K
To gain an atomistic-level understanding of the experimental observation that the cocrystal TNT/CL-20 leads to decreased sensitivity, we carried out reactive molecular dynamics (RMD) simulations using the ReaxFF reactive force field. We compared the thermal decomposition of the TNT/CL-20 cocrystal with that of pure crystals of TNT and CL-20 and with a simple physical mixture of TNT and CL-20. We find that cocrystal has a lower decomposition rate than CL-20 but higher than TNT, which is consistent with experimental observation. We find that the formation of carbon clusters arising from TNT, a carbon-rich molecule, plays an important role in the thermal decomposition process, explaining the decrease in sensitivity for the cocrystal. At low temperature and in the early stage of chemical reactions under high temperature, the cocrystal releases energy more slowly than the simple mixture of CL-20–TNT. These results confirm the expectation that co-crystallization is an effective way to decrease the sensitivity for energetic materials while retaining high performance.
Co-reporter:Michael S. Webster-Gardiner, Ross Fu, George C. Fortman, Robert J. Nielsen, T. Brent Gunnoe and William A. Goddard III
Catalysis Science & Technology 2015 vol. 5(Issue 1) pp:96-100
Publication Date(Web):20 Oct 2014
DOI:10.1039/C4CY00972J
The Rh(I) complexes [(FlDAB)Rh(coe)(TFA)] (1) and [(BOZO)Rh(coe)(TFA)] (2) [FlDAB = N,N-bis-(pentafluorophenyl)-2,3-dimethyl-1,4-diaza-1,3-butadiene, coe = cyclooctene, TFA = trifluoroacetate, BOZO = bis(2-oxazolin-2-yl)] are efficient catalyst precursors for H/D exchange between arenes and DTFA. Catalyst precursor 1 exhibits a TOF of 0.06 s−1 at 150 °C for benzene H/D exchange. DFT calculations revealed that H/D exchange through reversible oxidative addition or internal electrophilic substitution of benzene is a viable pathway.
Co-reporter:Yuan Ping, Ravishankar Sundararaman and William A. Goddard III
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 45) pp:30499-30509
Publication Date(Web):26 Oct 2015
DOI:10.1039/C5CP05740J
The band edge positions of photocatalysts relative to the redox potentials of water play an important role in determining the efficiency of photoelectrochemical cells. These band positions depend on the structure of the solid–liquid interface, but direct ab initio molecular dynamics calculations of these interfaces, while expected to be accurate, are too computationally demanding for high-throughput materials screening. Thus rapid theoretical screening of new photocatalyst materials requires simplified continuum solvation models that are suitable for treating solid–liquid interfaces. In this paper, we evaluate the accuracy of the recently developed CANDLE and SaLSA continuum solvation models for predicting solvation effects on the band positions of several well-studied surfaces [Si(111), TiO2(110), IrO2(110) and WO3(001)] in water. We find that the solvation effects vary considerably, ranging from <0.5 eV for hydrophobic surfaces, 0.5–1 eV for many hydrophilic oxide surfaces, to ∼2 eV for oxygen-deficient surfaces. The solvation model predictions are in excellent agreement (within ∼0.1 eV) with ab initio molecular dynamics results where available, and in good agreement (within ∼0.2–0.3 eV) with experimental measurements. We also predict the energetics for surface oxygen vacancies and their effect on the band positions of the hydrated WO3(001) surface, leading to an explanation for why the solvation shift observed experimentally is substantially larger than predicted for the ideal surface. Based on the correlation between solvation shift and the type of surface and solvent, we suggest approaches to engineer the band positions of surfaces in aqueous and non-aqueous solutions.
Co-reporter:Quanjie Li, Soo-Kyung Kim, William A. Goddard III, Guangju Chen, and Hongwei Tan
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 3) pp:614-627
Publication Date(Web):February 2, 2015
DOI:10.1021/ci500523z
Human kappa opioid receptor (κ-OR), a G protein-coupled receptor (GPCR), has been identified as a drug target for treatment of such human disorders as pain perception, neuroendocrine physiology, affective behavior, and cognition. In order to find more selective and active agonists, one would like to do structure based drug design. Indeed, there is an X-ray structure for an antagonist bound to κ-OR, but structures for activated GPCRs are quite different from those for the inactive GPCRs. Here we predict the ensemble of 24 low-energy structures of human kappa opioid receptor (κ-OR), obtained by application of the GEnSeMBLE (GPCR Ensemble of Structures in Membrane Bilayer Environment) complete sampling method, which evaluates 13 trillion combinations of tilt and rotation angles for κ-OR to select the best 24. To validate these structures, we used the DarwinDock complete sampling method to predict the binding sites for five known agonists (ethylketocyclazocine, bremazocine, pentazocine, nalorphine, and morphine) bound to all 24 κ-OR conformations. We find that some agonists bind selectively to receptor conformations that lack the salt bridge between transmembrane domains 3 and 6 as expected for active conformations. These 3D structures for κ-OR provide a structural basis for understanding ligand binding and activation of κ-OR, which should be useful for guiding subtype specific drug design.
Co-reporter:Ruijie D. Teo, Sijia S. Dong, Zeev Gross, Harry B. Gray and William A. Goddard
Molecular BioSystems 2015 vol. 11(Issue 11) pp:2907-2914
Publication Date(Web):31 Jul 2015
DOI:10.1039/C5MB00352K
Corroles have been shown experimentally to cause cell cycle arrest, and there is some evidence that this might be attributed to an inhibitory effect of corroles on Heat shock protein 90 (Hsp90), which is known to play a vital role in cancer cell proliferation. In this study, we used molecular dynamics to examine the interaction of gallium corroles with Hsp90, and found that they can bind preferentially to the ATP-binding N-terminal site. We also found that structural variations of the corrole ring can influence the binding energies and affinities of the corrole to Hsp90. We predict that both the bis-carboxylated corrole (4-Ga) and a proposed 3,17-bis-sulfonated corrole (7-Ga) are promising alternatives to Ga(III) 5,10,15-tris(pentafluorophenyl)-2,17-bis(sulfonic acid)-corrole (1-Ga) as anti-cancer agents.
Co-reporter:Sijia S. Dong;Dr. Ravinder Abrol; William A. Goddard III
ChemMedChem 2015 Volume 10( Issue 4) pp:650-661
Publication Date(Web):
DOI:10.1002/cmdc.201500023
Abstract
Human somatostatin receptor subtype 5 (hSSTR5) regulates cell proliferation and hormone secretion. However, the identification of effective therapeutic small-molecule ligands is impeded because experimental structures are not available for any SSTR subtypes. Here, we predict the ensemble of low-energy 3D structures of hSSTR5 using a modified GPCR Ensemble of Structures in Membrane BiLayer Environment (GEnSeMBLE) complete sampling computational method. We find that this conformational ensemble displays most interhelical interactions conserved in class A G protein-coupled receptors (GPCRs) plus seven additional interactions (e.g., Y2.43–D3.49, T3.38–S4.53, K5.64–Y3.51) likely conserved among SSTRs. We then predicted the binding sites for a series of five known antagonists, leading to predicted binding energies consistent with experimental results reported in the literature. Molecular dynamics (MD) simulation of 50 ns in explicit water and lipid retained the predicted ligand-bound structure and formed new interaction patterns (e.g. R3.50–T6.34) consistent with the inactive μ-opioid receptor X-ray structure. We suggest more than six mutations for experimental validation of our prediction. The final predicted receptor conformations and antagonist binding sites provide valuable insights for designing new small-molecule drugs targeting SSTRs.
Co-reporter:Sanja Pudar
The Journal of Physical Chemistry C 2015 Volume 119(Issue 49) pp:27370-27381
Publication Date(Web):October 15, 2015
DOI:10.1021/acs.jpcc.5b06224
In this paper, we report quantum mechanical studies (using the B3LYP flavor of density functional theory) for various pathways of ammonia activation on bismuth molybdates, a process required for ammoxidation of propene to acrylonitrile. Using a Mo3O9 cluster to model the bulk surface, we examined the activation of ammonia by both fully oxidized (MoIV) and reduced (MoIV) molybdenum sites. Our results show that ammonia activation does not take place on the fully oxidized Mo(VI) sites. Here the net barriers for the first hydrogen transfer (ΔE‡ = 44.6 kcal/mol, ΔG‡673K = 44.2 kcal/mol) and the second hydrogen transfer (ΔE‡ = 54.5 kcal/mol, ΔG‡673K = 51.7 kcal/mol) are prohibitively high for the reaction temperature of 400 °C. Instead, our calculations show that the reduced Mo(IV) surface sites are far more suitable for this process. Here, the calculated barrier for the first hydrogen transfer from a Mo(IV)–NH3 to an adjacent Mo(VI)═O is 18.2 kcal/mol (ΔG‡673K = 15.4 kcal/mol). For the second hydrogen transfer step, we explored three pathways, and found that the H transfer from a Mo–NH2 to an adjacent Mo(V)–OH to form water is more favorable (ΔE‡ = 26.2 kcal/mol (ΔG‡673K = 24.0 kcal/mol) than transfer to an adjacent Mo(VI)═O or Mo(V)═O group. These studies complement previous studies for activation and reaction of propene on these surfaces, completing the QM study into the fundamental mechanism.
Co-reporter:Yuan Ping
The Journal of Physical Chemistry C 2015 Volume 119(Issue 21) pp:11570-11577
Publication Date(Web):April 23, 2015
DOI:10.1021/acs.jpcc.5b00861
IrO2 is one of the most active catalysts for the oxygen evolution reaction (OER) and remains the only known stable OER catalyst in acidic conditions. As a first step in understanding the mechanism for OER we carried out detailed Density Functional Theory (DFT) studies of the electronic structure of IrO2. We compared the electronic states and magnetic properties of IrO2 using several density functionals. We found that DFT with hybrid functionals (B3PW and PBE0) leads to a weak ferromagnetic coupling, although IrO2 has often been reported as nonmagnetic. We also found a magnetic ground state for RuO2, whose electronic structure is similar to that of IrO2. Ru–Ru antiferromagnetic interaction has been observed experimentally in nanoparticle RuO2. Further low temperature measurements are necessary to confirm whether a weak magnetism may occur below 20 K in IrO2. We also found that PBE leads to a better agreement with the experimental XPS spectra, compared with hybrid functionals and PBE+U.
Co-reporter:Qi An
The Journal of Physical Chemistry C 2015 Volume 119(Issue 8) pp:4095-4103
Publication Date(Web):February 4, 2015
DOI:10.1021/jp5116405
Gallium nitride (GaN) is a wide bandgap semiconductor with many important applications in optoelectronics, photonics, and both high power and high temperature operation devices. Understanding the surface deposition mechanisms and energetics for different precursors is essential to improving thin-film crystalline quality and growth process requirements for extended engineering applications. Here, we use ab initio calculations to study the reaction mechanisms of GaN thin film growth on (0001) surface from ammonia (NH3) and hydrazine (N2H4), nitrogen precursors, and trimethylgallium (TMG) and triethylgallium (TEG), gallium precursors. We find that the initial dehydrogenation of N2H4 is more facile than that of NH3, at 1.15 versus 13.61 kcal/mol, respectively, and that neighboring adsorbed surface hydrogens reduce the barriers for further decomposition of NH2 and NH. We also find that the growth of nitrogen layers is a reaction-limited process rather than diffusion-limited at low adsorbate coverage. On the other hand, the deposition of Ga on a nitrogen rich surface via TMG is limited by the abstraction reaction of the second methyl (CH3) group in TMG, with a barrier of 42.54 kcal/mol. The mechanisms of adsorption of TMG and TEG are different, whereas TMG dissociatively chemisorbs releasing one methane group, the beta-hydrate elimination (C2H4 + H) in TEG is favored through surface interactions (without chemisorption) at a comparable energy barrier to the first CH3 dissociation in TMG. This does not suggest a more favorable thermodynamic route to low-temperature growth, but it does favor TEG for avoiding the explicit abstraction or insertion of C groups during metalorganic chemical vapor deposition (MOCVD) or atomic layer deposition (ALD) techniques.
Co-reporter:Cai-Chao Ye
The Journal of Physical Chemistry C 2015 Volume 119(Issue 5) pp:2290-2296
Publication Date(Web):January 8, 2015
DOI:10.1021/jp510328d
We investigated the initial chemical reactions of BCHMX [cis-1,3,4,6-tetranitrooctahydroimidazo-[4,5-d]imidazole] with the following procedure. First we used density functional theory molecular dynamics simulations (DFT-MD) on the periodic crystal to discover the initial reaction steps. This allowed us to determine the most important reactions through DFT-MD simulations at high temperatures. Then we started with the midpoint of the reaction (unimolecular or bimolecular) from the DFT-MD and carried out higher quality finite cluster DFT calculations to locate the true transition state of the reaction, followed by calculations along the reaction path to determine the initial and final states. We find that for the noncompressed BCHMX the nitro-aci isomerization reaction occurs earlier than the NO2-releasing reaction, while for compressed BCHMX intermolecular hydrogen-transfer and bimolecular NO2-releasing reactions occur earlier than the nitrous acid (HONO)-releasing reaction. At high pressures, the initial reaction involves intermolecular hydrogen transfer rather than intramolecular hydrogen transfer, and the intermolecular hydrogen transfer decreases the reaction barrier for release of NO2 by ∼7 kcal/mol. Thus, the HONO-releasing reaction takes place more easily in compressed BCHMX. We find that this reaction barrier is 10 kcal/mol lower than the unimolecular NO2 release and ∼3 kcal/mol lower than the bimolecular NO2 release. This rationalizes the origin of the higher sensitivity of BCHMX compared to RDX (1,3,5-trinitrohexahydro-1,3,5-triazine) and HMX (octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine). We suggest changes in BCHMX that might help decrease the sensitivity by avoiding the intermolecular hydrogen-transfer and HONO-releasing reaction.
Co-reporter:Qi An
The Journal of Physical Chemistry C 2015 Volume 119(Issue 4) pp:2196-2207
Publication Date(Web):December 29, 2014
DOI:10.1021/jp510951s
Dihydroxylammonium 5,5′-bis(tetrazole)-1,1′-diolate (TKX-50) is a newly synthesized energetic material with high energy storage, low impact sensitivity, and low toxicity. These features make it a viable candidate to replace such commonly used energetic materials as RDX and CL-20 in the next generation of explosives. Sensitivity determines the engineering application of energetic materials (EMs) and has been widely studied for various EMs. To understand the origin of the anisotropic sensitivity and properties of this new synthesized EM, we report a flexible classical force field for TKX-50 developed to reproduce the molecular properties (geometry, vibrational frequencies and torsion barriers) and the crystal properties (cell parameters and lattice energy). We then used this force field in molecular dynamics (MD) simulations to predict such thermodynamic and mechanical properties as isothermal compressibility, thermal expansion, elastic moduli, and heat capacity. Furthermore, we carried out large scale (∼a half million atoms) MD simulations to investigate the mechanical response to shocks in the [100], [010] and [001] directions. The predicted Hugoniot elastic limits (HELs) are 6.1 GPa for [100], 14.2 GPa for [010] and 9.1 GPa for [001] shocks. Thus, single crystal TKX-50 shows anisotropic impact sensitivity with [010] as the most sensitive direction and [100] as least sensitive. The plastic deformations in shock compression along the [100] direction primary arise from the (001)/[210] and (010)/[001] slip systems of. For the [010] shock, the primary slip systems are (100)/[021] and (001)/[210]. However, no obvious slip system was observed for [001] shock.
Co-reporter:Hyung Mo Jeong;Kyung Min Choi;Tao Cheng;Dong Ki Lee;Renjia Zhou;Il Woo Ock;Delia J. Milliron, III;Jeung Ku Kang
PNAS 2015 Volume 112 (Issue 26 ) pp:7914-7919
Publication Date(Web):2015-06-30
DOI:10.1073/pnas.1503546112
Nanocrystals are promising structures, but they are too large for achieving maximum energy storage performance. We show that
rescaling 3-nm particles through lithiation followed by delithiation leads to high-performance energy storage by realizing
high capacitance close to the theoretical capacitance available via ion-to-atom redox reactions. Reactive force-field (ReaxFF)
molecular dynamics simulations support the conclusion that Li atoms react with nickel oxide nanocrystals (NiO-n) to form lithiated
core–shell structures (Ni:Li2O), whereas subsequent delithiation causes Ni:Li2O to form atomic clusters of NiO-a. This is consistent with in situ X-ray photoelectron and optical spectroscopy results showing
that Ni2+ of the nanocrystal changes during lithiation–delithiation through Ni0 and back to Ni2+. These processes are also demonstrated to provide a generic route to rescale another metal oxide. Furthermore, assembling
NiO-a into the positive electrode of an asymmetric device enables extraction of full capacitance for a counter negative electrode,
giving high energy density in addition to robust capacitance retention over 100,000 cycles.
Co-reporter:Tao Cheng; Hai XiaoIII
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 23) pp:4767-4773
Publication Date(Web):November 12, 2015
DOI:10.1021/acs.jpclett.5b02247
The great interest in the photochemical reduction from CO2 to fuels and chemicals has focused attention on Cu because of its unique ability to catalyze formation of carbon-containing fuels and chemicals. A particular goal is to learn how to modify the Cu catalysts to enhance the production selectivity while reducing the energy requirements (overpotential). To enable such developments, we report here the free-energy reaction barriers and mechanistic pathways on the Cu(100) surface, which produces only CH4 (not C2H4 or CH3OH) in acid (pH 0). We predict a threshold potential for CH4 formation of −0.52 V, which compares well to experiments at low pH, −0.45 to −0.50 V. These quantum molecular dynamics simulations included ∼5 layers of explicit water at the water/electrode interface using enhanced sampling methodology to obtain the free energies. We find that that chemisorbed hydroxyl-methylene (CH–OH) is the key intermediate determining the selectivity for methane over methanol.
Co-reporter:Ross Fu;Matthew E. O'Reilly;Robert J. NielsenIII;T. Brent Gunnoe
Chemistry - A European Journal 2015 Volume 21( Issue 3) pp:1286-1293
Publication Date(Web):
DOI:10.1002/chem.201405460
Abstract
A series of rhodium(III) bis(quinolinyl)benzene (bisqx) complexes was studied as candidates for the homogeneous partial oxidation of methane. Density functional theory (DFT) (M06 with Poisson continuum solvation) was used to investigate a variety of (bisqx) ligand candidates involving different functional groups to determine the impact on RhIII(bisqx)-catalyzed methane functionalization. The free energy activation barriers for methane CH activation and Rh–methyl functionalization at 298 K and 498 K were determined. DFT studies predict that the best candidate for catalytic methane functionalization is RhIII coordinated to unsubstituted bis(quinolinyl)benzene (bisq). Support is also found for the prediction that the η2-benzene coordination mode of (bisqx) ligands on Rh encourages methyl group functionalization by serving as an effective leaving group for SN2 and SR2 attack.
Co-reporter:Matthew L. Gethers, John C. Thomas, Shan Jiang, Nathan O. Weiss, Xiangfang Duan, William A. Goddard III, and Paul S. Weiss
ACS Nano 2015 Volume 9(Issue 11) pp:10909
Publication Date(Web):October 1, 2015
DOI:10.1021/acsnano.5b03936
We demonstrate the use of “holey” graphene as a mask against molecular adsorption. Prepared porous graphene is transferred onto a Au{111} substrate, annealed, and then exposed to dilute solutions of 1-adamantanethiol. In the pores of the graphene lattice, we find islands of organized, self-assembled molecules. The bare Au in the pores can be regenerated by postdeposition annealing, and new molecules can be self-assembled in the exposed Au region. Graphene can serve as a robust, patternable mask against the deposition of self-assembled monolayers.Keywords: chemical patterning; graphene; mask; nanoscience; scanning tunneling microscopy; self-assembly;
Co-reporter:Jason M. Crowley; Jamil Tahir-KheliIII
The Journal of Physical Chemistry Letters 2015 Volume 6(Issue 19) pp:3792-3796
Publication Date(Web):September 4, 2015
DOI:10.1021/acs.jpclett.5b01586
It has been established experimentally that Bi2Te3 and Bi2Se3 are topological insulators, with zero band gap surface states exhibiting linear dispersion at the Fermi energy. Standard density functional theory (DFT) methods such as PBE lead to large errors in the band gaps for such strongly correlated systems, while more accurate GW methods are too expensive computationally to apply to the thin films studied experimentally. We show here that the hybrid B3PW91 density functional yields GW-quality results for these systems at a computational cost comparable to PBE. The efficiency of our approach stems from the use of Gaussian basis functions instead of plane waves or augmented plane waves. This remarkable success without empirical corrections of any kind opens the door to computational studies of real chemistry involving the topological surface state, and our approach is expected to be applicable to other semiconductors with strong spin-orbit coupling.
Co-reporter:Bin Tang
The Journal of Physical Chemistry C 2015 Volume 119(Issue 43) pp:24649-24656
Publication Date(Web):October 7, 2015
DOI:10.1021/acs.jpcc.5b08086
Boron carbide (B4C) is the third hardest material in nature, but applications are hindered by its brittle failure under impact. We found that this brittle failure of B4C arises from amorphous shear band formation due to deconstruction of icosahedral clusters, and on the basis of this model we suggest and validate with quantum mechanics (QM, PBE flavor of density function theory) that a laminated B4C–B6O composite structure will eliminate this brittle failure. Using QM to apply shear deformations along various slip systems, we find that the (001)/[100] slip system has the lowest maximum shear strength, indicating it to be the most plausible slip system. We find that this composite structure has a shear strength of 38.33 GPa, essentially the same as that of B4C (38.97 GPa), indicating the same intrinsic hardness as B4C. However, the critical failure strain for (001)/[100] slip in the composite is 0.465, which is 41% higher than B4C, indicating a dramatically improvement on ductility. This arises because incorporation of B6O prevents the failure mechanism of B4C in which the carbene formed during shear deformation reacts with the C–B–C chains. This suggests a new strategy for designing ductile superhard ceramics.
Co-reporter:Jaehyeok Jin
The Journal of Physical Chemistry C 2015 Volume 119(Issue 5) pp:2622-2629
Publication Date(Web):January 14, 2015
DOI:10.1021/jp511752n
Experimentally, quenching from warmer water leads to faster freezing than quenching from colder water—the Mpemba effect. Using molecular dynamics, we find that quenching water from 370 K and above leads to a 100 K density of states (DOS) closer to that of ice than quenching from 300 K and below. Especially we find that the biggest difference is for 80–160 cm–1 which upon quenching from colder water is much lower than that in ice, while it is much higher than in ice when quenching from warm water. We find that the range of 100–160 cm–1 corresponds to framework vibrations within a hexamer, suggesting that the water hexamer serves as a nucleus for crystallization. We tested this by fixing one hexamer and quenching slowly from 370 K, leading to increased correlation with pure ice. We also showed that the structure quenched from 370 K evolves to the ice faster than 300 K case. These results suggest that the higher population of water hexamer states in warm water is responsible for the faster crystallization underlying the Mpemba effect.
Co-reporter:Tao Cheng ; Andrés Jaramillo-Botero ; William A Goddard ; III;Huai Sun
Journal of the American Chemical Society 2014 Volume 136(Issue 26) pp:9434-9442
Publication Date(Web):June 2, 2014
DOI:10.1021/ja5037258
We develop here the methodology for dramatically accelerating the ReaxFF reactive force field based reactive molecular dynamics (RMD) simulations through use of the bond boost concept (BB), which we validate here for describing hydrogen combustion. The bond order, undercoordination, and overcoordination concepts of ReaxFF ensure that the BB correctly adapts to the instantaneous configurations in the reactive system to automatically identify the reactions appropriate to receive the bond boost. We refer to this as adaptive Accelerated ReaxFF Reactive Dynamics or aARRDyn. To validate the aARRDyn methodology, we determined the detailed sequence of reactions for hydrogen combustion with and without the BB. We validate that the kinetics and reaction mechanisms (that is the detailed sequences of reactive intermediates and their subsequent transformation to others) for H2 oxidation obtained from aARRDyn agrees well with the brute force reactive molecular dynamics (BF-RMD) at 2498 K. Using aARRDyn, we then extend our simulations to the whole range of combustion temperatures from ignition (798 K) to flame temperature (2998K), and demonstrate that, over this full temperature range, the reaction rates predicted by aARRDyn agree well with the BF-RMD values, extrapolated to lower temperatures. For the aARRDyn simulation at 798 K we find that the time period for half the H2 to form H2O product is ∼538 s, whereas the computational cost was just 1289 ps, a speed increase of ∼0.42 trillion (1012) over BF-RMD. In carrying out these RMD simulations we found that the ReaxFF-COH2008 version of the ReaxFF force field was not accurate for such intermediates as H3O. Consequently we reoptimized the fit to a quantum mechanics (QM) level, leading to the ReaxFF–OH2014 force field that was used in the simulations.
Co-reporter:Yuhua Zhou ; Jing Yang ; Haibin Su ; Jie Zeng ; San Ping Jiang
Journal of the American Chemical Society 2014 Volume 136(Issue 13) pp:4954-4964
Publication Date(Web):March 14, 2014
DOI:10.1021/ja411268q
We have developed for fuel cells a novel proton exchange membrane (PEM) using inorganic phosphotungstic acid (HPW) as proton carrier and mesoporous silica as matrix (HPW-meso-silica) . The proton conductivity measured by electrochemical impedance spectroscopy is 0.11 S cm–1 at 90 °C and 100% relative humidity (RH) with a low activation energy of ∼14 kJ mol–1. In order to determine the energetics associated with proton migration within the HPW-meso-silica PEM and to determine the mechanism of proton hopping, we report density functional theory (DFT) calculations using the generalized gradient approximation (GGA). These DFT calculations revealed that the proton transfer process involves both intramolecular and intermolecular proton transfer pathways. When the adjacent HPWs are close (less than 17.0 Å apart), the calculated activation energy for intramolecular proton transfer within a HPW molecule is higher (29.1–18.8 kJ/mol) than the barrier for intermolecular proton transfer along the hydrogen bond. We find that the overall barrier for proton movement within the HPW-meso-silica membranes is determined by the intramolecular proton transfer pathway, which explains why the proton conductivity remains unchanged when the weight percentage of HPW on meso-silica is above 67 wt %. In contrast, the activation energy of proton transfer on a clean SiO2 (111) surface is computed to be as high as ∼40 kJ mol–1, confirming the very low proton conductivity on clean silica surfaces observed experimentally.
Co-reporter:Wei-Guang Liu ; Anna V. Sberegaeva ; Robert J. Nielsen ; William A. Goddard ; III;Andrei N. Vedernikov
Journal of the American Chemical Society 2014 Volume 136(Issue 6) pp:2335-2341
Publication Date(Web):January 22, 2014
DOI:10.1021/ja409036c
The mechanism of the (dpms)PtIIMe(OHn)(2–n)− oxidation in water to form (dpms)PtIVMe(OH)2 and (dpms)PtIVMe2(OH) complexes was analyzed using DFT calculations. At pH < 10, (dpms)PtIIMe(OHn)(2–n)– reacts with O2 to form a methyl Pt(IV)–OOH species with the methyl group trans to the pyridine nitrogen, which then reacts with (dpms)PtIIMe(OHn)(2–n)– to form 2 equiv of (dpms)PtIVMe(OH)2, the major oxidation product. Both the O2 activation and the O–O bond cleavage are pH dependent. At higher pH, O–O cleavage is inhibited whereas the Pt-to-Pt methyl transfer is not slowed down, so making the latter reaction predominant at pH > 12. The pH-independent Pt-to-Pt methyl transfer involves the isomeric methyl Pt(IV)–OOH species with the methyl group trans to the sulfonate. This methyl Pt(IV)–OOH complex is more stable and more reactive in the Pt-to-Pt methyl-transfer reaction as compared to its isomer with the methyl group trans to the pyridine nitrogen. A similar structure–reactivity relationship is also observed for the SN2 functionalization to form methanol by two isomeric (dpms)PtIVMe(OH)2 complexes, one featuring the methyl ligand trans to the sulfonate group and another with the methyl trans to the pyridine nitrogen. The barrier to functionalize the former isomer with the CH3 group trans to the sulfonate group is 2–9 kcal/mol lower. The possibility of the involvement of Pt(III) species in the reactions studied was found to correspond to high-barrier reactions and is hence not viable. It is concluded that the dpms ligand facilitates Pt(II) oxidation both enthalpically and entropically.
Co-reporter:Matthew E. O’Reilly ; Ross Fu ; Robert J. Nielsen ; Michal Sabat ; William A. Goddard ; III;T. Brent Gunnoe
Journal of the American Chemical Society 2014 Volume 136(Issue 42) pp:14690-14693
Publication Date(Web):October 10, 2014
DOI:10.1021/ja508367m
Traditional C–H bond activation by a concerted metalation–deprotonation (CMD) mechanism involves precoordination of the C–H bond followed by deprotonation from an internal base. Reported herein is a “through-arene” activation of an uncoordinated benzylic C–H bond that is 6 bonds away from a RhIII ion. The mechanism, which was investigated by experimental and DFT studies, proceeds through a dearomatized xylene intermediate. This intermediate was observed spectroscopically upon addition of a pyridine base to provide a thermodynamic trap.
Co-reporter:Ross Fu, Robert J. Nielsen, and William A. Goddard III , George C. Fortman and T. Brent Gunnoe
ACS Catalysis 2014 Volume 4(Issue 12) pp:4455
Publication Date(Web):October 20, 2014
DOI:10.1021/cs5005322
In the search for new organometallic catalysts for low-temperature selective conversion of CH4 to CH3OH, we apply quantum mechanical virtual screening to select the optimum combination of ligand and solvent on rhodium to achieve low barriers for CH4 activation and functionalization to recommend for experimental validation. Here, we considered Rh because its lower electronegativity compared with Pt and Pd may allow it to avoid poisoning by coordinating media. We report quantum mechanical predictions (including implicit and explicit solvation) of the mechanisms for RhIII(NN) and RhIII(NNF) complexes [where (NN) = bis(N-phenyl)benzylamidinate and (NNF) = bis(N-pentafluorophenyl)pentafluorobenzylamidinate] to catalytically activate and functionalize methane using trifluoroacetic acid (TFAH) or water as a solvent. In particular, we designed the (NNF) ligand as a more electrophilic analogue to the (NN) ligand, and our results predict the lowest transition state barrier (ΔG‡ = 27.6 kcal/mol) for methane activation in TFAH from a pool of four different classes of ligands. To close the catalytic cycle, the functionalization of methylrhodium intermediates was also investigated, involving carbon–oxygen bond formation via SN2 attack by solvent, or SR2 attack by a vanadium oxo. Activation barriers for the functionalization of methylrhodium intermediates via nucleophilic attack are lower when the solvent is water, but CH4 activation barriers are higher. In addition, we have found a correlation between CH4 activation barriers and rhodium–methyl bond energies that allow us to predict the activation transition state energies for future ligands, as well.Keywords: amidinate; catalysis; C−H activation; fluorinated ligands; methane functionalization; quantum mechanical screening; rhodium
Co-reporter:Qi An, William A. Goddard III, Hai Xiao, and Tao Cheng
Chemistry of Materials 2014 Volume 26(Issue 14) pp:4289
Publication Date(Web):June 24, 2014
DOI:10.1021/cm5020114
We predict three new polymorphs of boron by applying density functional theory (PBE flavor) to large shear deformations starting from the recently discovered γ-B28 boron phase (stable above 9 GPa and 1000 K). We find that continuous deformation along the (100)/⟨001⟩ slip system leads to two new phases, named here as γ-B12–(B2)6 and γ-B12–(B···B)6. We show that these γ-B12–(B2)6 and γ-B12–(B···B)6 phases can also be obtained from uniaxial tensile and compressive deformations of the γ-B28 phase along the ⟨101⟩ direction, respectively. However, the reverse compressive loading on the newly formed γ-B12–(B2)6 phase transforms it to itself, not the γ-B28 phase, because of the transferability of the three-center two-electron bond under deformation. This makes the new phase γ-B12–(B2)6 a special type of superelastic material. In addition, application of reverse tensile deformation on the newly formed γ-B12–(B···B)6 phase, transforms it to a third new phase, named α-B12–BB, that is metallic, suggesting increased ductility that might make α-B12–BB important for applications in electronic devices. We compared the structural character, mechanical properties, and electronic properties of these new phases to each other and to other phases of boron. We show that the three new phases are dynamically stable at zero pressure. These results show how modifying the connections between boron icosahedra using one to two atom chains can lead to dramatically different mechanical and electronic properties.
Co-reporter:Yao Sha, Ted H. Yu, Boris V. Merinov, and William A. Goddard III
ACS Catalysis 2014 Volume 4(Issue 4) pp:1189
Publication Date(Web):February 24, 2014
DOI:10.1021/cs4009623
The high cost of proton exchange membrane fuel cells (PEMFCs) comes largely from the use of platinum-containing electrocatalysts. Despite significant progress made the past decade on reducing the platinum catalyst loading in the PEMFC electrodes, further substantial cost reductions require the replacement of platinum with less expensive nonplatinum electrocatalytic materials. In this study, PdCu alloys have computationally been investigated as possible non-Pt catalysts for oxygen reduction reaction (ORR) in PEMFCs. We used density functional theory (DFT) calculations to determine the structural preference and ORR activity as a function of the composition and structure. Five PdCu alloy surface structures, B2, L12, L10, L11-nonlayered, and L11-layered, were considered, and the layered L11 surface structure was found to exhibit significantly improved ORR kinetics compared to that of pure Pd.Keywords: DFT; ORR; PdCu alloys; reaction mechanisms; surfaces
Co-reporter:Mu-Jeng Cheng, Ross Fu and William A. Goddard, III
Chemical Communications 2014 vol. 50(Issue 14) pp:1748-1750
Publication Date(Web):18 Dec 2013
DOI:10.1039/C3CC47502F
We use our recent discovery of the reduction-coupled oxo activation (ROA) principle to design a series of organometallic molecules that activate C–H bonds through this unique proton/electron-decoupled hydrogen abstraction mechanism, in which the main group oxo moiety binds to the proton while the electron is transferred to the transition metal. Here we illustrate this general class of catalyst clusters with several examples that are validated through quantum mechanics calculations.
Co-reporter:Tingting Zhou, Lianchi Liu, William A. Goddard III, Sergey V. Zybin and Fenglei Huang
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 43) pp:23779-23791
Publication Date(Web):17 Sep 2014
DOI:10.1039/C4CP03781B
Recently quantum mechanical (QM) calculations on a single Si-PETN (silicon-pentaerythritol tetranitrate) molecule were used to explain its colossal sensitivity observed experimentally in terms of a unique Liu carbon-silyl nitro-ester rearrangement (R3Si–CH2–O–R2 → R3Si–O–CH2–R2). In this paper we expanded the study of Si-PETN from a single molecule to a bulk system by extending the ReaxFF reactive force field to describe similar Si–C–H–O–N systems with parameters optimized to reproduce QM results. The reaction mechanisms and kinetics of thermal decomposition of solid Si-PETN were investigated using ReaxFF reactive molecular dynamics (ReaxFF-RMD) simulations at various temperatures to explore the origin of the high sensitivity. We find that at lower temperatures, the decomposition of Si-PETN is initiated by the Liu carbon-silyl nitro-ester rearrangement forming Si–O bonds which is not observed in PETN. As the reaction proceeds, the exothermicity of Si–O bond formation promotes the onset of NO2 formation from N–OC bond cleavage which does not occur in PETN. At higher temperatures PETN starts to react by the usual mechanisms of NO2 dissociation and HONO elimination; however, Si-PETN remains far more reactive. These results validate the predictions from QM that the significantly increased sensitivity of Si-PETN arises from a unimolecular process involving the unusual Liu rearrangement but not from multi-molecular collisions. It is the very low energy barrier and the high exothermicity of the Si–O bond formation providing energy early in the decomposition process that is responsible.
Co-reporter:Dr. Soo-Kyung Kim; William A. Goddard III;Dr. Kyu Yang Yi;Dr. Byung Ho Lee;Chae Jo Lim; Bartosz Trzaskowski
ChemMedChem 2014 Volume 9( Issue 8) pp:1732-1743
Publication Date(Web):
DOI:10.1002/cmdc.201402087
Abstract
Human Urotensin-II (U-II) is the most potent mammalian vasoconstrictor known.1 Thus, a U-II antagonist would be of therapeutic value in a number of cardiovascular disorders.2 Here, we describe our work on the prediction of the structure of the human U-II receptor (hUT2R) using GEnSeMBLE (GPCR Ensemble of Structures in Membrane BiLayer Environment) complete sampling Monte Carlo method. With the validation of our predicted structures, we designed a series of new potential antagonists predicted to bind more strongly than known ligands. Next, we carried out R-group screening to suggest a new ligand predicted to bind with 7 kcal mol−1 better energy than 1-{2-[4-(2-bromobenzyl)-4-hydroxypiperidin-1-yl]ethyl}-3-(thieno[3,2-b]pyridin-7-yl)urea, the designed antagonist predicted to have the highest affinity for the receptor. Some of these predictions were tested experimentally, validating the computational results. Using the pharmacophore generated from the predicted structure for hUT2R bound to ACT-058362, we carried out virtual screening based on this binding site. The most potent hit compounds identified contained 2-(phenoxymethyl)-1,3,4-thiadiazole core, with the best derivative exhibiting an IC50 value of 0.581 μM against hUT2R when tested in vitro. Our efforts identified a new scaffold as a potential new lead structure for the development of novel hUT2R antagonists, and the computational methods used could find more general applicability to other GPCRs.
Co-reporter:Tod A. Pascal and William A. Goddard III
The Journal of Physical Chemistry B 2014 Volume 118(Issue 22) pp:5943-5956
Publication Date(Web):May 12, 2014
DOI:10.1021/jp410861h
We examine the thermodynamics of the liquid–vapor interface by direct calculation of the surface entropy, enthalpy, and free energy from extensive molecular dynamics simulations using the two-phase thermodynamics (2PT) method. Results for water, acetonitrile, cyclohexane, dimethyl sulfoxide, hexanol, N-methyl acetamide, and toluene are presented. We validate our approach by predicting the interfacial surface tensions (IFT—excess surface free energy per unit area) in excellent agreement with the mechanical calculations using Kirkwood–Buff theory. Additionally, we evaluate the temperature dependence of the IFT of water as described by the TIP4P/2005, SPC/Ew, TIP3P, and mW classical water models. We find that the TIP4P/2005 and SPC/Ew water models do a reasonable job of describing the interfacial thermodynamics; however, the TIP3P and mW are quite poor. We find that the underprediction of the experimental IFT at 298 K by these water models results from understructured surface molecules whose binding energies are too weak. Finally, we performed depth profiles of the interfacial thermodynamics which revealed long tails that extend far into what would be considered bulk from standard Gibbs theory. In fact, we find a nonmonotonic interfacial free energy profile for water, a unique feature that could have important consequences for the absorption of ions and other small molecules.
Co-reporter:Alessro Fortunelli;Yao Sha;Ted H. Yu;Luca Sementa;Giovanni Barcaro;Oliviero Andreussi
Angewandte Chemie International Edition 2014 Volume 53( Issue 26) pp:6669-6672
Publication Date(Web):
DOI:10.1002/anie.201403264
Abstract
Hydrogen fuel cells (FC) are considered essential for a sustainable economy based on carbon-free energy sources, but a major impediment are the costs. First-principles quantum mechanics (density functional theory including solvation) is used to predict how the energies and barriers for the mechanistic steps of the oxygen reduction reaction (ORR) over the fcc(111) platinum surface depend on the dielectric constant of the solvent. The ORR kinetics can be strongly accelerated by decreasing the effective medium polarizability from the high value it has in water. Possible ways to realize this experimentally are suggested. The calculated volcano structure for the dependence of rate on solvent polarization is considered to be general, and should be observed in other electrochemical systems.
Co-reporter:Ho-Cheng Tsai ; Ted H. Yu ; Yao Sha ; Boris V. Merinov ; Pu-Wei Wu ; San-Yuan Chen ; III
The Journal of Physical Chemistry C 2014 Volume 118(Issue 46) pp:26703-26712
Publication Date(Web):October 22, 2014
DOI:10.1021/jp507103c
Using quantum mechanics calculations, we have studied the segregation energy with adsorbed O and OH for 28 Pt3M alloys, where M is a transition metal. The calculations found surface segregation to become energetically unfavorable for Pt3Co and Pt3Ni, as well as for the most other Pt binary alloys, in the presence of adsorbed O and OH. However, Pt3Os and Pt3Ir remain surface segregated and show the best energy preference among the alloys studied for both adsorbed species on the surface. Binding energies of various oxygen reduction reaction (ORR) intermediates on the Pt(111) and Pt3Os(111) surfaces were calculated and analyzed. Energy barriers for different ORR steps were computed for Pt and Pt3Os catalysts, and the rate-determining steps (RDS) were identified. It turns out that the RDS barrier for the Pt3Os alloy catalyst is lower than the corresponding barrier for pure Pt. This result allows us to predict a better ORR performance of Pt3Os compared to that of pure Pt.
Co-reporter:Qi An ; Wei-Guang Liu ; William. A. Goddard ; III; Tao Cheng ; Sergey V. Zybin ;Hai Xiao
The Journal of Physical Chemistry C 2014 Volume 118(Issue 46) pp:27175-27181
Publication Date(Web):October 28, 2014
DOI:10.1021/jp509582x
Dihydroxylammonium 5,5′-bistetrazole-1,1′-diolate (TKX-50) is a recently synthesized energetic material (EM) with most promising performance, including high energy content, high density, low sensitivity, and low toxicity. TKX-50 forms an ionic crystal in which the unit cell contains two bistetrazole dianions {c-((NO)N3C)-[c-(CN3(NO)], formal charge of −2} and four hydroxylammonium (NH3OH)+ cations (formal charge of +1). We report here quantum mechanics (QM)-based reaction studies to determine the atomistic reaction mechanisms for the initial decompositions of this system. First we carried out molecular dynamics simulations on the periodic TKX-50 crystal using forces from density functional based tight binding calculations (DFTB-MD), which finds that the chemistry is initiated by proton transfer from the cation to the dianion. Continuous heating of this periodic system leads eventually to dissociation of the protonated or diprotonated bistetrazole to release N2 and N2O. To refine the mechanisms observed in the periodic DFTB-MD, we carried out finite cluster quantum mechanics studies (B3LYP) for the unimolecular decomposition of the bistetrazole. We find that for the bistetrazole dianion, the reaction barrier for release of N2 is 45.1 kcal/mol, while release of N2O is 72.2 kcal/mol. However, transferring one proton to the bistetrazole dianion decreases the reaction barriers to 37.2 kcal/mol for N2 release and 59.5 kcal/mol for N2O release. Thus, we predict that the initial decompositions in TKX-50 lead to N2 release, which in turn provides the energy to drive further decompositions. On the basis of this mechanism, we suggest changes to make the system less sensitive while retaining the large energy release. This may help improve the synthesis strategy of developing high nitrogen explosives with further improved performance.
Co-reporter:Qi An ; William A. Goddard ; III; Sergey V. Zybin ;Sheng-Nian Luo
The Journal of Physical Chemistry C 2014 Volume 118(Issue 34) pp:19918-19928
Publication Date(Web):August 8, 2014
DOI:10.1021/jp506501r
In order to elucidate how shocks in heterogeneous materials affect decomposition and reactive processes, we used the ReaxFF reactive force field in reactive molecules dynamics (RMD) simulations of the effects of strong shocks (2.5 and 3.5 km/s) on a prototype polymer bonded explosive (PBX) consisting of cyclotrimethylene trinitramine (RDX) bonded to hydroxyl-terminated polybutadiene (HTPB). We showed earlier that shock propagation from the high density RDX to the low density polymer (RDX → Poly) across a nonplanar periodic interface (sawtooth) leads to a hotspot at the initial asperity but no additional hotspot at the second asperity. This hotspot arises from shear along the interface induced by relaxation of the stress at the asperity. We now report the case for shock propagation from the low density polymer to the high density RDX (Poly → RDX) where we find a hotspot at the initial asperity and a second more dramatic hotspot at the second asperity. This second hotspot is enhanced due to shock wave convergence from shock wave interaction with nonplanar interfaces. We consider that this second hotspot is likely the source of the detonation in realistic PBX systems. We showed how these hotspots depend on the density mismatch between the RDX and polymer and found that decreasing the density by a factor of 2 dramatically reduces the hotspot. These results suggest that to make PBX less sensitive for propellants and explosives, the binder should be designed to provide low density at the asperity in contact with the RDX. Based on these simulations, we propose a new design for an insensitive PBX in which a low density polymer coating is deposited between the RDX and the usual polymer binder. To test this idea, we simulated shock wave propagation from two opposite directions (RDX → Poly and Poly → RDX) through the interface matched PBX (IM-PBX) material containing a 3 nm coating of low density (0.48 g/cm3) polymer. These simulations showed that this IM-PBX design dramatically suppresses hotspot formation.
Co-reporter:Jeon-Jin Choi;Heejin Kim;Soo Yeon Lim;Jaehoon Chung;Kyung Yoon Chung;Seung-Joo Kim III;Yousung Jung;Ji Hoon Lee;Byung Gon Kim;Woosuk Cho;Jang Wook Choi
PNAS 2014 Volume 111 (Issue 2 ) pp:599-604
Publication Date(Web):2014-01-14
DOI:10.1073/pnas.1316557110
Sodium ion batteries offer promising opportunities in emerging utility grid applications because of the low cost of raw materials,
yet low energy density and limited cycle life remain critical drawbacks in their electrochemical operations. Herein, we report
a vanadium-based ortho-diphosphate, Na7V4(P2O7)4PO4, or VODP, that significantly reduces all these drawbacks. Indeed, VODP exhibits single-valued voltage plateaus at 3.88 V
vs. Na/Na+ while retaining substantial capacity (>78%) over 1,000 cycles. Electronic structure calculations reveal that the remarkable
single plateau and cycle life originate from an intermediate phase (a very shallow voltage step) that is similar both in the
energy level and lattice parameters to those of fully intercalated and deintercalated states. We propose a theoretical scheme
in which the reaction barrier that arises from lattice mismatches can be evaluated by using a simple energetic consideration,
suggesting that the presence of intermediate phases is beneficial for cell kinetics by buffering the differences in lattice
parameters between initial and final phases. We expect these insights into the role of intermediate phases found for VODP
hold in general and thus provide a helpful guideline in the further understanding and design of battery materials.
Co-reporter:Mu-Jeng Cheng III;Ross Fu
Topics in Catalysis 2014 Volume 57( Issue 14-16) pp:1171-1187
Publication Date(Web):2014 September
DOI:10.1007/s11244-014-0284-6
We report here the results of density functional theory quantum mechanical (QM) studies of the detailed chemical mechanism underlying the n-butane selective oxidation to form maleic anhydride (MA) on vanadyl pyrophosphate [(VO)2P2O7] and vanadyl phosphate [VOPO4] surfaces. This QM-derived mechanism differs substantially from previous suggestions but is in excellent agreement with key experimental observations. We find that the O(1)=P bond of the oxidized X1 phase of the VOPO4 surface is the active site for initiating the VPO chemistry, by extracting the H from the n-butane C–H bond. This contrasts sharply with previous suggestions, all of which involved the V=O bonds. The ability of O(1)=P to cleave alkane C–H bonds arises from a new unique mechanism that decouples the proton transfer and electron transfer components of this H atom transfer reaction. We find that the juxtaposition of a highly reducible V+5 next to the P=O bond but coupled via a bridging oxygen dramatically enhances the activity of the P=O bond to extract the proton from an alkane, while simultaneously transferring the electron to the V to form V+4. This Reduction-Coupled Oxo Activation (ROA) mechanism had not been known prior to these QM studies, but we believe that it may lead to a new strategy in designing selective catalysts for alkane activation and functionalization, and indeed it may be responsible for the selective oxidation by a number of known mixed metal oxide catalysts. To demonstrate the viability of this new ROA mechanism, we examine step by step the full sequence of reactions from n-butane to MA via two independent pathways. We that find that every step is plausible, with a highest reaction barrier of 21.7 kcal/mol.
Co-reporter:Soo-Kyung Kim III
Journal of Computer-Aided Molecular Design 2014 Volume 28( Issue 12) pp:1175-1190
Publication Date(Web):2014 December
DOI:10.1007/s10822-014-9793-4
Olfactory receptors (ORs) are responsible for mediating the sense of smell; they allow humans to recognize an enormous number of odors but the connection between binding and perception is not known. We predict the ensemble of low energy structures for the human OR1G1 (hOR1G1) and also for six other diverse ORs, using the G protein-coupled receptor Ensemble of Structures in Membrane BiLayer Environment complete sampling method that samples 13 trillion different rotations and tilts using four different templates to predict the 24 structures likely to be important in binding and activation. Our predicted most stable structures of hOR1G1 have a salt-bridge between the conserved D3.49 and K6.30 in the D(E)RY region, that we expect to be associated with an inactive form. The hOR1G1 structure also has specific interaction in transmembrane domains (TMD) 3-6 (E3.39 and H6.40), which is likely an important conformational feature for all hORs because of the ~94 to 98 % conservation among all hOR sequences. Of the five ligands studied (nonanal, 9-decen-1-ol, 1-nonanol, camphor, and n–butanal), we find that the 4 expected to bind lead to similar binding energies with nonanol the strongest.
Co-reporter:Qi An and William A. Goddard III
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 23) pp:4169-4174
Publication Date(Web):November 18, 2014
DOI:10.1021/jz5022697
Co-reporter:Qi An, Hai Xiao, William A. Goddard III, and Xiangying Meng
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 3) pp:485-489
Publication Date(Web):January 6, 2014
DOI:10.1021/jz402682y
We combine the USPEX evolution searching method with density functional theory using dispersion corrections (DFT-ulg) to predict the crystal structure of the NNO extended solid at high pressures (from 100 to 500 GPa). We find that the NNO nanotube (with diameter ≈ 2.5 Å) is the most stable form above 180 GPa. We report here the stability, electronic properties, and mechanical properties of this novel nanotube and show that it is stable above 20 GPa. To find a similar structure that might be stable at ambient conditions, we considered the NPO tube and show that it is stable at zero pressure. The NPO phase leads to an insulator to metal transition at 25 GPa, where the PP van der Waals distance approaches the covalent bond distance. The energy content of this NPO nanotube crystal is 10.6 kJ/g, which is 152% higher than that of TNT and 86% higher than that of the HMX energetic material. This is the first example of a structural energetic material, which could have important applications in igniters, incendiaries, screening smoke ammunition, and similar devices. This process illustrates how materials discovery in extreme conditions can be used to discover and stabilize novel structures.Keywords: crystal structure prediction; DFT; PBE-ulg; stabilization of high-pressure phases; USPEX;
Co-reporter:Jiwon Jeon, Young Choon Park, Sang Soo Han, William A. Goddard III, Yoon Sup Lee, and Hyungjun Kim
The Journal of Physical Chemistry Letters 2014 Volume 5(Issue 24) pp:4285-4290
Publication Date(Web):November 25, 2014
DOI:10.1021/jz502197b
During the light-harvesting process of dye-sensitized solar cells (DSSCs), the hole localized on the dye after the charge separation yields an oxidized dye, D+. The fast regeneration of D+ using the redox pair (typically the I–/I3– couple) is critical for the efficient DSSCs. However, the kinetic processes of dye regeneration remain uncertain, still promoting vigorous debates. Here, we use molecular dynamics simulations to determine that the inner-sphere electron-transfer pathway provides a rapid dye regeneration route of ∼4 ps, where penetration of I− next to D+ enables an immediate electron transfer, forming a kinetic barrier. This explains the recently reported ultrafast dye regeneration rate of a few picoseconds determined experimentally. We expect that our MD based comprehensive understanding of the dye regeneration mechanism will provide a helpful guideline in designing TiO2−dye−electrolyte interfacial systems for better performing DSSCs.Keywords: dye regeneration; dye-sensitized solar cells; electron transfer; multiscale simulation;
Co-reporter:Bartosz Trzaskowski;Ravinder Abrol III;Alexandre Nesterov;Ivan Olave;Christopher Irons
PNAS 2014 Volume 111 (Issue 36 ) pp:13040-13045
Publication Date(Web):2014-09-09
DOI:10.1073/pnas.1413216111
We predicted the structural basis for pleiotropic signaling of the C-C chemokine type 5 (CCR5) G protein-coupled receptor
(GPCR) by predicting the binding of several ligands to the lower-energy conformations of the CCR5 receptor and 11 mutants.
For each case, we predicted the ∼20 most stable conformations for the receptor along with the binding sites for four anti-HIV
ligands. We found that none of the ligands bind to the lowest-energy apo-receptor conformation. The three ligands with a similar
pharmacophore (Maraviroc, PF-232798, and Aplaviroc) bind to a specific higher-energy receptor conformation whereas TAK-779
(with a different pharmacophore) binds to a different high-energy conformation. This result is in agreement with the very
different binding-site profiles for these ligands obtained by us and others. The predicted Maraviroc binding site agrees with
the recent structure of CCR5 receptor cocrystallized with Maraviroc. We performed 11 site-directed mutagenesis experiments
to validate the predicted binding sites. Here, we independently predicted the lowest 10 mutant protein conformations for each
of the 11 mutants and then docked the ligands to these lowest conformations. We found the predicted binding energies to be
in excellent agreement with our mutagenesis experiments. These results show that, for GPCRs, each ligand can stabilize a different
protein conformation, complicating the use of cocrystallized structures for ligand screening. Moreover, these results show
that a single-point mutation in a GPCR can dramatically alter the available low-energy conformations, which in turn alters
the binding site, potentially altering downstream signaling events. These studies validate the conformational selection paradigm
for the pleiotropic function and structural plasticity of GPCRs.
Co-reporter:Dezhou Guo ; Qi An ; William A. Goddard ; III; Sergey V. Zybin ;Fenglei Huang
The Journal of Physical Chemistry C 2014 Volume 118(Issue 51) pp:30202-30208
Publication Date(Web):November 20, 2014
DOI:10.1021/jp5093527
To gain an atomistic-level understanding of how compounding the TNT and CL-20 energetic materials into a TNT/CL-20 cocrystal might affect the sensitivity, we carried out the compressive–shear reactive molecular dynamics (CS-RMD) simulations. Comparing with the pure crystal of CL-20, we find that the cocrystal is much less sensitive. We find that the molecular origin of the energy barrier for anisotropic shear results from steric hindrance toward shearing of adjacent slip planes during shear deformation, which is decreased for the cocrystal. To compare the sensitivity for different crystals, we chose the shear slip system with lowest energy barrier as the most plausible one under external stresses for each crystal. Then we used the temperature rise and molecule decomposition as effective measures to distinguish sensitivities. Considering the criterion as number NO2 fragments produced, we find that the cocrystal has lower shear-induced initiation sensitivity by ∼70% under atmospheric pressure and ∼46% under high pressure (∼5 GPa) than CL-20. Based on the temperature increase rate, the cocrystal has initiation sensitivity lower by 22% under high pressure (∼5 GPa) than CL-20. These results are consistent with available experimental results, further validating the CS-RD model for distinguishing between sensitive and insensitive materials rapidly (within a few picoseconds of MD).
Co-reporter:Jiwon Jeon ; William A. Goddard ; III;Hyungjun Kim
Journal of the American Chemical Society 2013 Volume 135(Issue 7) pp:2431-2434
Publication Date(Web):February 5, 2013
DOI:10.1021/ja311714a
During the regeneration of the oxidized dye in dye-sensitized solar cells, the redox couple of I–/I3– reduces the photo-oxidized dye. The simplest mechanism would be a direct charge-transfer mechanism from I– to D+ [D+ + I– → D0 + I], called the single iodide process (SIP). However, this is an unfavorable equilibrium because the redox potential of I•/I– is 1.224 V vs SHE, which is 0.13 V higher than that of the dye. This led to the postulation of the two iodide process (TIP) [(D+···I–) + I– → (D···I2–) → D0 + I2–)] for a sufficiently high reducing power, but TIP is not consistent with either the recent experimental data suggesting the first-order kinetics or recent time-resolved spectroscopic measurements. To resolve this conundrum, we used quantum mechanics including Poisson–Boltzmann solvation to examine the electron-transfer process between I– and D+ for the Ru(dcb)2NCS2 or N3 dye. We find that I– is attracted to the oxidized dye, positioning I– next to the NCS. At this equilibrium position, the I– electron is already 40% transferred to the NCS, showing that the redox potential of I– is well matched with the dye. This matching of the redox potential occurs because I– is partially desolvated as it positions itself for the inner-sphere electron transfer (ISET). The previous analyses all assumed an outer-sphere electron-transfer process. Thus our ISET-SIP model is consistent with the known redox potentials and with recent experimental reports. With the ISET-SIP mechanism, one can start to consider how to enhance the dye regeneration kinetics by redesigning ligands to maximize the interaction with iodide.
Co-reporter:Mu-Jeng Cheng ; III
Journal of the American Chemical Society 2013 Volume 135(Issue 12) pp:4600-4603
Publication Date(Web):February 24, 2013
DOI:10.1021/ja3115746
We used density functional theory to study the mechanism of n-butane oxidation to maleic anhydride on the vanadium phosphorus oxide (VPO) surface. We found that O(1)═P on the VVOPO4 surface is the active center for initiating the VPO chemistry through extraction of H from alkane C–H bonds. This contrasts sharply with previous suggestions that the active center is either the V–O bonds or else a chemisorbed O2 on the (VIVO)2P2O7 surface. The ability of O(1)═P to cleave alkane C–H bonds is due to its strong basicity coupled with large reduction potentials of nearby VV ions. We examined several pathways for the subsequent functionalization of n-butane to maleic anhydride and found that the overall barrier does not exceed 21.7 kcal/mol.
Co-reporter:Michal Juríček ; Jonathan C. Barnes ; Edward J. Dale ; Wei-Guang Liu ; Nathan L. Strutt ; Carson J. Bruns ; Nicolaas A. Vermeulen ; Kala C. Ghooray ; Amy A. Sarjeant ; Charlotte L. Stern ; Youssry Y. Botros ; William A. Goddard ; III;J. Fraser Stoddart
Journal of the American Chemical Society 2013 Volume 135(Issue 34) pp:12736-12746
Publication Date(Web):July 18, 2013
DOI:10.1021/ja4052763
Incorporation of two biphenylene-bridged 4,4′-bipyridinium extended viologen units into a para-phenylene-based cyclophane results in a synthetic receptor that is ∼2 nm long and adopts a box-like geometry. This cyclophane, Ex2Box4+, possesses the ability to form binary and ternary complexes with a myriad of guest molecules ranging from long π-electron-rich polycyclic aromatic hydrocarbons, such as tetracene, tetraphene, and chrysene, to π-electron-poor 2,6-dinitrotoluene, 1,2,4-trichlorobenzene, and both the 9,10- and 1,4-anthraquinone molecules. Moreover, Ex2Box4+ is capable of forming one-to-one complexes with polyether macrocycles that consist of two π-electron-rich dioxynaphthalene units, namely, 1,5-dinaphtho[38]crown-10. This type of broad molecular recognition is possible because the electronic constitution of Ex2Box4+ is such that the pyridinium rings located at the “ends” of the cyclophane are electron-poor and prefer to enter into donor–acceptor interactions with π-electron-rich guests, while the “middle” of the cyclophane, consisting of the biphenylene spacer, is more electron-rich and can interact with π-electron-poor guests. In some cases, these different modes of binding can act in concert to generate one-to-one complexes which possess high stability constants in organic media. The binding affinity of Ex2Box4+ was investigated in the solid state by way of single-crystal X-ray diffraction and in solution by using UV–vis and NMR spectroscopy for 12 inclusion complexes consisting of the tetracationic cyclophane and the corresponding guests of different sizes, shapes, and electronic compositions. Additionally, density functional theory was carried out to elucidate the relative energetic differences between the different modes of binding of Ex2Box4+ with anthracene, 9,10-anthraquinone, and 1,4-anthraquinone in order to understand the degree with which each mode of binding contributes to the overall encapsulation of each guest.
Co-reporter:Ted H. Yu, Wei-Guang Liu, Yao Sha, Boris V. Merinov, Pezhman Shirvanian, William A. Goddard III
Journal of Membrane Science 2013 Volume 437() pp:276-285
Publication Date(Web):15 June 2013
DOI:10.1016/j.memsci.2013.02.060
•DFT barriers for Nafion main chain and side chain degradation.•Four environments: concentrated OH and Fuel cell with and without gas crossover.•Main chain degradation mechanisms including chain scission.•Side chain degradation favored over main chain in the actual fuel cell.Degradation of the Nafion electrolyte in Proton Exchange Membrane Fuel Cells (PEMFCs) limits the lifetime, motivating development of materials that resist degradation. The mechanism for degradation of Nafion under fuel cell conditions remains uncertain. Studies of Nafion degradation in concentrated OH environments, such as Fenton or vapor HOOH tests, show that the main chain significantly degrades in these conditions. However it has not been established whether this applies to fuel cell conditions. We have used quantum mechanics (Density Functional Theory with the B3LYP and M06 functionals) to determine the mechanism of Nafion degradation under both concentrated OH and fuel cell conditions. These studies confirm that under concentrated OH conditions Nafion degrades when peroxide radicals attack end groups (–COOH, –CFCF2, –CF2H); followed by degradation of Nafion along the polymer main chain, as proposed previously. However we find that under fuel cell conditions, Nafion degradation occurs along the polymer side chain starting with H attacking the side chain groups such as the sulfonic acid, –SO3−. We find that it is easier for OH to attack the main chain than H, while vice versa, it is easier for H radical to attack the side chain than OH.
Co-reporter:Young-Woo Son;Harry A. Atwater;Min Seok Jang III;Hyungjun Kim
PNAS 2013 Volume 110 (Issue 22 ) pp:8786-8789
Publication Date(Web):2013-05-28
DOI:10.1073/pnas.1305416110
Graphene is a room temperature ballistic electron conductor and also a very good thermal conductor. Thus, it has been regarded
as an ideal material for postsilicon electronic applications. A major complication is that the relativistic massless electrons
in pristine graphene exhibit unimpeded Klein tunneling penetration through gate potential barriers. Thus, previous efforts
to realize a field effect transistor for logic applications have assumed that introduction of a band gap in graphene is a
prerequisite. Unfortunately, extrinsic treatments designed to open a band gap seriously degrade device quality, yielding very
low mobility and uncontrolled on/off current ratios. To solve this dilemma, we propose a gating mechanism that leads to a
hundredfold enhancement in on/off transmittance ratio for normally incident electrons without any band gap engineering. Thus,
our saw-shaped geometry gate potential (in place of the conventional bar-shaped geometry) leads to switching to an off state
while retaining the ultrahigh electron mobility in the on state. In particular, we report that an on/off transmittance ratio
of 130 is achievable for a sawtooth gate with a gate length of 80 nm. Our switching mechanism demonstrates that intrinsic
graphene can be used in designing logic devices without serious alteration of the conventional field effect transistor architecture.
This suggests a new variable for the optimization of the graphene-based device—geometry of the gate electrode.
Co-reporter:Hai Xiao;Wei-Guang Liu;Sergey V. Zybin III;Qi An
PNAS 2013 Volume 110 (Issue 14 ) pp:5321-5325
Publication Date(Web):2013-04-02
DOI:10.1073/pnas.1222890110
A number of exotic structures have been formed through high-pressure chemistry, but applications have been hindered by difficulties
in recovering the high-pressure phase to ambient conditions (i.e., one atmosphere and 300 K). Here we use dispersion-corrected
density functional theory [PBE-ulg (Perdew-Burke-Ernzerhof flavor of DFT with the universal low gradient correction for long range London dispersion)] to predict
that above 60 gigapascal (GPa) the most stable form of N2O (the laughing gas in its molecular form) is a one-dimensional polymer with an all-nitrogen backbone analogous to cis-polyacetylene in which alternate N are bonded (ionic covalent) to O. The analogous trans-polymer is only 0.03∼0.10 eV/molecular unit less stable. Upon relaxation to ambient conditions, both polymers relax below 14 GPa to the same stable
nonplanar trans-polymer. The predicted phonon spectrum and dissociation kinetics validates the stability of this trans-poly-NNO at ambient conditions, which has potential applications as a type of conducting nonlinear optical polymer with all-nitrogen
chains and as a high-energy oxidizer for rocket propulsion. This work illustrates in silico materials discovery particularly in the realm of extreme conditions (very high pressure or temperature).
Co-reporter:Ted H. Yu, Timo Hofmann, Yao Sha, Boris V. Merinov, Deborah J. Myers, Clemens Heske, and William A. Goddard, III
The Journal of Physical Chemistry C 2013 Volume 117(Issue 50) pp:26598-26607
Publication Date(Web):November 25, 2013
DOI:10.1021/jp4071554
To facilitate a less empirical approach to developing improved catalysts, it is important to correlate catalytic performance to surrogate properties that can be measured or predicted accurately and quickly, allowing experimental synthesis and testing of catalysts to focus on the most promising cases. Particularly hopeful is correlating catalysis performance to the electronic density of states (DOS). Indeed, there has been success in using just the center of the d-electron density, which in some cases correlates linearly with oxygen atom chemisorption energy, leading to a volcano plot for catalytic performance versus “d-band center”. To test such concepts we calculated the barriers and binding energies for the various reactions and intermediates involved in the oxygen reduction reaction (ORR) for all 12 transition metals in groups 8–11 (Fe–Cu columns). Our results show that the oxygen binding energy can serve as a useful parameter in describing the catalytic activity for pure metals, but it does not necessarily correlate with the d-band center. In addition, we find that the d-band center depends substantially on the calculation method or the experimental setup, making it a much less reliable indicator for ORR activity than the oxygen binding energy. We further examine several surfaces of the same pure metals to evaluate how the d-band center and oxygen binding energy depend on the surface.
Co-reporter:Hyeyoung Shin, Tod A. Pascal, William A. Goddard III, and Hyungjun Kim
The Journal of Physical Chemistry B 2013 Volume 117(Issue 3) pp:916-927
Publication Date(Web):December 20, 2012
DOI:10.1021/jp310422q
Water-soluble polymers such as polyethylene glycol (PEG) are critical components of industrial processes ranging from drug delivery to water purification. However, the understanding of the microscopic structure of these polymers in water and of the thermodynamics of the mixtures is limited because available experimental techniques (such as SLS and SANS) give little information about conformations and provide even the radius of gyration only in the dilute limit (<∼5 wt % PEG). Computer simulations employing Monte Carlo (MC) and molecular dynamics (MD) techniques can provide an atomistic molecular structure; however, such approaches have difficulties in predicting the equilibrium polymer configurations of high-molecular-weight polymers at normal densities and in obtaining entropies and free energies directly from the MD. Here, we develop the scaled effective solvent (SES) method to predict the equilibrium ensemble of polymer configurations, which we illustrate for the case of a 20 kDa PEG (455 monomers) at a 25 wt % PEG aqueous solution (3339 waters per PEG chain). We evaluate the free energy and entropy of the members of this ensemble including explicit water, validating that it leads to average sizes (Rg) observed experimentally and that all members of the ensemble have favorable free energies. With the SES method validated to provide well-equilibrated polymer chains in water, it should be useful for predicting ensembles of polymer chains in polymer melts and in solvents.
Co-reporter:Qi An, William A. Goddard III, Sergey V. Zybin, Andres Jaramillo-Botero, and Tingting Zhou
The Journal of Physical Chemistry C 2013 Volume 117(Issue 50) pp:26551-26561
Publication Date(Web):December 2, 2013
DOI:10.1021/jp404753v
We report reactive molecular dynamics simulations using the ReaxFF reactive force field to examine shock-induced hot-spot formation followed by detonation initiation in realistic (2.7 million atoms) models of polymer bonded explosives (PBX) with nonplanar interfaces. We considered here two energetic materials (EMs) pentaerythritol tetranitrate (PETN), a common EM for PBX, and silicon pentaerythritol tetranitrate (Si-PETN), which is so extremely sensitive that it has not been possible to characterize its shock properties experimentally. In each case the EM was embedded in a hydroxyl-terminated polybutadiene (HTPB) based polymer binder matrix to form a model of PBX that has a periodic sawtooth nonplanar interface. For the cases in which the shock wave propagates from the EM to polymer (EM→poly), we observed that a hot spot arises from shear localization at the convex polymer asperity. For the case in which the shock direction is inverted (shock wave propagates from the polymer to the EM, EM←poly), we find that a hot spot is initiated at the concave polymer asperity and a second more significant hot spot forms at the convex polymer asperity. This second hot spot is enhanced due to converging shock wave interactions with the nonplanar interface. Under the same shock conditions, the first step in the Si-PETN decomposition is the Si–C–O–X rearrangement to Si–O–C–X through a five centered transition state on the Si that releases 45 kcal/mol of energy that leads to a continuous increase of temperature and pressure in the hot-spot region, until detonation. By contrast, the first step for PETN is NO2 release, which is endothermic by 39 kcal/mol, with the consequence that the hot spot is attenuated by the polymer binder, reaching a steady temperature state involving NO2 dissociation and HONO formation.
Co-reporter:Si-ping Han, Hareem T. Maune, Robert D. Barish, Marc Bockrath, and William A. Goddard III
Nano Letters 2012 Volume 12(Issue 3) pp:1129-1135
Publication Date(Web):February 9, 2012
DOI:10.1021/nl201818u
Ultrathin film preparations of single-walled carbon nanotube (SWNT) allow economical utilization of nanotube properties in electronics applications. Recent advances have enabled production of micrometer scale SWNT transistors and sensors but scaling these devices down to the nanoscale, and improving the coupling of SWNTs to other nanoscale components, may require techniques that can generate a greater degree of nanoscale geometric order than has thus far been achieved. Here, we introduce linker-induced surface assembly, a new technique that uses small structured DNA linkers to assemble solution dispersed nanotubes into parallel arrays on charged surfaces. Parts of our linkers act as spacers to precisely control the internanotube separation distance down to <3 nm and can serve as scaffolds to position components such as proteins between adjacent parallel nanotubes. The resulting arrays can then be stamped onto other substrates. Our results demonstrate a new paradigm for the self-assembly of anisotropic colloidal nanomaterials into ordered structures and provide a potentially simple, low cost, and scalable route for preparation of exquisitely structured parallel SWNT films with applications in high-performance nanoscale switches, sensors, and meta-materials.
Co-reporter:John A. Keith ; Douglas C. Behenna ; Nathaniel Sherden ; Justin T. Mohr ; Sandy Ma ; Smaranda C. Marinescu ; Robert J. Nielsen ; Jonas Oxgaard ; Brian M. Stoltz ; III
Journal of the American Chemical Society 2012 Volume 134(Issue 46) pp:19050-19060
Publication Date(Web):October 28, 2012
DOI:10.1021/ja306860n
We use first principles quantum mechanics (density functional theory) to report a detailed reaction mechanism of the asymmetric Tsuji allylation involving prochiral nucleophiles and nonprochiral allyl fragments, which is consistent with experimental findings. The observed enantioselectivity is best explained with an inner-sphere mechanism involving the formation of a 5-coordinate Pd species that undergoes a ligand rearrangement, which is selective with regard to the prochiral faces of the intermediate enolate. Subsequent reductive elimination generates the product and a Pd0 complex. The reductive elimination occurs via an unconventional seven-centered transition state that contrasts dramatically with the standard three-centered C–C reductive elimination mechanism. Although limitations in the present theory prevent the conclusive identification of the enantioselective step, we note that three different computational schemes using different levels of theory all find that inner-sphere pathways are lower in energy than outer-sphere pathways. This result qualitatively contrasts with established allylation reaction mechanisms involving prochiral nucleophiles and prochiral allyl fragments. Energetic profiles of all reaction pathways are presented in detail.
Co-reporter:Wei-Guang Liu ; III
Journal of the American Chemical Society 2012 Volume 134(Issue 31) pp:12970-12978
Publication Date(Web):July 11, 2012
DOI:10.1021/ja300545e
Experimental results, such as NO2 hydrolysis and the hypergolicity of hydrazine/nitrogen tetroxide pair, have been interpreted in terms of NO2 dimers. Such interpretations are complicated by the possibility of several forms for the dimer: symmetric N2O4, cis-ONO-NO2, and trans-ONO-NO2. Quantum mechanical (QM) studies of these systems are complicated by the large resonance energy in NO2 which changes differently for each dimer and changes dramatically as bonds are formed and broken. As a result, none of the standard methods for QM are uniformly reliable. We report here studies of these systems using density functional theory (B3LYP) and several ab initio methods (MP2, CCSD(T), and GVB-RCI). At RCCSD(T)/CBS level, the enthalpic barrier to form cis-ONO-NO2 is 1.9 kcal/mol, whereas the enthalpic barrier to form trans-ONO-NO2 is 13.2 kcal/mol, in agreement with the GVB-RCI result. However, to form symmetric N2O4, RCCSD(T) gives an unphysical barrier due to the wrong asymptotic behavior of its reference function at the dissociation limit, whereas GVB-RCI shows no barrier for such a recombination. The difference of barrier heights in these three recombination reactions can be rationalized in terms of the amount of B2 excitation involved in the bond formation process. We find that the enthalpic barrier for N2O4 isomerizing to trans-ONO-NO2 is 43.9 kcal/mol, ruling out the possibility of such an isomerization playing a significant role in gas-phase hydrolysis of NO2. A much more favored path is to form cis-ONO-NO2 first then convert to trans-ONO-NO2 with a 2.4 kcal/mol enthalpic barrier. We also propose that the isotopic oxygen exchange in NO2 gas is possibly via the formation of trans-ONO-NO2 followed by ON+ migration.
Co-reporter:Jacob S. Kanady ; Jose L. Mendoza-Cortes ; Emily Y. Tsui ; Robert J. Nielsen ; William A. Goddard ; III;Theodor Agapie
Journal of the American Chemical Society 2012 Volume 135(Issue 3) pp:1073-1082
Publication Date(Web):December 15, 2012
DOI:10.1021/ja310022p
The oxygen-evolving complex (OEC) of photosystem II contains a Mn4CaOn catalytic site, in which reactivity of bridging oxidos is fundamental to OEC function. We synthesized structurally relevant cuboidal Mn3MOn complexes (M = Mn, Ca, Sc; n = 3,4) to enable mechanistic studies of reactivity and incorporation of μ3-oxido moieties. We found that MnIV3CaO4 and MnIV3ScO4 were unreactive toward trimethylphosphine (PMe3). In contrast, our MnIII2MnIV2O4 cubane reacts with this phosphine within minutes to generate a novel MnIII4O3 partial cubane plus Me3PO. We used quantum mechanics to investigate the reaction paths for oxygen atom transfer to phosphine from MnIII2MnIV2O4 and MnIV3CaO4. We found that the most favorable reaction path leads to partial detachment of the CH3COO– ligand, which is energetically feasible only when Mn(III) is present. Experimentally, the lability of metal-bound acetates is greatest for MnIII2MnIV2O4. These results indicate that even with a strong oxygen atom acceptor, such as PMe3, the oxygen atom transfer chemistry from Mn3MO4 cubanes is controlled by ligand lability, with the MnIV3CaO4 OEC model being unreactive. The oxidative oxide incorporation into the partial cubane, MnIII4O3, was observed experimentally upon treatment with water, base, and oxidizing equivalents. 18O-labeling experiments provided mechanistic insight into the position of incorporation in the partial cubane structure, consistent with mechanisms involving migration of oxide moieties within the cluster but not consistent with selective incorporation at the site available in the starting species. These results support recent proposals for the mechanism of the OEC, involving oxido migration between distinct positions within the cluster.
Co-reporter:Jun Tan, Ravinder Abrol, Bartosz Trzaskowski, and William A. Goddard III
Journal of Chemical Information and Modeling 2012 Volume 52(Issue 7) pp:1875-1885
Publication Date(Web):June 1, 2012
DOI:10.1021/ci300133a
The G protein-coupled receptor (GPCR) TAS2R38 is a bitter taste receptor that can respond to bitter compounds such as phenylthiocarbamide (PTC) and 6-n-propylthiouracil (PROP). This receptor was chosen because its four haplotypes (based on three residue site polymorphism) hTAS2R38PAV, hTAS2R38AVI, hTAS2R38AAI, and hTAS2R38PVV are known to have dramatically different responses to PTC and PROP. We aimed to identify the protein–ligand interaction features that determine whether the bitter taste signal from this receptor is sent to the cortex. To do this we predicted the 3D structures of the TAS2R38 bitter taste receptor using our new BiHelix and SuperBiHelix Monte Carlo methods (No experimental determinations of the 3D structure have been reported for any taste receptors.). We find that residue 262 (2nd position in the polymorphism) is involved in the interhelical hydrogen bond network stabilizing the GPCR structure in tasters (hTAS2R38PAV, hTAS2R38AAI, and hTAS2R38PVV), while it is not in the nontaster (hTAS2R38AVI). This suggests that the hydrogen bond interactions between TM3 and TM6 or between TM5 and TM6 may play a role in activating this GPCR. To further validate these structures, we used the DarwinDock method to predict the binding sites and 3D structures for PTC and PROP bound to hTAS2R38PAV, hTAS2R38AVI, hTAS2R38AAI, and hTAS2R38PVV, respectively. Our results show that PTC and PROP can form H-bonds with the backbone of residue 262 in the tasters (hTAS2R38PAV, hTAS2R38AAI, and hTAS2R38PVV) but not in the nontaster (hTAS2R38AVI). Thus it appears that the hydrogen bond interaction between TM3 and TM6 may activate the receptor to pass the ligand binding signal to intracellular processes and that the H-bond between agonists and residue 262 in tasters is involved in the bitter tasting. This is in agreement with experimental observations, providing validation of the predicted ligand-protein complexes and also a potential activation mechanism for the TAS2R38 receptor.
Co-reporter:Hyungjun Kim, Jeong-Mo Choi, and William A. Goddard III
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 3) pp:360-363
Publication Date(Web):January 10, 2012
DOI:10.1021/jz2016395
Conventional density functional theory (DFT) fails to describe accurately the London dispersion essential for describing molecular interactions in soft matter (biological systems, polymers, nucleic acids) and molecular crystals. This has led to several methods in which atom-dependent potentials are added into the Kohn–Sham DFT energy. Some of these corrections were fitted to accurate quantum mechanical results, but it will be tedious to determine the appropriate parameters to describe all of the atoms of the periodic table. We propose an alternative approach in which a single parameter in the low-gradient (lg) functional form is combined with the rule-based UFF (universal force-field) nonbond parameters developed for the entire periodic table (up to Lr, Z = 103), named as a DFT-ulg method. We show that DFT-ulg method leads to a very accurate description of the properties for molecular complexes and molecular crystals, providing the means for predicting more accurate weak interactions across the periodic table.Keywords: DFT-ulg; low-gradient functional; PBE-ulg; soft matter; universal force field;
Co-reporter:Jiwon Jeon, Hyungjun Kim, William A. Goddard III, Tod A. Pascal, Ga-In Lee, and Jeung Ku Kang
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 4) pp:556-559
Publication Date(Web):February 3, 2012
DOI:10.1021/jz3000036
Ionic liquids (ILs) provide an attractive medium for various chemical and redox reactions, where they are generally regarded as hydrophobic. However, Seddon et al. discovered that 4–10 wt % water absorbs into ILs that contain bulky anions, and Cammarata et al. found that the molecular state of water in ILs is dramatically different from that of bulk liquid water or that of water vapor. To determine the microstructure of water incorporated into ILs and the impact on properties, we carried out first-principles-based molecular dynamics simulations. We find water in three distinct phases depending on water content, and that the transport properties depend on the nature of the water phases. These results suggest that the optimal water content is ∼10% mole fraction of water molecules (∼1.1 wt %) for applications such as nonvolatile electrolytes for dye-sensitized solar cells (DSSCs). This suggests a strategy for improving the performance of IL DSSC by replacing water with additives that would play the same role as water (since too much water can deteriorate performance at the anode–dye interface).Keywords: dye-sensitized solar cells; electrolytes; ionic liquids;
Co-reporter:Seokmin Jeon, Hyungjun Kim, William A. Goddard, III, and Harry A. Atwater
The Journal of Physical Chemistry C 2012 Volume 116(Issue 33) pp:17604-17612
Publication Date(Web):July 9, 2012
DOI:10.1021/jp3041555
We investigate the adsorption and decomposition states of a water molecule on a Ga-rich GaP(001)(2×4) surface using the PBE flavor of density functional theory (DFT). We selected the GaP(001)(2×4) mixed dimer surface reconstruction model to represent the Ga-rich GaP(001)(2×4) surface. Because our focus is on reactions between a single water molecule and the surface, the surface water coverage is kept at 0.125 ML, which corresponds to one water molecule in the (2×4) unit cell. We report here the geometries and energies for an exhaustive set of adsorption and decomposition states induced by a water molecule on the (2×4) unit cell. Our results support a mechanism in which (1) the first step is the molecular adsorption, with the water molecule forming a Lewis acid–Lewis base bond to the sp2 Ga atom of either the first-layer Ga–P mixed dimer or the second layer Ga–Ga dimers using an addition reaction, (2) which is followed by dissociation of the adsorbed H2O to form the HO/H decomposition state in which the hydroxyl moiety bonds with surface sp2 Ga atoms, while the hydrogen moiety binds with the first-layer P atom, (3) which is followed by the O/2H decomposition state, in which the oxygen moiety forms bridged Ga–O–Ga structures with surface Ga dimers while one H bonds with the first-layer P atom and the other to surface sp2 Ga atoms. (4) We find that driving off the hydrogen as H2 leads to the surface oxide state, bridged Ga–O–Ga structures. This surface oxide formation reaction is exothermic relative to the energy of H2O plus the reconstructed surface. These results provide guidelines for experiments and theory to validate the key steps and to obtain kinetics data for modeling the growth processes.
Co-reporter:Yao Sha, Ted H. Yu, Boris V. Merinov, Pezhman Shirvanian, and William A. Goddard, III
The Journal of Physical Chemistry C 2012 Volume 116(Issue 40) pp:21334-21342
Publication Date(Web):September 4, 2012
DOI:10.1021/jp303966u
We use quantum mechanics, density functional theory at the PBE level, to predict the binding-site preferences and reaction barriers for all intermediates involved in the oxygen reduction reaction (ORR) on the low energy surface of Pt3Ni alloy. Here we calculate that the surface layer is Ni depleted (100% Pt) while the second layer is Ni enriched (50% Pt) as shown by experiment. Even though the top layer is pure Pt, we find that the sublayer Ni imposes strong preferences in binding sites for most intermediates, which in turn strongly influences the reaction barriers. This strong preference leads to a strong site dependence of the barriers. Considering water as the solvent, we predict that, at low coverage of Oad and OHad, the barrier for the rate-determining step is 0.81 eV, whereas, at high coverage, this barrier decreases to 0.43 eV. It can be compared to a barrier of 0.50 eV for pure Pt, explaining the improved ORR rate for the Pt3Ni alloy. We report the results both for gas phase and for aqueous phase environments.
Co-reporter:Tod A. Pascal and William A Goddard III
The Journal of Physical Chemistry B 2012 Volume 116(Issue 47) pp:13905-13912
Publication Date(Web):November 5, 2012
DOI:10.1021/jp309693d
When water and methanol are mixed, the entropy of mixing decreases, whereas mixing simple liquids normally leads to an increase in entropy. One speculation on the origin of the anomaly involves formation of water icebergs next to the hydrophobic methanol group, while more recent theories point to nanoscale clustering of methanol molecules. To elucidate the origin of this effect, we carried out extensive molecular dynamics calculations on water/methanol mixtures ranging from 0 to 100% and applied the 2PT method to extract the entropy and free energy changes of each component as a function of concentration. We find that water molecules lose at most 1/35 of their liquid entropy in mixtures. Methanol molecules, on the other hand, lose 3 times as much entropy as the water molecules, and their rotational entropy contains the signature of the entropic loss. We find that methanol has a discontinuous specific heat profile in these mixtures with a maximum at 40% methanol. These results do not support the iceberg model of immobilized waters and instead suggests a molecular mechanism of hydrophobic segregation at low methanol concentration where ordering of the methanol molecules bury the hydrophobic group away from the water phase. For higher methanol concentrations, there is insufficient water to accomplish this effect, and the system freely mixes and transitions to one better described as water dissolved into methanol.
Co-reporter:Yao Sha ; Ted H. Yu ; Boris V. Merinov ; III
The Journal of Physical Chemistry C 2012 Volume 116(Issue 10) pp:6166-6173
Publication Date(Web):February 2, 2012
DOI:10.1021/jp207526u
To use density functional theory (DFT) to seek improved catalysts for the oxygen reduction reaction (ORR) in a proton exchange membrane fuel cell, we developed a systematic way to handle the barriers of electron transfer reactions (e.g., H+ + e– + Oad → OHad) within the DFT framework. We report applications of this new method to determining the dependence for the barriers of various ORR reaction steps on the operating electrochemical potential for the Pt-catalyzed fuel cell. This method is used to estimate the optimum operating potential. In the Article, we show how to estimate the change in efficiency from changes in the reaction barriers. On the basis of our mechanism and calculated barriers, the optimum operating voltage for the ORR on Pt is found to be 0.68 V/NHE, which is close to the standard operating voltage of ∼0.8 V/NHE, validating this analysis.
Co-reporter:Qi An ; Yi Liu ; Sergey V. Zybin ; Hyungjun Kim ; III
The Journal of Physical Chemistry C 2012 Volume 116(Issue 18) pp:10198-10206
Publication Date(Web):April 17, 2012
DOI:10.1021/jp300711m
We applied the compress-and-shear reactive dynamics (CS-RD) simulation model to study the anisotropic shock sensitivity of cyclotrimethylene trinitramine (RDX) crystals. We predict that, for mechanical shocks between 3 and 7 GPa, RDX is most sensitive to shocks perpendicular to the (100) and (210) planes, whereas it is insensitive for shocks perpendicular to the (120), (111), and (110) planes. These results are all consistent with available experimental information, further validating the CS-RD model for distinguishing between sensitive and insensitive shock directions. We find that, for sensitive directions, the shock impact triggers a slip system that leads to large shear stresses arising from steric hindrance, causing increased energy inputs that increase the temperature, leading to dramatically increased chemical reactions. Thus, our simulations demonstrate that the molecular origin of anisotropic shock sensitivity results from steric hindrance toward shearing of adjacent slip planes during shear deformation. Thus, strain energy density, temperature rise, and molecule decomposition are effective measures to distinguish anisotropic sensitivities. We should emphasize that CS-RD has been developed as a tool to distinguish rapidly (within a few picoseconds) between sensitive and insensitive shock directions of energetic materials. If the high stresses and rates used here continued much longer and for larger systems, it would ultimately result in detonation for all directions, but we have not demonstrated this.
Co-reporter:Andrea Kirkpatrick;Jiyoung Heo III;Ravinder Abrol
PNAS 2012 Volume 109 (Issue 49 ) pp:19988-19993
Publication Date(Web):2012-12-04
DOI:10.1073/pnas.1218051109
The glucagon-like peptide 1 receptor (GLP1R) is a G protein-coupled receptor (GPCR) involved in insulin synthesis and regulation;
therefore, it is an important drug target for treatment of diabetes. However, GLP1R is a member of the class B1 family of
GPCRs for which there are no experimental structures. To provide a structural basis for drug design and to probe class B GPCR
activation, we predicted the transmembrane (TM) bundle structure of GLP1R bound to the peptide Exendin-4 (Exe4; a GLP1R agonist
on the market for treating diabetes) using the MembStruk method for scanning TM bundle conformations. We used protein–protein
docking methods to combine the TM bundle with the X-ray crystal structure of the 143-aa N terminus coupled to the Exe4 peptide.
This complex was subjected to 28 ns of full-solvent, full-lipid molecular dynamics. We find 14 strong polar interactions of
Exe4 with GLP1R, of which 8 interactions are in the TM bundle (2 interactions confirmed by mutation studies) and 6 interactions
involve the N terminus (3 interactions found in the crystal structure). We also find 10 important hydrophobic interactions,
of which 4 interactions are in the TM bundle (2 interactions confirmed by mutation studies) and 6 interactions are in the
N terminus (6 interactions present in the crystal structure). Thus, our predicted structure agrees with available mutagenesis
studies. We suggest a number of mutation experiments to further validate our predicted structure. The structure should be
useful for guiding drug design and can provide a structural basis for understanding ligand binding and receptor activation
of GLP1R and other class B1 GPCRs.
Co-reporter:Tod A Pascal, Shiang-Tai Lin, William Goddard III, and Yousung Jung
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 3) pp:294-298
Publication Date(Web):January 7, 2012
DOI:10.1021/jz201612y
To improve the description of solvation thermodynamics of biomolecules, we report here the dependence of solvation on the curvature and surface charge of positively charged solutes in water based on extensive molecular dynamics simulations analyzed using the two-phase thermodynamic method. At a surface charge of +0.4e, the compensating forces of favorable electrostatic stabilization and entropic destabilization cancel almost exactly, representing a molecular crossover point from hydrophobic to hydrophilic behavior, independent of curvature. These results suggest that one should include charge-dependent entropic corrections to continuum models aimed at predicting the solvation free energies of large biomolecules.Keywords: electrostatics; free-energy simulations; interfacial effects; molecular dynamics; surface tension;
Co-reporter:Qi An, Konrad Samwer, William A. Goddard III, William L. Johnson, Andres Jaramillo-Botero, Glenn Garret, and Marios D. Demetriou
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 21) pp:3143-3148
Publication Date(Web):October 10, 2012
DOI:10.1021/jz3014425
Metallic glasses have been established to have unique properties such as ductility, toughness, and soft magnetism with promising engineering applications. However, the glass-forming ability (GFA) has not been sufficient to synthesize the bulk metallic glasses (BMGs) required for many engineering applications. Attempts to develop the understanding of the GFA required to predict the optimum alloys have not yet been proven successful. We develop here a computational model based on molecular dynamics simulations that explains the dramatic change of GFA with alloying small amounts of Al into Cu–Zr. We find that the high GFA to form BMGs depends on a combination of three factors, (a) a low thermodynamic driving force for crystallization, (b) a high melt viscosity, and (c) large ratios of icosahedral structures in the liquid phase. These computational methods to predict these factors that suppress formation of crystal nuclei and slow the dynamic motions in the liquids are practical for in silico prediction of new alloys with optimal GFA.Keywords: icosahedral structure; melt viscosity; metallic glass; microalloying; thermodynamic driving force;
Co-reporter:Jose L. Mendoza-Cortes and William A. Goddard III, Hiroyasu Furukawa and Omar M. Yaghi
The Journal of Physical Chemistry Letters 2012 Volume 3(Issue 18) pp:2671-2675
Publication Date(Web):August 13, 2012
DOI:10.1021/jz301000m
Physisorption in porous materials is a promising approach for meeting H2 storage requirements for the transportation industry, because it is both fully reversible and fast at mild conditions. However, most current candidates lead to H2 binding energies that are too weak (leading to volumetric capacity at 298 K of <10 g/L compared to the DOE 2015 Target of 40 g/L). Using accurate quantum mechanical (QM) methods, we studied the H2 binding energy to 48 compounds based on various metalated analogues of five common linkers for covalent organic frameworks (COFs). Considering the first transition row metals (Sc though Cu) plus Pd and Pt, we find that the new COF-301-PdCl2 reaches 60 g total H2/L at 100 bar, which is 1.5 times the DOE 2015 target of 40 g/L and close to the ultimate (2050) target of 70 g/L. The best current materials, MOF-200 and MOF-177, are predicted to store 7.6 g/L (0.54 wt % excess) and 9.6 g/L (0.87 wt % excess), respectively, at 298 K and 100 bar compared with 60 g/L (4.2 wt % excess) for COF-301-PdCl2.Keywords: COF; force field; Grand Canonical Monte Carlo; Hydrogen storage; metalation; MP2; multiscale; quantum mechanics; sorption isotherm; transition metals;
Co-reporter:Yi Liu, Sergey V. Zybin, Jiaqi Guo, Adri C. T. van Duin, and William A. Goddard III
The Journal of Physical Chemistry B 2012 Volume 116(Issue 48) pp:14136-14145
Publication Date(Web):November 13, 2012
DOI:10.1021/jp308351g
To gain an atomistic-level understanding on physical and chemical processes occurring at the interfaces of hypergolic propellants, we carried out the first reactive dynamic (ReaxFF) simulations to study the reactive hypergolic mixture of monomethylhydrazine (MMH) and dinitrogen tetroxide (NTO), in comparison with the ethanol (EtOH) and NTO mixture that is reactive but nonhypergolic. Our studies show that the MMH–NTO mixture releases energy more rapidly than the EtOH–NTO mixture upon mixing the fuels and oxidizers. We found that the major early chemical reactions between MMH and NTO are hydrogen abstractions and N–N bond scissions. The MMH–NTO mixture has more reaction channels than EtOH–NTO based on statistical analyses of chemical reaction events and channels at early stages of the dynamics. Analyzing the evolution of product distribution over reaction time shows that the oxidizer (NO2) diffuses into the fuels (MMH or EtOH) for the occurrence of reactions, demonstrating the influence of physical mixing on chemical reactions. Our simulations suggest that effective hypergolic systems require fuels with low energy barriers of H abstractions and/or bond scissions and oxidizers with large diffusion mobility for efficient physical mixing.
Co-reporter:Jason M. Keith and William A. Goddard III
Organometallics 2012 Volume 31(Issue 2) pp:545-552
Publication Date(Web):January 11, 2012
DOI:10.1021/om200809u
We elucidate here the mechanism for the reaction of molecular oxygen with palladium-hydride complexes in toluene using quantum mechanics (B3LYP/LACVP** with the PBF polarizable continuum solvent model) for ((−)sparteine)-PdII(H)OAc. Here we focus specifically on two classes of pathways: (1) those proceeding through Pd0 and (2) those involving direct insertion of oxygen into the Pd–H bond. In particular, we present calculated potential energy surfaces and structures of the ((−)-sparteine)PdH system in which the OAc ion is substituted for Cl. We find that the acetate ligand has the ability to act as a base while chelating the Pd, making an external base unnecessary and significantly lowering the energy barrier involved in the Pd0 pathway. This switches the calculated preference to the reductive elimination pathway proceeding through Pd0 (ΔΔH⧧ = 1.0 kcal/mol, ΔΔG⧧ = −7.7 kcal/mol). The results presented herein demonstrate the ability to steer the reaction pathway through the choice of ancillary ligands. We expect that this strategy might contribute significantly to the development of new catalytic systems utilizing molecular oxygen as the stoichiometric oxidant.
Co-reporter:Tod A. Pascal, William A. Goddard III, Prabal K. Maiti, and Nagarajan Vaidehi
The Journal of Physical Chemistry B 2012 Volume 116(Issue 40) pp:12159-12167
Publication Date(Web):September 21, 2012
DOI:10.1021/jp306473u
DNA three-way junctions (TWJs) are important intermediates in various cellular processes and are the simplest of a family of branched nucleic acids being considered as scaffolds for biomolecular nanotechnology. Branched nucleic acids are stabilized by divalent cations such as Mg2+, presumably due to condensation and neutralization of the negatively charged DNA backbone. However, electrostatic screening effects point to more complex solvation dynamics and a large role of interfacial waters in thermodynamic stability. Here, we report extensive computer simulations in explicit water and salt on a model TWJ and use free energy calculations to quantify the role of ionic character and strength on stability. We find that enthalpic stabilization of the first and second hydration shells by Mg2+ accounts for 1/3 and all of the free energy gain in 50% and pure MgCl2 solutions, respectively. The more distorted DNA molecule is actually destabilized in pure MgCl2 compared to pure NaCl. Notably, the first shell, interfacial waters have very low translational and rotational entropy (i.e., mobility) compared to the bulk, an entropic loss that is overcompensated by increased enthalpy from additional electrostatic interactions with Mg2+. In contrast, the second hydration shell has anomalously high entropy as it is trapped between an immobile and bulklike layer. The nonmonotonic entropic signature and long-range perturbations of the hydration shells to Mg2+ may have implications in the molecular recognition of these motifs. For example, we find that low salt stabilizes the parallel configuration of the three-way junction, whereas at normal salt we find antiparallel configurations deduced from the NMR. We use the 2PT analysis to follow the thermodynamics of this transition and find that the free energy barrier is dominated by entropic effects that result from the decreased surface area of the antiparallel form which has a smaller number of low entropy waters in the first monolayer.
Co-reporter:Lianchi Liu, Andres Jaramillo-Botero, William A. Goddard III, and Huai Sun
The Journal of Physical Chemistry A 2012 Volume 116(Issue 15) pp:3918-3925
Publication Date(Web):March 13, 2012
DOI:10.1021/jp210135j
Ettringite is a hexacalcium aluminate trisulfate hydrate mineral that forms during Portland cement hydration. Its presence plays an important role in controlling the setting rate of the highly reactive aluminate phases in cement paste and has also been associated with severe cracking in cured hardened cement. To understand how it forms and how its properties influence those of hardened cement and concrete, we have developed a first-principles-based ReaxFF reactive force field for Ca/Al/H/O/S. Here, we report on the development of this ReaxFF force field and on its validation and application using reactive molecular dynamics (RMD) simulations to characterize and understand the elastic, plastic, and failure response of ettringite at the atomic scale. The ReaxFF force field was validated by comparing the lattice parameters, pairwise distribution functions, and elastic constants of an ettringite crystal model obtained from RMD simulations with those from experiments. The predicted results are in close agreement with published experimental data. To characterize the atomistic failure modes of ettringite, we performed stress–strain simulations to find that Ca–O bonds are responsible for failure of the calcium sulfate and tricalcium aluminate (C3A) column in ettringite during uniaxial compression and tension and that hydrogen bond re-formation during compression induces an increase in plastic strain beyond the material’s stress–strain proportionality limit. These results provide essential insight into understanding the mechanistic role of this mineral in cement and concrete degradation, and the ReaxFF potential developed in this work serves as a fundamental tool to further study the kinetics of hydration in cement and concrete.
Co-reporter:José L. Mendoza-Cortés, Sang Soo Han, and William A. Goddard III
The Journal of Physical Chemistry A 2012 Volume 116(Issue 6) pp:1621-1631
Publication Date(Web):December 21, 2011
DOI:10.1021/jp206981d
The Yaghi laboratory has developed porous covalent organic frameworks (COFs), COF102, COF103, and COF202, and metal–organic frameworks (MOFs), MOF177, MOF180, MOF200, MOF205, and MOF210, with ultrahigh porosity and outstanding H2 storage properties at 77 K. Using grand canonical Monte Carlo (GCMC) simulations with our recently developed first principles based force field (FF) from accurate quantum mechanics (QM), we calculated the molecular hydrogen (H2) uptake at 298 K for these systems, including the uptake for Li-, Na-, and K-metalated systems. We report the total, delivery and excess amount in gravimetric and volumetric units for all these compounds. For the gravimetric delivery amount from 1 to 100 bar, we find that eleven of these compounds reach the 2010 DOE target of 4.5 wt % at 298 K. The best of these compounds are MOF200-Li (6.34) and MOF200-Na (5.94), both reaching the 2015 DOE target of 5.5 wt % at 298 K. Among the undoped systems, we find that MOF200 gives a delivery amount as high as 3.24 wt % while MOF210 gives 2.90 wt % both from 1 to 100 bar and 298 K. However, none of these compounds reach the volumetric 2010 DOE target of 28 g H2/L. The best volumetric performance is for COF102-Na (24.9), COF102-Li (23.8), COF103-Na (22.8), and COF103-Li (21.7), all using delivery g H2/L units for 1–100 bar. These are the highest volumetric molecular hydrogen uptakes for a porous material under these thermodynamic conditions. Thus, one can obtain outstanding H2 uptakes with Li, Na, and K doping of simple frameworks constructed from simple, cheap organic linkers. We present suggestions for strategies for synthesis of alkali metal-doped MOFs or COFs.
Co-reporter:Seung Geol Lee, Tod A. Pascal, Wonsang Koh, Giuseppe F. Brunello, William A. Goddard, III, and Seung Soon Jang
The Journal of Physical Chemistry C 2012 Volume 116(Issue 30) pp:15974-15985
Publication Date(Web):July 11, 2012
DOI:10.1021/jp301610b
Technologies ranging from solvent extraction and drug delivery to tissue engineering are beginning to benefit from the unique ability of “smart polymers” to undergo controllable structural changes in response to external stimuli. The prototype is poly(N-isopropylacrylamide) (P(NIPAAm)) which exhibits an abrupt and reversible hydrophilic to hydrophobic transition above its lower critical solution temperature (LCST) of ∼305 K. We report here molecular dynamics simulations to show the deswelling mechanisms of the hydrated surface-grafted P(NIPAAm) brush at various temperatures such as 275, 290, 320, 345, and 370 K. The deswelling of the P(NIPAAm) brush is clearly observed above the lower critical solution temperature below which the P(NIPAAm) brush is associated with water molecules stably. By simulating the poly(acrylamide) brush as a reference system having the upper critical solution temperature (UCST) behavior with the same conditions employed in the P(NIPAAm) brush simulations, we confirmed that the deswelling of P(NIPAAm) brush does not take place at a given range of temperatures, which validates our simulation procedure. By analyzing the pair correlation functions and the coordination numbers, we found that the dissociation of water from the P(NIPAAm) brush occurs mainly around the isopropyl group of the P(NIPAAm) above the LCST because of its hydrophobicity. We also found that the NH of the amide group in NIPAAm does not actively participate in the hydrogen bonding with water molecules because of the steric hindrance caused by the attached isopropyl group, and thereby the hydrogen bonding interactions between amide groups and water molecules are significantly weakened with increasing temperature, leading to deswelling of the hydrated P(NIPAAm) brush above the LCST through favorable entropic change. These results explain the experimental observations in terms of a simple molecular mechanism for polymer function.
Co-reporter:Ekaterina Tkatchouk ; Neal P. Mankad ; Diego Benitez ; William A. Goddard ; III;F. Dean Toste
Journal of the American Chemical Society 2011 Volume 133(Issue 36) pp:14293-14300
Publication Date(Web):August 23, 2011
DOI:10.1021/ja2012627
We present a detailed study of the mechanism for oxidative heteroarylation, based on DFT calculations and experimental observations. We propose binuclear Au(II)–Au(II) complexes to be key intermediates in the mechanism for gold catalyzed oxidative heteroarylation. The reaction is thought to proceed via a gold redox cycle involving initial oxidation of Au(I) to binuclear Au(II)–Au(II) complexes by Selectfluor, followed by heteroauration and reductive elimination. While it is tempting to invoke a transmetalation/reductive elimination mechanism similar to that proposed for other transition metal complexes, experimental and DFT studies suggest that the key C–C bond forming reaction occurs via a bimolecular reductive elimination process (devoid of transmetalation). In addition, the stereochemistry of the elimination step was determined experimentally to proceed with complete retention. Ligand and halide effects played an important role in the development and optimization of the catalyst; our data provides an explanation for the ligand effects observed experimentally, useful for future catalyst development. Cyclic voltammetry data is presented that supports redox synergy of the Au···Au aurophilic interaction. The monometallic reductive elimination from mononuclear Au(III) complexes is also studied from which we can predict a ∼15 kcal/mol advantage for bimetallic reductive elimination.
Co-reporter:Ted H. Yu ; Yao Sha ; Wei-Guang Liu ; Boris V. Merinov ; Pezhman Shirvanian ; III
Journal of the American Chemical Society 2011 Volume 133(Issue 49) pp:19857-19863
Publication Date(Web):October 21, 2011
DOI:10.1021/ja2074642
We report results of quantum mechanics (QM) mechanistic studies of Nafion membrane degradation in a polymer electrolyte membrane (PEM) fuel cell. Experiments suggest that Nafion degradation is caused by generation of trace radical species (such as OH●, H●) only when in the presence of H2, O2, and Pt. We use density functional theory (DFT) to construct the potential energy surfaces for various plausible reactions involving intermediates that might be formed when Nafion is exposed to H2 (or H+) and O2 in the presence of the Pt catalyst. We find a barrier of 0.53 eV for OH radical formation from HOOH chemisorbed on Pt(111) and of 0.76 eV from chemisorbed OOHad, suggesting that OH might be present during the ORR, particularly when the fuel cell is turned on and off. Based on the QM, we propose two chemical mechanisms for OH radical attack on the Nafion polymer: (1) OH attack on the S–C bond to form H2SO4 plus a carbon radical (barrier: 0.96 eV) followed by decomposition of the carbon radical to form an epoxide (barrier: 1.40 eV). (2) OH attack on H2 crossover gas to form hydrogen radical (barrier: 0.04 eV), which subsequently attacks a C–F bond to form HF plus carbon radicals (barrier as low as 1.00 eV). This carbon radical can then decompose to form a ketone plus a carbon radical with a barrier of 0.86 eV. The products (HF, OCF2, SCF2) of these proposed mechanisms have all been observed by F NMR in the fuel cell exit gases along with the decrease in pH expected from our mechanism.
Co-reporter:Sijia S. Dong ; Robert J. Nielsen ; Joshua H. Palmer ; Harry B. Gray ; Zeev Gross ; Siddharth Dasgupta ; III
Inorganic Chemistry 2011 Volume 50(Issue 3) pp:764-770
Publication Date(Web):January 7, 2011
DOI:10.1021/ic1005902
The electronic structures of metallocorroles (tpfc)M(NH3)2 and (tfc)M(NH3)2 (tpfc is the trianion of 5,10,15-(tris)pentafluorophenylcorrole, tfc is the trianion of 5,10,15-trifluorocorrole, and M = Co, Rh, Ir) have been computed using first principles quantum mechanics [B3LYP flavor of Density Functional Theory (DFT) with Poisson−Boltzmann continuum solvation]. The geometry was optimized for both the neutral systems (formal MIII oxidation state) and the one-electron oxidized systems (formally MIV). As expected, the MIII systems have a closed shell d6 configuration; for all three metals, the one-electron oxidation was calculated to occur from a ligand-based orbital (highest occupied molecular orbital (HOMO) of B1 symmetry). The ground state of the formal MIV system has MIII-Cπ character, indicating that the metal remains d6, with the hole in the corrole π system. As a result the calculated MIV/III reduction potentials are quite similar (0.64, 0.67, and 0.56 V vs SCE for M = Ir, Rh and Co, respectively), whereas the differences would have been large for purely metal-based oxidations. Vertically excited states with substantial metal character are well separated from the ground state in one-electron-oxidized cobalt (0.27 eV) and rhodium (0.24 eV) corroles, but become closer in energy in the iridium (0.15 eV) analogues. The exact splittings depend on the chosen functional and basis set combination and vary by ∼0.1 eV.
Co-reporter:Tod A. Pascal, Shiang-Tai Lin and William A. Goddard III
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 1) pp:169-181
Publication Date(Web):23 Nov 2010
DOI:10.1039/C0CP01549K
We validate here the Two-Phase Thermodynamics (2PT) method for calculating the standard molar entropies and heat capacities of common liquids. In 2PT, the thermodynamics of the system is related to the total density of states (DoS), obtained from the Fourier Transform of the velocity autocorrelation function. For liquids this DoS is partitioned into a diffusional component modeled as diffusion of a hard sphere gas plus a solid component for which the DoS(υ) → 0 as υ → 0 as for a Debye solid. Thermodynamic observables are obtained by integrating the DoS with the appropriate weighting functions. In the 2PT method, two parameters are extracted from the DoS self-consistently to describe diffusional contributions: the fraction of diffusional modes, f, and DoS(0). This allows 2PT to be applied consistently and without re-parameterization to simulations of arbitrary liquids. We find that the absolute entropy of the liquid can be determined accurately from a single short MD trajectory (20 ps) after the system is equilibrated, making it orders of magnitude more efficient than commonly used perturbation and umbrella sampling methods. Here, we present the predicted standard molar entropies for fifteen common solvents evaluated from molecular dynamics simulations using the AMBER, GAFF, OPLS AA/L and Dreiding II forcefields. Overall, we find that all forcefields lead to good agreement with experimental and previous theoretical values for the entropy and very good agreement in the heat capacities. These results validate 2PT as a robust and efficient method for evaluating the thermodynamics of liquid phase systems. Indeed 2PT might provide a practical scheme to improve the intermolecular terms in forcefields by comparing directly to thermodynamic properties.
Co-reporter:Mu-Jeng Cheng, Robert J. Nielsen, Jamil Tahir-Kheli and William A. Goddard III
Physical Chemistry Chemical Physics 2011 vol. 13(Issue 20) pp:9831-9838
Publication Date(Web):18 Apr 2011
DOI:10.1039/C0CP02777D
We have studied the magnetic structure of the high symmetry vanadyl pyrophosphate ((VO)2P2O7, VOPO), focusing on the spin exchange couplings, using density functional theory (B3LYP) with the full three-dimensional periodicity. VOPO involves four distinct spin couplings: two larger couplings exist along the chain direction (a-axis), which we predict to be antiferromagnetic, JOPO = −156.8 K and JO = −68.6 K, and two weaker couplings appear along the c (between two layers) and b directions (between two chains in the same layer), which we calculate to be ferromagnetic, Jlayer = 19.2 K and Jchain = 2.8 K. Based on the local density of states and the response of spin couplings to varying the cell parameter a, we found that JOPO originates from a super-exchange interaction through the bridging –O–P–O– unit. In contrast, JO results from a direct overlap of 3dx2 − y2 orbitals on two vanadium atoms in the same V2O8 motif, making it very sensitive to structural fluctuations. Based on the variations in V–O bond length as a function of strain along a, we found that the V–O bonds of V–(OPO)2–V are covalent and rigid, whereas the bonds of V–(O)2–V are fragile and dative. These distinctions suggest that compression along the a-axis would have a dramatic impact on JO, changing the magnetic structure and spin gap of VOPO. This result also suggests that assuming JO to be a constant over the range of 2–300 K whilst fitting couplings to the experimental magnetic susceptibility is an invalid method. Regarding its role as a catalyst, the bonding pattern suggests that O2 can penetrate beyond the top layers of the VOPO surface, converting multiple V atoms from the +4 to +5 oxidation state, which seems crucial to explain the deep oxidation of n-butane to maleic anhydride.
Co-reporter:Xiang Y. Liu, Kapil S. Lokare, Somesh K. Ganesh, Jason M. Gonzales, Jonas Oxgaard, William A. Goddard III and Roy A. Periana
Dalton Transactions 2011 vol. 40(Issue 1) pp:301-304
Publication Date(Web):11 Nov 2010
DOI:10.1039/C0DT00997K
Using tetradentate, dianionic ligands, several new rhodium complexes have been prepared. Some of these diamine-bis(phenolate) compounds, are active for C–H activation of benzene. These complexes are air and thermally stable. All four complexes were characterized by X-ray diffraction.
Co-reporter:Soo-Kyung Kim, Peter Fristrup, Ravinder Abrol, and William A. Goddard III
Journal of Chemical Information and Modeling 2011 Volume 51(Issue 12) pp:3262-3274
Publication Date(Web):October 29, 2011
DOI:10.1021/ci200435b
Histamine receptors (HRs) are excellent drug targets for the treatment of diseases, such as schizophrenia, psychosis, depression, migraine, allergies, asthma, ulcers, and hypertension. Among them, the human H3 histamine receptor (hH3HR) antagonists have been proposed for specific therapeutic applications, including treatment of Alzheimer’s disease, attention deficit hyperactivity disorder (ADHD), epilepsy, and obesity.(1) However, many of these drug candidates cause undesired side effects through the cross-reactivity with other histamine receptor subtypes. In order to develop improved selectivity and activity for such treatments, it would be useful to have the three-dimensional structures for all four HRs. We report here the predicted structures of four HR subtypes (H1, H2, H3, and H4) using the GEnSeMBLE (GPCR ensemble of structures in membrane bilayer environment) Monte Carlo protocol,(2) sampling ∼35 million combinations of helix packings to predict the 10 most stable packings for each of the four subtypes. Then we used these 10 best protein structures with the DarwinDock Monte Carlo protocol to sample ∼50 000 × 1020 poses to predict the optimum ligand–protein structures for various agonists and antagonists. We find that E2065.46 contributes most in binding H3 selective agonists (5, 6, 7) in agreement with experimental mutation studies. We also find that conserved E5.46/S5.43 in both of hH3HR and hH4HR are involved in H3/ H4 subtype selectivity. In addition, we find that M3786.55 in hH3HR provides additional hydrophobic interactions different from hH4HR (the corresponding amino acid of T3236.55 in hH4HR) to provide additional subtype bias. From these studies, we developed a pharmacophore model based on our predictions for known hH3HR selective antagonists in clinical study [ABT-239 1, GSK-189,254 2, PF-3654746 3, and BF2.649 (tiprolisant) 4] that suggests critical selectivity directing elements are: the basic proton interacting with D1143.32, the spacer, the aromatic ring substituted with the hydrophilic or lipophilic groups interacting with lipophilic pockets in transmembranes (TMs) 3–5–6 and the aliphatic ring located in TMs 2–3–7. These 3D structures for all four HRs should help guide the rational design of novel drugs for the subtype selective antagonists and agonists with reduced side effects.
Co-reporter:Soo-Kyung Kim, Youyong Li, Ravinder Abrol, Jiyoung Heo, and William A. Goddard III
Journal of Chemical Information and Modeling 2011 Volume 51(Issue 2) pp:420-433
Publication Date(Web):February 7, 2011
DOI:10.1021/ci100375b
Subtype 2 serotonin (5-hydroxytryptamine, 5-HT) receptors are major drug targets for schizophrenia, feeding disorders, perception, depression, migraines, hypertension, anxiety, hallucinogens, and gastrointestinal dysfunctions.(1) We report here the predicted structure of 5-HT2B and 5-HT2C receptors bound to highly potent and selective 5-HT2B antagonist PRX-08066 3, (pKi: 30 nM), including the key binding residues [V103 (2.53), L132 (3.29), V190 (4.60), and L347 (6.58)] determining the selectivity of binding to 5-HT2B over 5-HT2A. We also report structures of the endogenous agonist (5-HT) and a HT2B selective antagonist 2 (1-methyl-1−1,6,7,8-tetrahydro-pyrrolo[2,3-g]quinoline-5-carboxylic acid pyridine-3-ylamide). We examine the dynamics for the agonist- and antagonist-bound HT2B receptors in explicit membrane and water finding dramatically different patterns of water migration into the NPxxY motif and the binding site that correlates with the stability of ionic locks in the D(E)RY region.
Co-reporter:Yao Sha, Ted H. Yu, Boris V. Merinov, Pezhman Shirvanian, and William A. Goddard III
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 6) pp:572-576
Publication Date(Web):February 24, 2011
DOI:10.1021/jz101753e
We report the reaction pathways and barriers for the oxygen reduction reaction (ORR) on platinum, both for gas phase and in solution, based on quantum mechanics calculations (PBE-DFT) on semi-infinite slabs. We find a new mechanism in solution: O2 → 2Oad (Eact = 0.00 eV), Oad + H2Oad → 2OHad (Eact = 0.50 eV), OHad + Had → H2Oad (Eact = 0.24 eV), in which OHad is formed by the hydration of surface Oad. For the gas phase (hydrophilic phase of Nafion), we find that the favored step for activation of the O2 is Had + O2ad → HOOad (Eact = 0.30 eV) → HOad + Oad (Eact = 0.12 eV) followed by Oad + H2Oad → 2OHad (Eact = 0.23 eV), OHad + Had → H2Oad (Eact = 0.14 eV). This suggests that to improve the efficiency of ORR catalysts, we should focus on decreasing the barrier for Oad hydration while providing hydrophobic conditions for the OH and H2O formation steps.Keywords: DFT; fuel cells; ORR; PBE; platinum cathode; Poisson−Boltzmann solvation;
Co-reporter:Yuki Matsuda, Jamil Tahir-Kheli, and William A. Goddard , III
The Journal of Physical Chemistry C 2011 Volume 115(Issue 25) pp:12586-12591
Publication Date(Web):May 17, 2011
DOI:10.1021/jp106048u
The calculated band gaps reported previously for silicon nanowires (SiNW) have disagreed with the experimental values both in magnitude and in the behavior with radius. We resolve this discrepancy here. We report ab initio quantum mechanical calculations of hydrogen terminated Si [001] nanowires (H–SiNWs) as a function of diameter (d) and hydrogen coverage using the B3LYP density functional. For smaller diameters (d ≤ 1.9 nm) we find that the most stable surface is fully saturated with hydrogen leading to direct band gaps. For larger diameters, the surface dangling bonds are not saturated, leading to surface LUMO and HOMO states that lower the gap and lead to an indirect band gap. This transition from direct to indirect gap resolves the previous disagreement in the scaling of band gap with diameter. We conclude that the electronic properties of Si NW depend sensitively on controlling the diameter and surface hydrogen coverage.
Co-reporter:Albert C. Fahrenbach;Jonathan C. Barnes;Hao Li;Diego Benítez;Ashish N. Basuray;Lei Fang;Chi-Hau Sue;Gokhan Barin;Sanjeev K. Dey III;J. Fraser Stoddart
PNAS 2011 108 (51 ) pp:
Publication Date(Web):2011-12-20
DOI:10.1073/pnas.1109795108
In donor–acceptor mechanically interlocked molecules that exhibit bistability, the relative populations of the translational
isomers—present, for example, in a bistable [2]rotaxane, as well as in a couple of bistable [2]catenanes of the donor–acceptor
vintage—can be elucidated by slow scan rate cyclic voltammetry. The practice of transitioning from a fast scan rate regime
to a slow one permits the measurement of an intermediate redox couple that is a function of the equilibrium that exists between
the two translational isomers in the case of all three mechanically interlocked molecules investigated. These intermediate
redox potentials can be used to calculate the ground-state distribution constants, K. Whereas, (i) in the case of the bistable [2]rotaxane, composed of a dumbbell component containing π-electron-rich tetrathiafulvalene and dioxynaphthalene recognition sites for the ring component (namely, a tetracationic cyclophane,
containing two π-electron-deficient bipyridinium units), a value for K of 10 ± 2 is calculated, (ii) in the case of the two bistable [2]catenanes—one containing a crown ether with tetrathiafulvalene
and dioxynaphthalene recognition sites for the tetracationic cyclophane, and the other, tetrathiafulvalene and butadiyne recognition
sites—the values for K are orders (one and three, respectively) of magnitude greater. This observation, which has also been probed by theoretical
calculations, supports the hypothesis that the extra stability of one translational isomer over the other is because of the
influence of the enforced side-on donor–acceptor interactions brought about by both π-electron-rich recognition sites being part of a macrocyclic polyether.
Co-reporter:Wei-Guang Liu, Siddharth Dasgupta, Sergey V. Zybin, and William A. Goddard III
The Journal of Physical Chemistry A 2011 Volume 115(Issue 20) pp:5221-5229
Publication Date(Web):April 28, 2011
DOI:10.1021/jp202021s
We report quantum mechanics calculations (B3LYP flavor of density functional theory) to determine the chemical reaction mechanism underlying the hypergolic reaction of pure HNO3 with N,N,N′,N′-tetramethylethylenediamine (TMEDA) and N,N,N′,N′-tetramethylmethylenediamine (TMMDA). TMEDA and TMMDA are dimethyl amines linked by two CH2 groups or one CH2 group, respectively, but ignite very differently with HNO3. We explain this dramatic difference in terms of the role that N lone-pair electrons play in activating adjacent chemical bonds. We identify two key atomistic level factors that affect the ignition delay: (1) The exothermicity for formation of the dinitrate salt from TMEDA or TMMDA. With only a single CH2 group between basic amines, the diprotonation of TMMDA results in much stronger electrostatic repulsion, reducing the heat of dinitrate salt formation by 6.3 kcal/mol. (2) The reaction of NO2 with TMEDA or TMMDA, which is the step that releases the heat and reactive species required to propagate the reaction. Two factors of TMEDA promote the kinetics by providing routes with low barriers to oxidize the C: (a) formation of a stable intermediate with a C–C double bond and (b) the lower bond energy for breaking the C–C single bond (by 18 kcal/mol comparing to alkane) between two amines. Both factors would decrease the ignition delay for TMEDA versus TMMDA. The same factors also explain the shorter ignition delay of 1,4-dimethylpiperazine (DMPipZ) versus 1,3,5-trimethylhexahydro-1,3,5-triazine (TMTZ). These results indicate that TMEDA and DMPipZ are excellent green replacements for hydrazines as the fuel in bipropellants.
Co-reporter:Jose L. Mendoza-Cortes, Tod A. Pascal, and William A. Goddard III
The Journal of Physical Chemistry A 2011 Volume 115(Issue 47) pp:13852-13857
Publication Date(Web):October 12, 2011
DOI:10.1021/jp209541e
We designed 14 new covalent organic frameworks (COFs), which are expected to adsorb large amounts of methane (CH4) at 298 K and up to 300 bar. We have calculated their delivery uptake using grand canonical Monte Carlo (GCMC) simulations. We also report their thermodynamic stability based on 7.5 ns molecular dynamics simulations. Two new frameworks, COF-103-Eth-trans and COF-102-Ant, are found to exceed the DOE target of 180 v(STP)/v at 35 bar for methane storage. Their performance is comparable to the best previously reported materials: PCN-14 and Ni-MOF-74. Our results indicate that using thin vinyl bridging groups aid performance by minimizing the interaction methane-COF at low pressure. This is a new feature that can be used to enhance loading in addition to the common practice of adding extra fused benzene rings. Most importantly, this report shows that pure nonbonding interactions, van der Waals (vdW) and electrostatic forces in light elements (C, O, B, H, and Si), can rival the enhancement in uptake obtained for microporous materials derived from early transition metals.
Co-reporter:Sang Soo Han ; Seung-Hoon Choi ; III
The Journal of Physical Chemistry C 2011 Volume 115(Issue 8) pp:3507-3512
Publication Date(Web):February 9, 2011
DOI:10.1021/jp200321y
We use grand canonical Monte Carlo simulations with first principles based force fields to show that alkali metal (Li+, Na+, and K+)-doped zeolitic imidazolate frameworks (ZIFs) lead to significant improvement of H2 uptake at room temperature. For example, at 298 K and 100 bar, Li-ZIF-70 totally binds to 3.08 wt % H2, Na-ZIF-70 to 2.19 wt % H2, and K-ZIF-70 to 1.62 wt % H2, much higher than 0.74 wt % H2 for pristine ZIF-70. Thus, the dopant effect follows the order of Li-ZIF > Na-ZIF > K-ZIF, which correlates with the H2 binding energies to the dopants. Moreover, the total H2 uptake is higher at lower temperatures: 243 K > 273 K > 298 K. On the other hand, delivery H2 uptake, which is the difference between the total adsorption at the charging pressure (say 100 bar) and the discharging pressure (say 5 bar), is the important factor for practical on-board hydrogen storage in vehicles. We show that delivery H2 uptake leads to Na-ZIF-70 (1.37 wt %) > K-ZIF-70 (1.25 wt %) > Li-ZIF-70 (1.07 wt %) > ZIF-70 (0.68 wt %), which is different from the trend from the total and excess uptake. Moreover, the delivery uptake increases with increasing temperatures (i.e., 298 K > 273 K > 243 K)! To achieve high delivery H2 uptake at room temperature, the large free volume of ZIFs is required. We find that higher H2 binding energy needs not always lead to higher delivery H2 uptake.
Co-reporter:Lianchi Liu, Yi Liu, Sergey V. Zybin, Huai Sun, and William A. Goddard III
The Journal of Physical Chemistry A 2011 Volume 115(Issue 40) pp:11016-11022
Publication Date(Web):September 2, 2011
DOI:10.1021/jp201599t
The practical levels of density functional theory (DFT) for solids (LDA, PBE, PW91, B3LYP) are well-known not to account adequately for the London dispersion (van der Waals attraction) so important in molecular solids, leading to equilibrium volumes for molecular crystals ∼10–15% too high. The ReaxFF reactive force field is based on fitting such DFT calculations and suffers from the same problem. In the paper we extend ReaxFF by adding a London dispersion term with a form such that it has low gradients (lg) at valence distances leaving the already optimized valence interactions intact but behaves as 1/R6 for large distances. We derive here these lg corrections to ReaxFF based on the experimental crystal structure data for graphite, polyethylene (PE), carbon dioxide, and nitrogen and for energetic materials: hexahydro-1,3,5-trinitro-1,3,5-s-triazine (RDX), pentaerythritol tetranitrate (PETN), 1,3,5-triamino-2,4,6-trinitrobenzene (TATB), and nitromethane (NM). After this dispersion correction the average error of predicted equilibrium volumes decreases from 18.5 to 4.2% for the above systems. We find that the calculated crystal structures and equation of state with ReaxFF-lg are in good agreement with experimental results. In particular, we examined the phase transition between α-RDX and γ-RDX, finding that ReaxFF-lg leads to excellent agreement for both the pressure and volume of this transition occurring at ∼4.8 GPa and ∼2.18 g/cm3 density from ReaxFF-lg vs 3.9 GPa and ∼2.21 g/cm3 from experiment. We expect ReaxFF-lg to improve the descriptions of the phase diagrams for other energetic materials.
Co-reporter:Lianchi Liu, Chen Bai, and Huai Sun and William A. Goddard III
The Journal of Physical Chemistry A 2011 Volume 115(Issue 19) pp:4941-4950
Publication Date(Web):April 21, 2011
DOI:10.1021/jp110435p
We report the kinetic analysis and mechanism for the initial steps of pyrolysis and combustion of a new fuel material, 1,6-dicyclopropane-2,4-hexyne, that has enormous heats of pyrolysis and combustion, making it a potential high-energy fuel or fuel additive. These studies employ the ReaxFF force field for reactive dynamics (RD) simulations of both pyrolysis and combustion processes for both unimolecular and multimolecular systems. We find that both pyrolysis and combustion initiate from unimolecular reactions, with entropy-driven reactions being most important in both processes. Pyrolysis initiates with extrusion of an ethylene molecule from the fuel molecule and is followed quickly by isomerization of the fuel molecule, which induces additional radicals that accelerate the pyrolysis process. In the combustion process, we find three distinct mechanisms for the O2 attack on the fuel molecule: (1) attack on the cyclopropane, ring expanding to form the cyclic peroxide which then decomposes; (2) attack onto the central single bond of the diyne which then fissions to form two C5H5O radicals; (3) attack on the alkyne-cyclopropane moiety to form a seven-membered ring peroxide which then decomposes. Each of these unimolecular combustion processes releases energy that induces additional radicals to accelerate the combustion process. Here oxygen has major effects both as the radical acceptor and as the radical producer. We extract both the effective activation energy and the effective pre-exponential factor by kinetic analysis of pyrolysis and combustion from these ReaxFF simulations. The low value of the derived effective activation energy (26.18 kcal/mol for pyrolysis and 16.40 kcal/mol for combustion) reveals the high activity of this fuel molecule.
Co-reporter:Hyungjun Kim ; Seung Soon Jang ; Richard A. Kiehl
The Journal of Physical Chemistry C 2011 Volume 115(Issue 9) pp:3722-3730
Publication Date(Web):February 16, 2011
DOI:10.1021/jp1114916
We investigate here a possible mechanism for the room temperature negative differential resistance (NDR) in the Au/AN-OPE/RS/Hg self-assembled monolayer (SAM) system, where AN-OPE = 2′-amino,5′-nitro-oligo(phenylene ethynylene) and RS is a C14 alkyl thiolate. Kiehl and co-workers showed that this molecular system leads to NDR with hysteresis and sweep-rate-dependent position and amplitude in the NDR peak. To investigate a molecular basis for this interesting behavior, we combine first-principles quantum mechanics (QM) and mesoscale lattice Monte Carlo methods to simulate the switching as a function of voltage and voltage rate, leading to results consistent with experimental observations. This simulation shows how the structural changes at the microscopic level lead to the NDR and sweep-rate-dependent macroscopic I−V curve observed experimentally, suggesting a microscopic model that might aid in designing improved NDR systems.
Co-reporter:Guoyun Zhang ; Qi An ; III
The Journal of Physical Chemistry C 2011 Volume 115(Issue 5) pp:2320-2331
Publication Date(Web):January 10, 2011
DOI:10.1021/jp107382u
Enormous progress has been made over the past decade in developing bulk amorphous alloys with high strength and other mechanical properties while possessing improved glass forming and processing abilities. Particularly important in developing new systems is identifying the compositions most likely to lead to glass formation. We illustrate here for the Al−Ni system how Molecular Dynamics (MD) using a force field derived from first-principles quantum mechanics (QM) can be used to determine the optimum compositions for glass forming ability (GFA). Using the Two-Phase Thermodynamics (2PT) method to extract entropy and free energy directly from MD, we find that the GFA is closely related to (ΔGlc)Tg, the Gibbs free energy difference between the liquid state and crystal state at the glass transition temperature Tg. We find that the glass phase is preferred at compositions where (ΔGlc)Tg is small and where the equilibrium crystal structure is complex. For the AlNi alloys, our calculations suggest that the best glass-forming composition is 87.5% Al and 12.5% Ni. On the basis of the Honeycutt−Andersen type of local structure analysis of alloys in the liquid state, we propose an atomic scale explanation of GFA. Large GFA arises when there is a great difference in the atom bonding preference between different atom species.
Co-reporter:Julius T. Su;Hyungjun Kim III
PNAS 2011 Volume 108 (Issue 37 ) pp:
Publication Date(Web):2011-09-13
DOI:10.1073/pnas.1110322108
We recently developed the electron force field (eFF) method for practical nonadiabatic electron dynamics simulations of materials
under extreme conditions and showed that it gave an excellent description of the shock thermodynamics of hydrogen from molecules
to atoms to plasma, as well as the electron dynamics of the Auger decay in diamondoids following core electron ionization.
Here we apply eFF to the shock thermodynamics of lithium metal, where we find two distinct consecutive phase changes that
manifest themselves as a kink in the shock Hugoniot, previously observed experimentally, but not explained. Analyzing the
atomic distribution functions, we establish that the first phase transition corresponds to (i) an fcc-to-cI16 phase transition that was observed previously in diamond anvil cell experiments at low temperature and (ii) a second phase transition that corresponds to the formation of a new amorphous phase (amor) of lithium that is distinct
from normal molten lithium. The amorphous phase has enhanced valence electron-nucleus interactions due to localization of
electrons into interstitial locations, along with a random connectivity distribution function. This indicates that eFF can
characterize and compute the relative stability of states of matter under extreme conditions (e.g., warm dense matter).
Co-reporter:Tod A. Pascal;Yousung Jung
PNAS 2011 Volume 108 (Issue 29 ) pp:
Publication Date(Web):2011-07-19
DOI:10.1073/pnas.1108073108
The spontaneous filling of hydrophobic carbon nanotubes (CNTs) by water observed both experimentally and from simulations
is counterintuitive because confinement is generally expected to decrease both entropy and bonding, and remains largely unexplained.
Here we report the entropy, enthalpy, and free energy extracted from molecular dynamics simulations of water confined in CNTs
from 0.8 to 2.7-nm diameters. We find for all sizes that water inside the CNTs is more stable than in the bulk, but the nature
of the favorable confinement of water changes dramatically with CNT diameter. Thus we find (i) an entropy (both rotational and translational) stabilized, vapor-like phase of water for small CNTs (0.8–1.0 nm), (ii) an enthalpy stabilized, ice-like phase for medium-sized CNTs (1.1–1.2 nm), and (iii) a bulk-like liquid phase for tubes larger than 1.4 nm, stabilized by the increased translational entropy as the waters sample
a larger configurational space. Simulations with structureless coarse-grained water models further reveal that the observed
free energies and sequence of transitions arise from the tetrahedral structure of liquid water. These results offer a broad
theoretical basis for understanding water transport through CNTs and other nanostructures important in nanofluidics, nanofiltrations,
and desalination.
Co-reporter:William A. Goddard III;Lianchi Liu;Jonathan E. Mueller
Topics in Catalysis 2011 Volume 54( Issue 10-12) pp:659-668
Publication Date(Web):2011 July
DOI:10.1007/s11244-011-9688-8
We report here first-principles-based predictions of the structures, mechanisms, and activation barriers for propane activation by the M2 phase of the MoVNbTeO multi-metal oxide catalysts capable of the direct conversion of propane to acrylonitrile. Our approach is to combine extensive quantum mechanical (QM) calculations to establish the mechanisms for idealized representations of the surfaces for these catalytic systems and then to modify the parameters in the ReaxFF reactive force field for molecular dynamics (MD) calculations to describe accurately the activation barriers and reaction mechanisms of the chemical reactions over complex mixed metal oxides. The parameters for ReaxFF are derived entirely from QM without the use of empirical data so that it can be applied to novel systems on which there is little or no data. To understand the catalysis in these systems it is essential to determine the surface structures that control the surface chemistry. High quality three-dimensional (3D) Rietveld structures are now available for the M1 and M2 phases of the MoVNbTeO catalysts. However the details of the chemical mechanisms controlling selectivity and activity have remained elusive because the catalytically important sites in these Rietveld structures are occupied by mixtures of Mo and V atoms, obscuring the actual distributions of the metals and oxides at the active sites. To solve this problem we use a supercell of the Rietveld structure sufficiently large that all atoms can be whole, then we use Monte Carlo techniques based on ReaxFF to resolve these partial occupations into the optimum configuration of whole atoms still consistent with the X-ray data. We will report the ReaxFF resolved 3D structures for the M2 phase of the MoVNbTeO system. Using the resolved 3D structures we consider the distribution of sites on the important surfaces and carry out ReaxFF Reactive Dynamics (RD) calculations to follow the initial steps of the reactions. Such studies provide insights into the chemical reaction steps on MMO catalysts that should be useful in designing more selective and more active systems. We find that this suggests the critical role of the TeIV oxo chains for activating propene but not propane in the M2 phase. This suggests a new mechanism for this phase.
Co-reporter:Tod A. Pascal, Yi He, Shaoyi Jiang, and William A. Goddard III
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 14) pp:1757-1760
Publication Date(Web):June 29, 2011
DOI:10.1021/jz200760n
Hydrogels are highly flexible network polymers being developed as scaffolds for tissue engineering and joint replacement. Their mechanical properties depend largely on their water content. To determine the associated mechanical and thermodynamic properties, we apply the new two-phase thermodynamics method (2PT) to short, molecular dynamics (MD) trajectories of solvated carboxybetaine methacrylate (CBMA) hydrogels. The calculated optimum water content agrees well with recent experiments. We find that the thermodynamics is dominated by a competition between the enthalpy of tightly bound water molecules (which enhance the population of low-energy states of the hydrogel) and the entropy-driven formation of a quasi-liquid water phase in the void volume. These new insights into the role of water in stabilizing hydrophilic motifs is expected to guide design strategies aimed at creating hydrogels with improved performance.Keywords: biomaterials; confinement; entropy; free energy simulations; molecular dynamics; polymer matrix;
Co-reporter:Qi An, Glenn Garrett, Konrad Samwer, Yi Liu, Sergey V. Zybin, Sheng-Nian Luo, Marios D. Demetriou, William L. Johnson, and William A. Goddard III
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 11) pp:1320-1323
Publication Date(Web):May 12, 2011
DOI:10.1021/jz200351m
We demonstrate the stochastic nature of cavitation in a binary metallic liquid Cu46Zr54 during hydrostatic expansion by employing molecular dynamics (MD) simulations using a quantum mechanics (QM)-derived potential. The activation volume is obtained from MD simulations and transition-state theory. Extrapolation of the pressure dependence of the activation volume from our MD simulations to low tensile pressure agrees remarkably with macroscale cavitation experiments. We find that classical nucleation theory can predict the cavitation rate if we incorporate the Tolman length derived from the MD simulations.Keywords: activation volume; cavitation; CNT; stochastic; tolman;
Co-reporter:Hai Xiao, Jamil Tahir-Kheli, and William A. Goddard III
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 3) pp:212-217
Publication Date(Web):January 18, 2011
DOI:10.1021/jz101565j
Co-reporter:Jamil Tahir-Kheli and William A. Goddard III
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 18) pp:2326-2330
Publication Date(Web):August 25, 2011
DOI:10.1021/jz200916t
Cuprate high-temperature superconductors exhibit a pseudogap in the normal state that decreases monotonically with increasing hole doping and closes at x ≈ 0.19 holes per planar CuO2 while the superconducting doping range is 0.05 < x < 0.27 with optimal Tc at x ≈ 0.16. Using ab initio quantum calculations at the level that leads to accurate band gaps, we found that four-Cu-site plaquettes are created in the vicinity of dopants. At x ≈ 0.05, the plaquettes percolate, so that the Cu dx2y2/O pσ orbitals inside the plaquettes now form a band of states along the percolating swath. This leads to metallic conductivity and, below Tc, to superconductivity. Plaquettes disconnected from the percolating swath are found to have degenerate states at the Fermi level that split and lead to the pseudogap. The pseudogap can be calculated by simply counting the spatial distribution of isolated plaquettes, leading to an excellent fit to experiment. This provides strong evidence in favor of inhomogeneous plaquettes in cuprates.Keywords: cuprates; percolation; plaquette; pseudogap; QM; superconductivity; theory;
Co-reporter:Sang Soo Han, Ted H. Yu, Boris V. Merinov, Adri C. T. van Duin, Rachid Yazami and William A. Goddard III
Chemistry of Materials 2010 Volume 22(Issue 6) pp:2142
Publication Date(Web):February 12, 2010
DOI:10.1021/cm903760t
Using density functional theory (DFT) calculations, we have studied structural models of graphite fluorides for five fluorine compositions; C1F (CF1), C2F (CF0.5), C3F (CF0.33), C4F (CF0.25), and C16F (CF0.0625). For each composition, we considered several possible structural models and calculated heat of formation relative to the pristine graphite and F2 molecule. We also simulated X-ray diffraction patterns for each structural model and compared those with experiments. We find, in agreement with earlier experiments, that the most stable structure of the C1F (CF1) has an infinite array of trans-linked cyclohexane chairs of covalent C−F bonds (1.38 Å). We also find that the effects of the layer stacking sequence such as AB or AA′ is not significant. For the C2F (CF0.5) system, an earlier model in the literature indicated that all carbon atoms have only sp3 hybridization due to coexistence of C−C and C−F covalent bonds. However, in this work, we propose a new C2F (CF0.5) crystal structure in which half of the carbon atoms has sp3 hybridization due to C−F covalent bonds and the other half has sp2 hybridization as found in pristine graphite. Besides, with the structural models of graphite fluorides considered in this work, their formation mechanism is also clarified. Initially, two fluorines are positioned at adjacent carbon atoms with the trans geometry, and then, one graphene layer is fully covered with fluorine while other layers are still pristine. After full coverage of the graphene layer, newly added fluorine will get located on this pristine graphene layer, and then, finally, all carbon layers are covered with fluorine leading to formation of C1F crystal. Through this mechanism, we can explain phase transition from the C2F to the C1F through further fluorination, which was demonstrated by an earlier experiment.
Co-reporter:Yuki Matsuda ; Wei-Qiao Deng ; III
The Journal of Physical Chemistry C 2010 Volume 114(Issue 41) pp:17845-17850
Publication Date(Web):September 23, 2010
DOI:10.1021/jp806437y
In this paper, we predict the current−voltage (I−V) characteristics and contact resistance of “end-contacted” metal electrode−graphene and metal electrode−carbon nanotube (CNT) interfaces for five metals, Ti, Pd, Pt, Cu, and Au, based on the first-principles quantum mechanical (QM) density functional and matrix Green’s function methods. We find that the contact resistance (normalized to surface C atoms) is 107 kΩ for Ti, 142 kΩ for Pd, 149 kΩ for Pt, 253 kΩ for Cu, and 187 kΩ for Au. This can be compared with the contact resistance (per C) for “side-contacted” metal−graphene or metal−CNT interfaces of 8.6 MΩ for Pd, 34.7 MΩ for Pt, 630 MΩ for Cu, etc. Those are in good agreement with available experimental results, 40.5 MΩ for Pt, for example. Thus, compared to the values for side-contacted interfaces from QM, we find a decrease in contact resistance by factors ranging from 6751 for Au and 2488 for Cu, to 233 for Pt and 60 Pd, to 8.8 for Ti. This suggests a strong advantage for developing technology to achieve “end-contacted” configurations.
Co-reporter:Sanja Pudar, Jonas Oxgaard, and William A. Goddard III
The Journal of Physical Chemistry C 2010 Volume 114(Issue 37) pp:15678-15694
Publication Date(Web):2017-2-22
DOI:10.1021/jp103054x
In order to understand the mechanism for selective ammoxidation of propene to acrylonitrile by bismuth molybdates, we report quantum mechanical studies (using the B3LYP flavor of density functional theory) for the various steps involved in converting the allyl-activated intermediate to acrylonitrile over molybdenum oxide (using a Mo3O9 cluster model) under conditions adjusted to describe both high and low partial pressures of NH3 in the feed. We find that the rate-determining step in converting of allyl to acrylonitrile at all feed partial pressures is the second hydrogen abstraction from the nitrogen-bound allyl intermediate (Mo−NH−CH2−CH═CH2) to form Mo−NH═CH−CH═CH2). We find that imido groups (Mo═NH) have two roles: (1) a direct effect on H abstraction barriers, H abstraction by an imido moiety is (∼8 kcal/mol) more favorable than abstraction by an oxo moiety (Mo═O), and (2) an indirect effect, the presence of spectator imido groups decreases the H abstraction barriers by an additional ∼15 kcal/mol. Therefore, at higher NH3 pressures (which increases the number of Mo═NH groups), the second H abstraction barrier decreases significantly, in agreement with experimental observations that propene conversion is higher at higher partial pressures of NH3. At high NH3 pressures we find that the final hydrogen abstraction has a high barrier [ΔH‡fourth-ab = 31.6 kcal/mol compared to ΔH‡second-ab = 16.4 kcal/mol] due to formation of low Mo oxidation states in the final state. However, we find that reoxidizing the surface prior to the last hydrogen abstraction leads to a significant reduction of this barrier to ΔH‡fourth-ab = 15.9 kcal/mol, so that this step is no longer rate determining. Therefore, we conclude that reoxidation during the reaction is necessary for facile conversion of allyl to acrylonitrile.
Co-reporter:Jonathan E. Mueller, Adri C. T. van Duin and William A. Goddard III
The Journal of Physical Chemistry C 2010 Volume 114(Issue 12) pp:5675-5685
Publication Date(Web):January 27, 2010
DOI:10.1021/jp9089003
We report here reactive dynamics (RD) simulations of the adsorption and decomposition of a gas of 20−120 methane, ethyne, ethene, benzene, cyclohexane, or propene molecules interacting with a 21 Å diameter nickel nanoparticle (468 atoms). These RD simulations use the recently developed ReaxFF reactive force field to describe decomposition, reactivity, and desorption of hydrocarbons as they interact with nickel surfaces. We carried out 100 ps of RD as the temperature is ramped at a constant rate from 500 to 2500 K (temperature programmed reactions). We find that all four unsaturated hydrocarbon species chemisorb to the catalyst particle with essentially no activation energy (attaching to the surface through π electrons) and then proceed to decompose by breaking C−H bonds to form partially dehydrogenated species prior to decomposition to lower order hydrocarbons. The eventual breaking of C−C bonds usually involves a surface Ni atom inserting into the C−C bond to produce an atomic C that simultaneously with C−C cleavage moves into the subsurface layer of the particle. The greater stability of this subsurface atomic C (forming up to four Ni−C bonds) over adatom C on the particle surface (forming at most three Ni−C bonds) is critical for favorable cleaving of C−C bonds. For the two saturated hydrocarbon species (methane and cyclohexane), we observe an activation energy associated with dissociative chemisorption. These results are consistent with available experimental reactivity data and quantum mechanics (QM) energy surfaces, validating the accuracy of ReaxFF for studying hydrocarbon decomposition on nickel clusters.
Co-reporter:Yao Sha, Ted H. Yu, Yi Liu, Boris V. Merinov and William A. Goddard III
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 5) pp:856-861
Publication Date(Web):February 15, 2010
DOI:10.1021/jz9003153
Co-reporter:Yuki Matsuda, Jamil Tahir-Kheli, and William A. Goddard III
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 19) pp:2946-2950
Publication Date(Web):September 20, 2010
DOI:10.1021/jz100889u
We report ab initio quantum mechanical calculations of band structures of single-walled carbon nanotubes (SWNTs) using the B3LYP flavor of density functional theory. In particular, we find excellent agreement with the small band gaps in “metallic” zigzag SWNTs observed by Lieber et al. [0.079 vs 0.080 eV for (9,0), 0.041 vs 0.042 eV for (12,0), and 0.036 eV vs 0.029 eV for (15,0)]. This contrasts with the results from LDA and PBE, which lead to band gaps 70−100% too small, and with those from the GW correction to LDA, which leads to a gap two times too large. Interestingly we find that the (5,0) system, expected to be a large gap semiconductor, is metallic. These results show that B3LYP leads to very accurate band gaps for CNTs, suggesting its use in designing CNT devices. We find that the effective mass of the CNT (significant in designing CNT devices) scales inversely proportional to the square of the diameter.Keywords (keywords): B3LYP; band gaps; chiral; DFT; nanoelectronics;
Co-reporter:Jamil Tahir-Kheli and William A. Goddard III
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 8) pp:1290-1295
Publication Date(Web):April 1, 2010
DOI:10.1021/jz100265k
We report that four properties of cuprates and their evolution with doping are consequences of simply counting four-site plaquettes arising from doping, (1) the universal Tc phase diagram (superconductivity between ∼0.05 and ∼0.27 doping per CuO2 plane and optimal Tc at ∼0.16), (2) the universal doping dependence of the room-temperature thermopower, (3) the superconducting neutron spin resonance peak (the “41 meV peak”), and (4) the dispersionless scanning tunneling conductance incommensurability. Properties (1), (3), and (4) are explained with no adjustable parameters, and (2) is explained with exactly one. The successful quantitative interpretation of four very distinct aspects of cuprate phenomenology by a simple counting rule provides strong evidence for four-site plaquette percolation in these materials. This suggests that inhomogeneity, percolation, and plaquettes play an essential role in cuprates. This geometric analysis may provide a useful guide to search for new compositions and structures with improved superconducting properties.Keywords (keywords): cuprates; incommensurability; percolation; QM; Seebeck; superconductivity;
Co-reporter:Steven M. Bischof, Daniel H. Ess, Steven K. Meier, Jonas Oxgaard, Robert J. Nielsen, Gaurav Bhalla, William A. Goddard III and Roy A. Periana
Organometallics 2010 Volume 29(Issue 4) pp:742-756
Publication Date(Web):January 26, 2010
DOI:10.1021/om900036j
The mechanism of benzene C−H bond activation by [Ir(μ-acac-O,O,C3)(acac-O,O)(OAc)]2 (4) and [Ir(μ-acac-O,O,C3)(acac-O,O)(TFA)]2 (5) complexes (acac = acetylacetonato, OAc = acetate, and TFA = trifluoroacetate) was studied experimentally and theoretically. Hydrogen−deuterium (H/D) exchange between benzene and CD3COOD solvent catalyzed by 4 (ΔH‡ = 28.3 ± 1.1 kcal/mol, ΔS‡ = 3.9 ± 3.0 cal K−1 mol−1) results in a monotonic increase of all benzene isotopologues, suggesting that once benzene coordinates to the iridium center, there are multiple H/D exchange events prior to benzene dissociation. B3LYP density functional theory (DFT) calculations reveal that this benzene isotopologue pattern is due to a rate-determining step that involves acetate ligand dissociation and benzene coordination, which is then followed by heterolytic C−H bond cleavage to generate an iridium-phenyl intermediate. A synthesized iridium-phenyl intermediate was also shown to be competent for H/D exchange, giving similar rates to the proposed catalytic systems. This mechanism nicely explains why hydroarylation between benzene and alkenes is suppressed in the presence of acetic acid when catalyzed by [Ir(μ-acac-O,O,C3)(acac-O,O)(acac-C3)]2 (3) (Matsumoto et al. J. Am. Chem. Soc. 2000, 122, 7414). Benzene H/D exchange in CF3COOD solvent catalyzed by 5 (ΔH‡ = 15.3 ± 3.5 kcal/mol, ΔS‡ = −30.0 ± 5.1 cal K−1 mol−1) results in significantly elevated H/D exchange rates and the formation of only a single benzene isotopologue, (C6H5D). DFT calculations show that this is due to a change in the rate-determining step. Now equilibrium between coordinated and uncoordinated benzene precedes a single rate-determining heterolytic C−H bond cleavage step.
Co-reporter:Jonathan E. Mueller, Adri C. T. van Duin, and William A. Goddard, III
The Journal of Physical Chemistry C 2010 Volume 114(Issue 47) pp:20028-20041
Publication Date(Web):November 4, 2010
DOI:10.1021/jp105513g
Competing, coverage-dependent pathways for ethane (CH3CH3) decomposition on Ni(111) are proposed on the basis of quantum mechanics (QM) calculations, performed by using the PBE flavor of density functional theory (DFT), for all C2Hy species adsorbed to a periodically infinite Ni(111) surface. For CH2CH3, CHCH3, and CCH3, we find that the surface C is tetrahedral in each case, with the surface C forming bonds to one, two, or three Ni atoms with bond energies scaling nearly linearly (Ebond = 32.5, 82.7, and 130.8 kcal/mol, respectively). In each of the remaining six C2Hy species, both C atoms are able to form bonds to the surface. Three of these (CH2CH2, CHCH2, and CCH2) adsorb most favorably at a fcc-top site with the methylene C located at an on-top site and the other C at an adjacent fcc site. The bond energies for these species are Ebond = 19.7, 63.2, and 93.6 kcal/mol, respectively. The remaining species (CHCH, CCH, and C2) all prefer binding at fcc-hcp sites, where the C atoms sit in a pair of adjacent fcc and hcp sites, with binding energies of Ebond = 57.7, 120.4, and 162.8 kcal/mol, respectively. We find that CHCHad is the most stable surface species (ΔHeth = −18.6), and an important intermediate along the lowest-energy decomposition pathway for ethane on Ni(111). The second most stable species, CCH3, is a close competitor (ΔHeth = −18.2 kcal/mol), lying along an alternative decomposition pathway that is preferred for high-surface-coverage conditions. The existence of these competing, low- and high-coverage decomposition pathways is consistent with the experiments. The QM results reported here were used as training data in the development of the ReaxFF reactive force field describing hydrocarbon reactions on nickel surfaces [Mueller, J. E.; van Duin, A: C. T.; Goddard, W. A. J. Phys. Chem. C 2010, 114, 4939−4949]. This has enabled Reactive dynamics studying the chemisorption and decomposition of systems far too complex for quantum mechanics. Thus we reported recently, the chemisorption and decomposition of six different hydrocarbon species on a Ni468 nanoparticle catalysts using this ReaxFF description [Mueller, J. E.; van Duin, A: C. T.; Goddard, W. A. J. Phys. Chem. C 2010, 114, 5675−5685].
Co-reporter:Soo-Kyung Kim Dr.;Youyong Li Dr.;Changmoon Park ;Ravinder Abrol Dr.;WilliamA. Goddard III
ChemMedChem 2010 Volume 5( Issue 9) pp:1594-1608
Publication Date(Web):
DOI:10.1002/cmdc.201000175
Abstract
Urotensin-II (U-II) has been shown to be the most potent mammalian vasoconstrictor known. Thus, a U-II antagonist might be of therapeutic value in a number of cardiovascular disorders. However, interspecies variability of several nonpeptidic ligands complicates the interpretation of in vivo studies of such antagonists in preclinical animal disease models. ACT058362 is a selective antagonist for the human U-II receptor (hUT2R) with a reported Kd value of ∼4 nM in a molecular binding assay, but it is reported to bind weakly to rat UT2R (rUT2R), with a Kd value of ∼1 500 nM. In contrast, the arylsulphonamide SB706375 is a selective antagonist against both hUT2R (Kd=∼9 nM) and rUT2R (Kd=∼21 nM). To understand the species selectivity of the UT2R, we investigated the binding site of ACT058362 and SB706375 in both hUT2R and rUT2R to explain the dramatically lower (∼400-fold) affinity of ACT058362 for rUT2R and the similar affinity (∼10 nM) of SB706375 for both UT2Rs. These studies used MembStruk and MSCDock to predict the UT2R structure and the binding site of ACT058362 and SB706375. Based on binding energies, we found two binding modes each with D1303.32 as the crucial anchoring point (Ballesteros–Weinstein numbering given in superscript). We predict that ACT058362 (an aryl–amine–aryl or ANA ligand) binds in the transmembrane (TM) 3456 region, while SB706375 (an aryl–aryl–amine or AAN ligand) binds in the TM 1237 region. These predicted sites explain the known differences in binding of the ANA ligand to rat and human receptors, while explaining the similar binding of the AAN compound to rat and human receptors. Moreover the predictions explain currently available structure–activity relationship (SAR) data. To further validate the predicted binding sites of these ligands in hUT2R and rUT2R, we propose several mutations that would help define the structural origins of differential responses between UT2R of different species, potentially indicating novel UT2R antagonists with cross-species high affinity.
Co-reporter:José L. Mendoza-Cortés, Sang Soo Han, Hiroyasu Furukawa, Omar M. Yaghi, and William A. Goddard III
The Journal of Physical Chemistry A 2010 Volume 114(Issue 40) pp:10824-10833
Publication Date(Web):September 16, 2010
DOI:10.1021/jp1044139
We determined the methane (CH4) uptake (at 298 K and 1 to 100 bar pressure) for a variety of covalent organic frameworks (COFs), including both two-dimensional (COF-1, COF-5, COF-6, COF-8, and COF-10) and three-dimensional (COF-102, COF-103, COF-105, and COF-108) systems. For all COFs, the CH4 uptake was predicted from grand canonical Monte Carlo (GCMC) simulations based on force fields (FF) developed to fit accurate quantum mechanics (QM) [second order Møller−Plesset (MP2) perturbation theory using doubly polarized quadruple-ζ (QZVPP) basis sets]. This FF was validated by comparison with the equation of state for CH4 and by comparison with the experimental uptake isotherms at 298 K (reported here for COF-5 and COF-8), which agrees well (within 2% for 1−100 bar) with the GCMC simulations. From our simulations we have been able to observe, for the first time, multilayer formation coexisting with a pore filling mechanism. The best COF in terms of total volume of CH4 per unit volume COF absorbent is COF-1, which can store 195 v/v at 298 K and 30 bar, exceeding the U.S. Department of Energy target for CH4 storage of 180 v/v at 298 K and 35 bar. The best COFs on a delivery amount basis (volume adsorbed from 5 to 100 bar) are COF-102 and COF-103 with values of 230 and 234 v(STP: 298 K, 1.01 bar)/v, respectively, making these promising materials for practical methane storage.
Co-reporter:Daniel H. Ess, William A. Goddard III, and Roy A. Periana
Organometallics 2010 Volume 29(Issue 23) pp:6459-6472
Publication Date(Web):October 29, 2010
DOI:10.1021/om100879y
The potential energy and interaction energy profiles for metal- and metal−ligand-mediated alkane C−H bond activation were explored using B3LYP density functional theory (DFT) and the absolutely localized molecular orbital energy decomposition analysis (ALMO-EDA). The set of complexes explored range from late transition metal group 10 (Pt and Pd) and group 11 (Au) metal centers to group 7−9 (Ir, Rh, Ru, and W) metal centers as well as a group 3 Sc complex. The coordination geometries, electron metal count (d8, d6, d4, and d0), and ligands (N-heterocycles, O-donor, phosphine, and Cp*) are also diverse. Quantitative analysis using ALMO-EDA of both directions of charge-transfer stabilization (occupied to unoccupied orbital stabilization) energies between the metal−ligand fragment and the coordinated C−H bond in the transition state for cleavage of the C−H bond allows classification of C−H activation reactions as electrophilic, ambiphilic, or nucleophilic on the basis of the net direction of charge-transfer energy stabilization. This bonding pattern transcends any specific mechanistic or bonding paradigm, such as oxidative addition, σ-bond metathesis, or substitution. Late transition metals such as Au(III), Pt(II), Pd(II), and Rh(III) metal centers with N-heterocycle, halide, or O-donor ligands show electrophilically dominated reaction profiles with forward charge-transfer from the C−H bond to the metal, leading to more stabilization than reverse charge transfer from the metal to the C−H bond. Transition states and reaction profiles for d6 Ru(II) and Ir(III) metals with Tp and acac ligands were found to have nearly equal forward and reverse charge-transfer energy stabilization. This ambiphilic region also includes the classically labeled electrophilic cationic species Cp*(PMe3)Ir(Me). Nucleophilic character, where the metal to C−H bond charge-transfer interaction is most stabilizing, was found in metathesis reactions with W(II) and Sc(III) metal center complexes in reactions as well as late transition metal Ir(I) and Rh(I) pincer complexes that undergo C−H bond insertion. Comparison of pincer ligands shows that the PCP ligand imparts more nucleophilic character to an Ir metal center than a deprotonated PNP ligand. The PCP and POCOP ligands do not show a substantial difference in the electronics of C−H activation. It was also found that Rh(I) is substantially more nucleophilic than Ir(I). Lastly, as a qualitative approximation, investigation of transition-state fragment orbital energies showed that relative frontier orbital energy gaps correctly reflect electrophilic, ambiphilic, or nucleophilic charge-transfer stabilization patterns.
Co-reporter:Yi Liu and William A. Goddard III
The Journal of Physical Chemistry Letters 2010 Volume 1(Issue 17) pp:2550-2555
Publication Date(Web):August 11, 2010
DOI:10.1021/jz100615g
Standard implementations of density functional theory (DFT) describe well strongly bound molecules and solids but fail to describe long-range van der Waals attractions. We propose here first-principles-based augmentation to DFT that leads to the proper long-range 1/R6 attraction of the London dispersion while leading to low gradients (small forces) at normal valence distances so that it preserves the accurate geometries and thermochemistry of standard DFT methods. The DFT-low gradient (DFT-lg) formula differs from previous DFT-D methods by using a purely attractive dispersion correction while not affecting valence bond distances. We demonstrate here that the DFT-lg model leads to good descriptions for graphite, benzene, naphthalene, and anthracene crystals, using just three parameters fitted to reproduce the full potential curves of high-level ab initio quantum mechanics [CCSD(T)] on gas-phase benzene dimers. The additional computational costs for this DFT-lg formalism are negligible.Keywords (keywords): density functional theory; DFT-D; dispersion; graphene; graphite; molecular crystals; van der Waals;
Co-reporter:Sang Soo Han, José L. Mendoza-Cortés and William A. Goddard III
Chemical Society Reviews 2009 vol. 38(Issue 5) pp:1460-1476
Publication Date(Web):24 Mar 2009
DOI:10.1039/B802430H
This critical review covers the application of computer simulations, including quantum calculations (ab initio and DFT), grand canonical Monte-Carlo simulations, and molecular dynamics simulations, to the burgeoning area of the hydrogen storage by metal–organic frameworks and covalent-organic frameworks. This review begins with an overview of the theoretical methods obtained from previous studies. Then strategies for the improvement of hydrogen storage in the porous materials are discussed in detail. The strategies include appropriate pore size, impregnation, catenation, open metal sites in metal oxide parts and within organic linker parts, doping of alkali elements onto organic linkers, substitution of metal oxide with lighter metals, functionalized organic linkers, and hydrogen spillover (186 references).
Co-reporter:Wei-Guang Liu ; Sergey V. Zybin ; Siddharth Dasgupta ; Thomas M. Klapötke III
Journal of the American Chemical Society 2009 Volume 131(Issue 22) pp:7490-7491
Publication Date(Web):May 12, 2009
DOI:10.1021/ja809725p
DFT calculations have identified the novel rearrangement shown here for decomposition of the Si derivative of the PETN explosive [PentaErythritol TetraNitrate (PETN), C(CH2ONO2)4] that explains the very dramatic increase in sensitivity observed experimentally. The critical difference is that Si-PETN allows a favorable five-coordinate transition state in which the new Si−O and C−O bonds form simultaneously, leading to a transition state barrier of 33 kcal/mol (it is 80 kcal/mol for PETN) and much lower than the normal O−NO2 bond fission observed in other energetic materials (∼40 kcal/mol). In addition this new mechanism is very exothermic (45 kcal/mol) leading to a large net energy release at the very early stages of Si-PETN decomposition that facilitates a rapid temperature increase and expansion of the reaction zone.
Co-reporter:Daniel H. Ess ; Robert J. Nielsen ; William A. Goddard III ;Roy A. Periana
Journal of the American Chemical Society 2009 Volume 131(Issue 33) pp:11686-11688
Publication Date(Web):August 4, 2009
DOI:10.1021/ja902748c
Absolutely localized molecular orbital energy decomposition analysis of C−H activation transition states (TSs), including Pt, Au, Ir, Ru, W, Sc, and Re metal centers, shows an electrophilic, ambiphilic, and nucleophilic charge transfer (CT) continuum irrespective of the bonding paradigm (oxidative addition, σ-bond metathesis, oxidative hydrogen migration, 1,2-substitution). Pt(II) insertion and Au(III) substitution TSs are highly electrophilic and dominated by C−H bond to metal/ligand orbital stabilization, while Ir−X and Ru−X (X = R, NH2, OR, or BOR2) substitution TSs are ambiphilic in nature. In this ambiphilic activation regime, an increase in one direction of CT typically leads to a decrease in the reverse direction. Comparison of Tp(CO)Ru−OH and Tp(CO)Ru−NH2 complexes showed no evidence for the classic dπ−pπ repulsion model. Complexes such as and Cp(CO)2W−B(OR)2, (PNP)Ir(I), Cp2ScMe, and (acac-κO,κO)2Re(III)−OH were found to mediate nucleophilic C−H activation, where the CT is dominated by the metal/ligand orbital to C−H antibonding orbital interaction. This CT continuum ultimately affects the metal−alkyl intermediate polarization and possible functionalization reactions. This analysis will impact the design of new activation reactions and stimulate the discovery of more nucleophilic activation complexes.
Co-reporter:Kenneth J. H. Young, Jonas Oxgaard, Daniel H. Ess, Steven K. Meier, Timothy Stewart, William A. Goddard, III and Roy A. Periana
Chemical Communications 2009 (Issue 22) pp:3270-3272
Publication Date(Web):23 Apr 2009
DOI:10.1039/B823303A
A discrete, air, protic, and thermally stable (NNC)Ir(III) pincer complex was synthesized that catalytically activates the CH bond of methane in trifluoroacetic acid; functionalization using NaIO4 and KIO3 gives the oxy-ester.
Co-reporter:Mårten Ahlquist, Roy A. Periana and William A. Goddard III
Chemical Communications 2009 (Issue 17) pp:2373-2375
Publication Date(Web):12 Mar 2009
DOI:10.1039/B821854D
Quantum mechanical (QM) results are used to establish the role of sulfuric acid solvent in facilitating the reaction between PtII(bpym)Cl2 (bpym = 2,2′-bipyrimidinyl) and methane; coordination of methane to the platinum catalyst is found to be catalyzed by the acidic medium.
Co-reporter:Diego Benitez, Ekaterina Tkatchouk, Ana Z. Gonzalez, William A. Goddard III and F. Dean Toste
Organic Letters 2009 Volume 11(Issue 21) pp:4798-4801
Publication Date(Web):September 25, 2009
DOI:10.1021/ol9018002
It is shown that [4 + 3] and [4 + 2] cycloaddition pathways are accessible in the Au(I) catalysis of allene−dienes. Seven-membered ring gold-stabilized carbenes, originating from the [4 + 3] cycloaddition process, are unstable and can rearrange via a 1,2-H or a 1,2-alkyl shift to yield six- and seven-membered products. Both steric and electronic properties of the AuL+ catalyst affect the electronic structure of the intermediate gold-stabilized carbene and its subsequent reactivity.
Co-reporter:Vyacheslav S. Bryantsev, Mamadou S. Diallo, Adri C. T. van Duin and William A. Goddard III
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 4) pp:1016-1026
Publication Date(Web):March 12, 2009
DOI:10.1021/ct800549f
In this paper we assess the accuracy of the B3LYP, X3LYP, and newly developed M06-L, M06-2X, and M06 functionals to predict the binding energies of neutral and charged water clusters including (H2O)n, n = 2−8, 20), H3O+(H2O)n, n = 1−6, and OH−(H2O)n, n = 1−6. We also compare the predicted energies of two ion hydration and neutralization reactions on the basis of the calculated binding energies. In all cases, we use as benchmarks calculated binding energies of water clusters extrapolated to the complete basis set limit of the second-order Møller−Plesset perturbation theory with the effects of higher order correlation estimated at the coupled-cluster theory with single, double, and perturbative triple excitations in the aug-cc-pVDZ basis set. We rank the accuracy of the functionals on the basis of the mean unsigned error (MUE) between calculated benchmark and density functional theory energies. The corresponding MUE (kcal/mol) for each functional is listed in parentheses. We find that M06-L (0.73) and M06 (0.84) give the most accurate binding energies using very extended basis sets such as aug-cc-pV5Z. For more affordable basis sets, the best methods for predicting the binding energies of water clusters are M06-L/aug-cc-pVTZ (1.24), B3LYP/6-311++G(2d,2p) (1.29), and M06/aug-cc-PVTZ (1.33). M06-L/aug-cc-pVTZ also gives more accurate energies for the neutralization reactions (1.38), whereas B3LYP/6-311++G(2d,2p) gives more accurate energies for the ion hydration reactions (1.69).
Co-reporter:Kimberly Chenoweth, David Chenoweth and William A. Goddard III
Organic & Biomolecular Chemistry 2009 vol. 7(Issue 24) pp:5255-5258
Publication Date(Web):09 Nov 2009
DOI:10.1039/B911482C
With the goal of identifying alkyne-like reagents for use in click chemistry, but without Cu catalysts, we used B3LYP density function theory (DFT) to investigate the trends in activation barriers for the 1,3-dipolar cycloadditions of azides with various cyclooctyne, dibenzocyclooctyne, and azacyclooctyne compounds. Based on these trends, we find monobenzocyclooctyne-based reagents that are predicted to have dramatically improved reactivity over currently employed reagents.
Co-reporter:Elodie Salmon, Adri C.T. van Duin, François Lorant, Paul-Marie Marquaire, William A. Goddard III
Organic Geochemistry 2009 Volume 40(Issue 3) pp:416-427
Publication Date(Web):March 2009
DOI:10.1016/j.orggeochem.2008.08.012
This paper reports ReaxFF MD simulation results on pyrolysis of a molecular model of the algaenan Botryococcus braunii race L biopolymer, specifically, ReaxFF predictions on the pyrolysis of prototypical chemical structures involving aliphatic chain esters and aldehydes. These preliminary computational experiments are then used to analyze the thermal cracking process within algaenan race L biopolymers. The simulations indicate that the thermal decomposition of the algaenan biopolymer is initiated by the cleavage of a C–O bond in the ester group, followed by the release of carbon dioxide. We also observe a significant, strongly temperature dependent, release of ethylene. This degradation mechanism leads to products similar to those observed in pyrolysis experiments, validating this computational approach.
Co-reporter:Jamil Tahir-Kheli, William A. Goddard III
Chemical Physics Letters 2009 Volume 472(4–6) pp:153-165
Publication Date(Web):20 April 2009
DOI:10.1016/j.cplett.2009.02.025
Abstract
A scientific revolution occurred in 1986–1994 in which the Tc for the best superconductors exploded from 23 K (Nb3Ge) to 138 K (Hg0.2Tl0.8Ba2Ca2Cu3O8.33). Despite enormous effort over the last 21 years, the superconducting mechanism remains unknown. All previous attempts assumed that the doped holes were in the CuO2 plane. We showed recently with improved quantum mechanics (QM) calculations that the hole is out of the CuO2 plane and delocalized over four Cu atoms in a square Plaquette that forms the basis of our Chiral Plaquette Polaron Paradigm (CPPP). Here, we show how very simple geometric arguments provide a qualitative understanding of the broad range of cuprate phenomenology. This simple geometric analysis may be useful in guiding the development of materials for improved superconductors. The CPPP suggests that judicious control of the dopant distribution could possibly lead to a room-temperature Tc.
Co-reporter:Julius T. Su III
PNAS 2009 Volume 106 (Issue 4 ) pp:1001-1005
Publication Date(Web):2009-01-27
DOI:10.1073/pnas.0812087106
To understand how core ionization and subsequent Auger decay lead to bond breaking in large systems, we simulate the wave
packet dynamics of electrons in the hydrogenated diamond nanoparticle C197H112. We find that surface core ionizations cause emission of carbon fragments and protons through a direct Auger mechanism, whereas
deeper core ionizations cause hydrides to be emitted from the surface via remote heating, consistent with results from photon-stimulated
desorption experiments [Hoffman A, Laikhtman A, (2006) J Phys Condens Mater 18:S1517–S1546]. This demonstrates that it is feasible to study the chemistry of highly excited large-scale systems using
simulation and analysis tools comparable in simplicity to those used for classical molecular dynamics.
Co-reporter:Kimberly Chenoweth, Adri C. T. van Duin, Siddharth Dasgupta and William A. Goddard III
The Journal of Physical Chemistry A 2009 Volume 113(Issue 9) pp:1740-1746
Publication Date(Web):February 11, 2009
DOI:10.1021/jp8081479
In order to investigate the initiation mechanisms and kinetics associated with the pyrolysis of JP-10 (exo-tricyclo[5.2.1.02,6]decane), a single-component hydrocarbon jet fuel, we carried out molecular dynamics (MD) simulations employing the ReaxFF reactive force field. We found that the primary decomposition reactions involve either (1) dissociation of ethylene from JP-10, resulting in the formation of a C8 hydrocarbon intermediate, or (2) the production of two C5 hydrocarbons. ReaxFF MD leads to good agreement with experiment for the product distribution as a function of temperature. On the basis of the rate of consumption of JP-10, we calculate an activation energy of 58.4 kcal/mol for the thermal decomposition of this material, which is consistent with a strain-facilitated C−C bond cleavage mechanism in JP-10. This compares well with the experimental value of 62.4 kcal/mol. In addition, we carried out ReaxFF MD studies of the reactive events responsible for oxidation of JP-10. Here we found overall agreement between the thermodynamic energies obtained from ReaxFF and quantum-mechanical calculations, illustrating the usefulness of ReaxFF for studying oxidation of hydrocarbons. The agreement of these results with available experimental observations demonstrates that ReaxFF can provide useful insights into the complicated thermal decomposition and oxidation processes of important hydrocarbon fuels.
Co-reporter:Hyungjun Kim, Wei-Qiao Deng and William A. Goddard, III, Seung Soon Jang, Mark E. Davis, Yushan Yan
The Journal of Physical Chemistry C 2009 Volume 113(Issue 3) pp:819-826
Publication Date(Web):2017-2-22
DOI:10.1021/jp804873s
To investigate the effect of hydration on the diffusion of sodium ions through the aluminum-doped zeolite BEA system (Si/Al = 30), we used the grand canonical Monte Carlo (GCMC) method to predict the water absorption into aluminosilicate zeolite structure under various conditions of vapor pressure and temperature, followed by molecular dynamics (MD) simulations to investigate how the sodium diffusion depends on the concentration of water molecules. The predicted absorption isotherm shows first-order-like transition, which is commonly observed in hydrophobic porous systems. The MD trajectories indicate that the sodium ions diffuse through zeolite porous structures via hopping mechanism, as previously discussed for similar solid electrolyte systems. These results show that above 15 wt % hydration (good solvation regime) the formation of the solvation cage dramatically increases sodium diffusion by reducing the hopping energy barrier by 25% from the value of 3.8 kcal/mol observed in the poor solvation regime.
Co-reporter:Kimberly Chenoweth ;AdriC.T. vanDuin ;WilliamA. Goddard III Dr.
Angewandte Chemie International Edition 2009 Volume 48( Issue 41) pp:7630-7634
Publication Date(Web):
DOI:10.1002/anie.200902574
Co-reporter:Steven K. Meier, Kenneth J. H. Young, Daniel H. Ess, William J. Tenn III, Jonas Oxgaard, William A. Goddard III and Roy A. Periana
Organometallics 2009 Volume 28(Issue 18) pp:5293-5304
Publication Date(Web):September 2, 2009
DOI:10.1021/om900039s
We report the synthesis of the pincer-cyclometalated (NNCt-Bu)Ir(III) dihydroxo pyridyl complex 6, which catalyzes hydrogen−deuterium (H/D) exchange between water and benzene in the presence of base (TOF = ∼6 × 10−3 s−1 at 190 °C). Experimental and density functional theory (B3LYP) studies suggest that H/D exchange occurs through loss of pyridine followed by benzene coordination and C−H bond activation by a heterolytic substitution mechanism to give a phenyl aquo complex, which may dimerize. Exchange of H2O for D2O followed by the microscopic reverse of CH activation leads to deuterium incorporation into benzene. Synthesis of the μ-hydroxo phenyl dinuclear complex [(NNCt-Bu)Ir(Ph)(μ-OH)]2 (9) also catalyzes H/D exchange with a turnover frequency (TOF = ∼7 × 10−3 s−1 at 190 °C) similar to that for 6.
Co-reporter:John A. Keith, Robert J. Nielsen, Jonas Oxgaard and William A. Goddard III, Patrick M. Henry
Organometallics 2009 Volume 28(Issue 6) pp:1618-1619
Publication Date(Web):February 20, 2009
DOI:10.1021/om800013p
Co-reporter:Kimberly Chenoweth ;AdriC.T. vanDuin ;WilliamA. Goddard III Dr.
Angewandte Chemie 2009 Volume 121( Issue 41) pp:7766-7770
Publication Date(Web):
DOI:10.1002/ange.200902574
Co-reporter:Elodie Salmon, Adri C.T. van Duin, François Lorant, Paul-Marie Marquaire, William A. Goddard III
Organic Geochemistry 2009 40(12) pp: 1195-1209
Publication Date(Web):
DOI:10.1016/j.orggeochem.2009.09.001
Co-reporter:Si-ping Han and William A. Goddard III
The Journal of Physical Chemistry B 2009 Volume 113(Issue 20) pp:7199-7204
Publication Date(Web):April 23, 2009
DOI:10.1021/jp805828g
Measurements of the radial breathing modes from Raman Spectroscopy have been most useful in characterizing the diameters of single-wall carbon nanotubes (SWNT), where there is a simple monotonic relationship between frequency and diameter. Similar correlations have also been used to predict sizes for double and multiple wall nanotubes and for bundles of SWNT. However this can lead to significant errors because the relationship between frequencies and diameter is much more complicated for DWNT. This is because of couplings between the vibrations of various walls. To provide guidance in such assignments we used the GraFF atomistic force field to predict the in-phase and counter-phase radial breathing modes (RBMs) of double wall carbon nanotubes (DWNTs) over a broad range of inner and outer diameters and chiralities. We then developed an analytical model to describe the RBMs of dispersed DWNTs. This enables the inner and outer shell diameters to be extracted from pairs of RBM peaks. We find that nanotubes bundles show significant dependent peak broadening and shifting compared to dispersed nanotubes. For bundles of SWNT and DWNT, the relationships are much more complicated.
Co-reporter:Shu-Hao Wen, An Li, Junling Song, Wei-Qiao Deng, Ke-Li Han and William A. Goddard III
The Journal of Physical Chemistry B 2009 Volume 113(Issue 26) pp:8813-8819
Publication Date(Web):June 9, 2009
DOI:10.1021/jp900512s
We report a simple first-principles-based simulation model (combining quantum mechanics with Marcus−Hush theory) that provides the quantitative structural relationships between angular resolution anisotropic hole mobility and molecular structures and packing. We validate that this model correctly predicts the anisotropic hole mobilities of ruberene, pentacene, tetracene, 5,11-dichlorotetracene (DCT), and hexathiapentacene (HTP), leading to results in good agreement with experiment.
Carbon Cluster Formation during Thermal Decomposition of Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine and 1,3,5-Triamino-2,4,6-trinitrobenzene High Explosives from ReaxFF Reactive Molecular Dynamics Simulations
Co-reporter:Luzheng Zhang, Sergey V. Zybin, Adri C. T. van Duin, Siddharth Dasgupta and William A. Goddard III, Edward M. Kober
The Journal of Physical Chemistry A 2009 Volume 113(Issue 40) pp:10619-10640
Publication Date(Web):October 1, 2009
DOI:10.1021/jp901353a
We report molecular dynamics (MD) simulations using the first-principles-based ReaxFF reactive force field to study the thermal decomposition of 1,3,5-triamino-2,4,6-trinitrobenzene (TATB) and octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) at various densities and temperatures. TATB is known to produce a large amount (15−30%) of high-molecular-weight carbon clusters, whereas detonation of nitramines such as HMX and RDX (1,3,5-trinitroperhydro-1,3,5-triazine) generate predominantly low-molecular-weight products. In agreement with experimental observation, these simulations predict that TATB decomposition quickly (by 30 ps) initiates the formation of large carbonaceous clusters (more than 4000 amu, or ∼15−30% of the total system mass), and HMX decomposition leads almost exclusively to small-molecule products. We find that HMX decomposes readily on this time scale at lower temperatures, for which the decomposition rate of TATB is about an order of magnitude slower. Analyzing the ReaxFF MD results leads to the detailed atomistic structure of this carbon-rich phase of TATB and allows characterization of the kinetics and chemistry related to this phase and their dependence on system density and temperature. The carbon-rich phase formed from TATB contains mainly polyaromatic rings with large oxygen content, leading to graphitic regions. We use these results to describe the initial reaction steps of thermal decomposition of HMX and TATB in terms of the rates for forming primary and secondary products, allowing comparison to experimentally derived models. These studies show that MD using the ReaxFF reactive force field provides detailed atomistic information that explains such macroscopic observations as the dramatic difference in carbon cluster formation between TATB and HMX. This shows that ReaxFF MD captures the fundamental differences in the mechanisms of such systems and illustrates how the ReaxFF may be applied to model complex chemical phenomena in energetic materials. The studies here illustrate this for modestly sized systems and modest periods; however, ReaxFF calculations of reactive processes have already been reported on systems with ∼106 atoms. Thus, with suitable computational facilities, one can study the atomistic level chemical processes in complex systems under extreme conditions.
Co-reporter:Vyacheslav S. Bryantsev, Mamadou S. Diallo and William A. Goddard III
The Journal of Physical Chemistry A 2009 Volume 113(Issue 34) pp:9559-9567
Publication Date(Web):August 5, 2009
DOI:10.1021/jp904816d
We use density functional theory (B3LYP) and the COSMO continuum solvent model to characterize the structure and stability of the hydrated Cu(II) complexes [Cu(MeNH2)(H2O)n−1]2+ and [Cu(OH)x(H2O)n−x]2−x (x = 1−3) as a function of metal coordination number (4−6) and cluster size (n = 4−8, 18). The small clusters with n = 4−8 are found to be the most stable in the nearly square-planar four-coordinate configuration, except for [Cu(OH)3(H2O)]−, which is three-coordinate. In the presence of the two full hydration shells (n = 18), however, the five-coordinate square-pyramidal geometry is the most favorable for Cu(MeNH2)2+ (5, 6) and Cu(OH)+ (5, 4, 6), and the four-coordinate geometry is the most stable for Cu(OH)2 (4, 5) and Cu(OH)3− (4). (Other possible coordination numbers for these complexes in the aqueous phase are shown in parentheses.) A small energetic difference between these structures (0.23−2.65 kcal/mol) suggests that complexes with different coordination numbers may coexist in solution. Using two full hydration shells around the Cu2+ ion (18 ligands) gives Gibbs free energies of aqueous reactions that are in excellent agreement with experiment. The mean unsigned error is 0.7 kcal/mol for the three consecutive hydrolysis steps of Cu2+ and the complexation of Cu2+ with methylamine. Conversely, calculations for the complexes with only one coordination shell (four equatorial ligands) lead to a mean unsigned error that is >6.0 kcal/mol. Thus, the explicit treatment of the first and the second shells is critical for the accurate prediction of structural and thermodynamic properties of Cu(II) species in aqueous solution.
Co-reporter:Yi Liu, Styliani Consta, Yujun Shi, R. H. Lipson and William A. Goddard III
The Journal of Physical Chemistry A 2009 Volume 113(Issue 25) pp:6865-6875
Publication Date(Web):June 2, 2009
DOI:10.1021/jp900487x
The size distributions and geometries of vapor clusters equilibrated with methanol−ethanol (Me−Et) liquid mixtures were recently studied by vacuum ultraviolet (VUV) laser time-of-flight (TOF) mass spectrometry and density functional theory (DFT) calculations (Liu, Y.; Consta, S.; Ogeer, F.; Shi, Y. J.; Lipson, R. H. Can. J. Chem. 2007, 85, 843−852). On the basis of the mass spectra recorded, it was concluded that the formation of neutral tetramers is particularly prominent. Here we develop grand canonical Monte Carlo (GCMC) and molecular dynamics (MD) frameworks to compute cluster size distributions in vapor mixtures that allow a direct comparison with experimental mass spectra. Using the all-atom optimized potential for liquid simulations (OPLS-AA) force field, we systematically examined the neutral cluster size distributions as functions of pressure and temperature. These neutral cluster distributions were then used to derive ionized cluster distributions to compare directly with the experiments. The simulations suggest that supersaturation at 12 to 16 times the equilibrium vapor pressure at 298 K or supercooling at temperature 240 to 260 K at the equilibrium vapor pressure can lead to the relatively abundant tetramer population observed in the experiments. Our simulations capture the most distinct features observed in the experimental TOF mass spectra: Et3H+ at m/z = 139 in the vapor corresponding to 10:90% Me−Et liquid mixture and Me3H+ at m/z = 97 in the vapors corresponding to 50:50% and 90:10% Me−Et liquid mixtures. The hybrid GCMC scheme developed in this work extends the capability of studying the size distributions of neat clusters to mixed species and provides a useful tool for studying environmentally important systems such as atmospheric aerosols.
Co-reporter:Jonathan E. Mueller, Adri C. T. van Duin and William A. Goddard III
The Journal of Physical Chemistry C 2009 Volume 113(Issue 47) pp:20290-20306
Publication Date(Web):October 29, 2009
DOI:10.1021/jp810555y
To provide a basis for understanding and improving such reactive processes on nickel surfaces as the catalytic steam reforming of hydrocarbons, the decomposition of hydrocarbons at fuel cell anodes, and the growth of carbon nanotubes, we report quantum mechanics calculations (PBE flavor of density functional theory, DFT) of the structures, binding energies, and reaction barriers for all CHx species on the Ni(111) surface using periodically infinite slabs. We find that all CHx species prefer binding to μ3 (3-fold) sites leading to bond energies ranging from 42.7 kcal/mol for CH3 to 148.9 kcal/mol for CH (the number of Ni−C bonds is not well-defined). We find reaction barriers of 18.3 kcal/mol for CH3,ad → CH2,ad + Had (with ΔE = +1.3 kcal/mol), 8.2 kcal/mol for CH2,ad → CHad + Had (with ΔE = −10.2 kcal/mol) and 32.3 kcal/mol for CHad → Cad + Had (with ΔE = 11.6 kcal/mol). Thus, CHad is the stable form of CHx on the surface. These results are in good agreement with the experimental data for the thermodynamic stability of small hydrocarbon species following dissociation of methane on Ni(111) and with the intermediates isolated during the reverse methanation process.
Co-reporter:Xin Xu III;Ying Zhang
PNAS 2009 Volume 106 (Issue 13 ) pp:4963-4968
Publication Date(Web):2009-03-31
DOI:10.1073/pnas.0901093106
We develop and validate a density functional, XYG3, based on the adiabatic connection formalism and the Görling–Levy coupling-constant
perturbation expansion to the second order (PT2). XYG3 is a doubly hybrid functional, containing 3 mixing parameters. It has
a nonlocal orbital-dependent component in the exchange term (exact exchange) plus information about the unoccupied Kohn–Sham
orbitals in the correlation part (PT2 double excitation). XYG3 is remarkably accurate for thermochemistry, reaction barrier
heights, and nonbond interactions of main group molecules. In addition, the accuracy remains nearly constant with system size.
Co-reporter:Diego Benitez, Ekaterina Tkatchouk and William A. Goddard III
Organometallics 2009 Volume 28(Issue 8) pp:2643-2645
Publication Date(Web):March 26, 2009
DOI:10.1021/om900041j
We settle a long-standing disagreement of DFT with experiment (both solution and gas phase) for the phosphine dissociation process in Grubbs metathesis catalysis. Our findings with the M06 functional provide further support to gas-phase experimental work, concluding that for the ring-closing metathesis of norbornene, the resting state is the alkylidene−olefin complex and the rate-determining step is the loss of norbornene as a ligand and generation of the 14-electron activated species. Comparing to recent solution NMR data on olefin−carbene Ru complexes relevant to olefin metathesis, we find that the M06 density functional leads to accurate predictions for the stability of conformers, ∼0.5 kcal/mol better than is found by B3LYP. Using this methodology, we suggest that Piers and co-workers observed the cis-dichloro “down” isomer exclusively following the ring opening of acenaphthalene.
Co-reporter:Luzheng Zhang, Adri C. T. van Duin, Sergey V. Zybin and William A. Goddard III
The Journal of Physical Chemistry B 2009 Volume 113(Issue 31) pp:10770-10778
Publication Date(Web):July 14, 2009
DOI:10.1021/jp900194d
We report reactive dynamics (RD) studies on: the decomposition of bulk hydrazine (N2H4); the decomposition of bulk monomethyl-hydrazine (CH3N2H3), hereafter referred to simply as methyl-hydrazine; the decomposition of hydrazine in the presence of hydrogen peroxide (H2O2); and decomposition hydrazine on catalytic surfaces Pt[100] and Pt[111] under various conditions. These studies use the ReaxFF reactive force field to describe the multitude of chemical reactions in these systems for a variety of reaction conditions in order to show that this approach leads to realistic decomposition mechanisms and rates. In particular, we determined how the decomposition of hydrazine is affected by temperature, pressure, and heating rate. We analyzed chemical reaction mechanism of the decomposition of hydrazine at the studied conditions and found that at lower temperatures the initial product from hydrazine decomposition is NH3, whereas at higher temperatures H2 and N2 are the dominant early products. Prominent intermediates observed during these decompositions include N2H3, N2H2, and NH2, in agreement with quantum mechanical studies (7.3 ps at 3000 K). As the heating rate is decreased, the onset for hydrazine decomposition shifts to lower temperatures. Using a constant heating rate, we found that higher pressure (increased density) favors formation of NH3 over N2 and H2. In studies of the catalytic decomposition of hydrazine on surfaces Pt[100] and Pt[111], we found that the presence of a Pt-catalyst reduces the initial decomposition temperature of hydrazine by about 50%. We found that the Pt[100]-surface is 20 times more active for hydrazine decomposition than the Pt[111]-surface, in qualitative agreement with experiments. These studies indicate how ReaxFF RD can be useful in understanding the chemical processes involved in bulk and catalytic decomposition and in oxidation of reactive species under various reaction conditions.
Co-reporter:Hyungjun Kim, William A. Goddard III, Seung Soon Jang, William R. Dichtel, James R. Heath and J. Fraser Stoddart
The Journal of Physical Chemistry A 2009 Volume 113(Issue 10) pp:2136-2143
Publication Date(Web):February 18, 2009
DOI:10.1021/jp809213m
Donor−acceptor binding of the π-electron-poor cyclophane cyclobis(paraquat-p-phenylene) (CBPQT4+) with the π-electron-rich tetrathiafulvalene (TTF) and 1,5-dioxynaphthalene (DNP) stations provides the basis for electrochemically switchable, bistable [2]rotaxanes, which have been incorporated and operated within solid-state devices to form ultradense memory circuits ( ChemPhysChem 2002, 3, 519−525; Nature 2007, 445, 414−417) and nanoelectromechanical systems. The rate of CBPQT4+ shuttling at each oxidation state of the [2]rotaxane dictates critical write-and-retention time parameters within the devices, which can be tuned through chemical synthesis. To validate how well computational chemistry methods can estimate these rates for use in designing new devices, we used molecular dynamics simulations to calculate the free energy barrier for the shuttling of the CBPQT4+ ring between the TTF and the DNP. The approach used here was to calculate the potential of mean force along the switching pathway, from which we calculated free energy barriers. These calculations find a turn-on time after the rotaxane is doubly oxidized of ∼10−7 s (suggesting that the much longer experimental turn-on time is determined by the time scale of oxidization). The return barrier from the DNP to the TTF leads to a predicted lifetime of 2.1 s, which is compatible with experiments.
Co-reporter:Diego Benitez, Ekaterina Tkatchouk and William A. Goddard III
Chemical Communications 2008 (Issue 46) pp:6194-6196
Publication Date(Web):23 Oct 2008
DOI:10.1039/B815665D
Using density functional theory with the B3LYP and M06 functionals, we show conclusively that the (H2IMes)(Cl)2Ru olefin metathesis mechanism is bottom-bound with the chlorides remaining trans throughout the reaction, thus attempts to effect diastereo- and enantioselectivity should focus on manipulations that maintain the trans-dichloro Ru geometry.
Co-reporter:Victor Wai Tak Kam and William A. Goddard III
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 12) pp:2160-2169
Publication Date(Web):November 19, 2008
DOI:10.1021/ct800196k
We present a new strategy for protein side-chain placement that uses flat-bottom potentials for rotamer scoring. The extent of the flat bottom depends on the coarseness of the rotamer library and is optimized for libraries ranging from diversities of 0.2 Å to 5.0 Å. The parameters reported here were optimized for forcefields using Lennard-Jones 12−6 van der Waals potential with DREIDING parameters but are expected to be similar for AMBER, CHARMM, and other forcefields. This Side-Chain Rotamer Excitation Analysis Method is implemented in the SCREAM software package. Similar scoring function strategies should be useful for ligand docking, virtual ligand screening, and protein folding applications.
Co-reporter:Jiyoung Heo, William W. Ja, Seymour Benzer and William A. Goddard III
Biochemistry 2008 Volume 47(Issue 48) pp:12740-12749
Publication Date(Web):November 7, 2008
DOI:10.1021/bi801335p
Peptide inhibitors of Methuselah (Mth), a G protein-coupled receptor (GPCR), were reported that can extend the life span of Drosophila melanogaster. Mth is a class B GPCR, which is characterized by a large, N-terminal ectodomain that is often involved with ligand recognition. The crystal structure of the Mth ectodomain, which binds to the peptide inhibitors with high affinity, was previously determined. Here we report the predicted structures for RWR motif peptides in complex with the Mth ectodomain. We studied representatives of both Pro-class and Arg-class RWR motif peptides and identified ectodomain residues Asp139, Phe130, Asp127, and Asp78 as critical in ligand binding. To validate these structures, we predicted the effects of various ligand mutations on the structure and binding to Mth. The binding of five mutant peptides to Mth was characterized experimentally by surface plasmon resonance, revealing measured affinities that are consistent with predictions. The electron density map calculated from our MD structure compares well with the experimental map of a previously determined peptide/Mth crystal structure and could be useful in refining the current low-resolution data. The elucidation of the ligand binding site may be useful in analyzing likely binding sites in other class B GPCRs.
Co-reporter:BrianL. Conley;SomeshK. Ganesh;JasonM. Gonzales Dr.;DanielH. Ess Dr.;RobertJ. Nielsen;VadimR. Ziatdinov Dr.;Jonas Oxgaard Dr.;WilliamA. Goddard III ;RoyA. Periana
Angewandte Chemie 2008 Volume 120( Issue 41) pp:7967-7970
Publication Date(Web):
DOI:10.1002/ange.200802575
Co-reporter:BrianL. Conley;SomeshK. Ganesh;JasonM. Gonzales Dr.;DanielH. Ess Dr.;RobertJ. Nielsen;VadimR. Ziatdinov Dr.;Jonas Oxgaard Dr.;WilliamA. Goddard III ;RoyA. Periana
Angewandte Chemie International Edition 2008 Volume 47( Issue 41) pp:7849-7852
Publication Date(Web):
DOI:10.1002/anie.200802575
Co-reporter:Christopher George, Hidehiro Yoshida and William A. Goddard III, Seung Soon Jang, Yong-Hoon Kim
The Journal of Physical Chemistry B 2008 Volume 112(Issue 47) pp:14888-14897
Publication Date(Web):October 23, 2008
DOI:10.1021/jp061759l
We combine first-principles density-functional theory with matrix Green’s function calculations to predict the structures and charge transport characteristics of self-assembled monolayers (SAMs) of four classes of systems in contact with Au(111) electrodes: conjugated polyene chains (n = 4, 8, 12, 16, and 30) thiolated at one or both ends and saturated alkane chains (n = 4, 8, 12, and 16) thiolated at one or both ends. For the polyene SAMs, we find no decay in the current as a function of chain length and conclude that these 1−3 nm long polyene SAMs act as metallic wires. We also find that the polyene-monothiolate leads to a contact resistance only 2.8 times higher than that for the polyene-dithiolate chains, indicating that the device conductance is dominated by the properties of the molecular connector with less importance in having a second molecule−electrode contact. For the alkane SAMs, we observe the normal exponential decay in the current as a function of the chain length with a decay constant of βn = 0.82 for the alkane-monothiolate and 0.88 for the alkane-dithiolate. We find that the contact resistance for the alkane-monothiolate is 12.5 times higher than that for the alkane-dithiolate chains, reflecting the extra resistance due to the weak contact on the nonthiolated end. These contrasting charge transport characteristics of alkane and polyene SAMs and their contact dependence are explained in terms of the atomic projected density of states.
Co-reporter:Vyacheslav S. Bryantsev, Mamadou S. Diallo and William A. Goddard III
The Journal of Physical Chemistry B 2008 Volume 112(Issue 32) pp:9709-9719
Publication Date(Web):July 23, 2008
DOI:10.1021/jp802665d
We derive a consistent approach for predicting the solvation free energies of charged solutes in the presence of implicit and explicit solvents. We find that some published methodologies make systematic errors in the computed free energies because of the incorrect accounting of the standard state corrections for water molecules or water clusters present in the thermodynamic cycle. This problem can be avoided by using the same standard state for each species involved in the reaction under consideration. We analyze two different thermodynamic cycles for calculating the solvation free energies of ionic solutes: (1) the cluster cycle with an n water cluster as a reagent and (2) the monomer cycle with n distinct water molecules as reagents. The use of the cluster cycle gives solvation free energies that are in excellent agreement with the experimental values obtained from studies of ion−water clusters. The mean absolute errors are 0.8 kcal/mol for H+ and 2.0 kcal/mol for Cu2+. Conversely, calculations using the monomer cycle lead to mean absolute errors that are >10 kcal/mol for H+ and >30 kcal/mol for Cu2+. The presence of hydrogen-bonded clusters of similar size on the left- and right-hand sides of the reaction cycle results in the cancelation of the systematic errors in the calculated free energies. Using the cluster cycle with 1 solvation shell leads to errors of 5 kcal/mol for H+ (6 waters) and 27 kcal/mol for Cu2+ (6 waters), whereas using 2 solvation shells leads to accuracies of 2 kcal/mol for Cu2+ (18 waters) and 1 kcal/mol for H+ (10 waters).
Co-reporter:Adri C. T. van Duin, Boris V. Merinov, Sang Soo Han, Claudio O. Dorso and William A. Goddard III
The Journal of Physical Chemistry A 2008 Volume 112(Issue 45) pp:11414-11422
Publication Date(Web):October 16, 2008
DOI:10.1021/jp801082q
Proton-conducting perovskites such as Y-doped BaZrO3 (BYZ) are promising candidates as electrolytes for a proton ceramic fuel cell (PCFC) that might permit much lower temperatures (from 400 to 600 °C). However, these materials lead to relatively poor total conductivity (∼10−4 S/cm) because of extremely high grain boundary resistance. In order to provide the basis for improving these materials, we developed the ReaxFF reactive force field to enable molecular dynamics (MD) simulations of proton diffusion in the bulk phase and across grain boundaries of BYZ. This allows us to elucidate the atomistic structural details underlying the origin of this poor grain boundary conductivity and how it is related to the orientation of the grains. The parameters in ReaxFF were based entirely on the results of quantum mechanics (QM) calculations for systems related to BYZ. We apply here the ReaxFF to describe the proton diffusion in crystalline BYZ and across grain boundaries in BYZ. The results are in excellent agreement with experiment, validating the use of ReaxFF for studying the transport properties of these membranes. Having atomistic structures for the grain boundaries from simulations that explain the overall effect of the grain boundaries on diffusion opens the door to in silico optimization of these materials. That is, we can now use theory and simulation to examine the effect of alloying on both the interfacial structures and on the overall diffusion. As an example, these calculations suggest that the reduced diffusion of protons across the grain boundary results from the increased average distances between oxygen atoms in the interface, which necessarily leads to larger barriers for proton hopping. Assuming that this is the critical issue in grain boundary diffusion, the performance of BYZ for multigranular systems might be improved using additives that would tend to precipitate to the grain boundary and which would tend to pull the oxygens atoms together. Possibilities might be to use a small amount of larger trivalent ions, such as La or Lu or of tetravalent ions such as Hf or Th. Since ReaxFF can also be used to describe the chemical processes on the anode and cathode and the migration of ions across the electrode-membrane interface, ReaxFF opens the door to the possibility of atomistic first principles predictions on models of a complete fuel cell.
Co-reporter:Vyacheslav S. Bryantsev, Mamadou S. Diallo, Adri C. T. van Duin and William A. Goddard III
The Journal of Physical Chemistry A 2008 Volume 112(Issue 38) pp:9104-9112
Publication Date(Web):September 3, 2008
DOI:10.1021/jp804373p
The hydrated structure of the Cu(II) ion has been a subject of ongoing debate in the literature. In this article, we use density functional theory (B3LYP) and the COSMO continuum solvent model to characterize the structure and stability of [Cu(H2O)n]2+ clusters as a function of coordination number (4, 5, and 6) and cluster size (n = 4−18). We find that the most thermodynamically favored Cu(II) complexes in the gas phase have a very open four-coordinate structure. They are formed from a stable square-planar [Cu(H2O)8]2+ core stabilized by an unpaired electron in the Cu(II) ion dx2−y2 orbital. This is consistent with cluster geometries suggested by recent mass-spectrometric experiments. In the aqueous phase, we find that the more compact five-coordinate square-pyramidal geometry is more stable than either the four-coordinate or six-coordinate clusters in agreement with recent combined EXAFS and XANES studies of aqueous solutions of Cu(II). However, a small energetic difference (∼1.4 kcal/mol) between the five- and six-coordinate models with two full hydration shells around the metal ion suggests that both forms may coexist in solution.
Co-reporter:Vyacheslav S. Bryantsev, Wibe A. de Jong, Kevin C. Cossel, Mamadou S. Diallo, William A. Goddard III, Gary S. Groenewold, Winnie Chien and Michael J. Van Stipdonk
The Journal of Physical Chemistry A 2008 Volume 112(Issue 26) pp:5777-5780
Publication Date(Web):June 10, 2008
DOI:10.1021/jp804202q
Theoretical calculations suggest a novel two-electron three-atom bonding scheme for complexation of O2 with U(V) compounds, leading to the stabilization of superoxo complexes in the side-on (η2) configuration. This binding motif is likely to play an important role in the oxidative processes involving trans-uranium compounds having valence 5fφ electrons.
Co-reporter:Jenelle K. Bray, William A. Goddard III
Journal of Molecular Graphics and Modelling 2008 Volume 27(Issue 1) pp:66-81
Publication Date(Web):August 2008
DOI:10.1016/j.jmgm.2008.02.006
We used the MembStruk computational procedure to predict the three-dimensional (3D) structure for the serotonin 5-HT2C G-protein-coupled receptor (GPCR). Using this structure, we used the MSCDock computational procedure to predict the 3D structures for bound ligand–protein complexes for agonists such as serotonin and antagonists such as ritanserin, metergoline, and methiothepin. In addition, we predicted the SAR data for a series of psilocybin analogs, both agonists and antagonists. We performed molecular dynamics (MD) on serotonin bound to 5-HT2C and we find the protein and binding site to be stable after 5 ns. We find good agreement with the currently known experimental data and we predict a number of new mutations which could be used to validate further our predicted structures. This agreement between theory and experiment suggests that our 3D structure is sufficiently accurate for use in drug design. We also compare a preliminary prediction for 5-HT2B with our prediction for 5-HT2C and find a difference in TM5 that contributes to different serotonin binding modes in 5-HT2B and 5-HT2C.
Co-reporter:Kimberly Chenoweth ; Adri C.T. van Duin ; Petter Persson ; Mu-Jeng Cheng ; Jonas Oxgaard ; III
The Journal of Physical Chemistry C 2008 Volume 112(Issue 37) pp:14645-14654
Publication Date(Web):July 25, 2008
DOI:10.1021/jp802134x
We have developed a new ReaxFF reactive force field to describe accurately reactions of hydrocarbons with vanadium oxide catalysts. The ReaxFF force field parameters have been fit to a large quantum mechanics (QM) training set containing over 700 structures and energetics related to bond dissociations, angle and dihedral distortions, and reactions between hydrocarbons and vanadium oxide clusters. In addition, the training set contains charge distributions for small vanadium oxide clusters and the stabilities of condensed-phase systems. We find that ReaxFF reproduces accurately the QM training set for structures and energetics of small clusters. Most important is that ReaxFF describes accurately the energetics for various oxidation states of the condensed phases, including V2O5, VO2, and V2O3 in addition to metallic V (V0). To demonstrate the capability of the ReaxFF force field for describing catalytic processes involving vanadium oxides, we performed molecular dynamics (MD) simulation for reactions of a gas of methanol exposed to the (001) surface of V2O5. We find that formaldehyde is the major product, in agreement with experiment. These studies find that water desorption from surface VIII sites is facilitated by interlayer bonding.
Co-reporter:Sang Soo Han and William A. Goddard III
The Journal of Physical Chemistry C 2008 Volume 112(Issue 35) pp:13431-13436
Publication Date(Web):2017-2-22
DOI:10.1021/jp800832b
Stimulated by the recent report by Yaghi and co-workers of hexagonal metal−organic frameworks (MOF) exhibiting reversible binding of up to 7.5 wt % at 77 K and 70 bar for MOF-177 (called here IRMOF-2-24), we have predicted additional trigonal organic linkers, including IRMOF-2-60, which we calculate to bind 9.7 wt % H2 storage at 77 K and 70 bar, the highest known value for 77 K. These calculations are based on grand canonical Monte Carlo (GCMC) simulations using force fields that match accurate quantum mechanical calculations on the binding of H2 to prototypical systems. These calculations were validated by comparison to the experimental loading curve for IRMOF-2-24 at 77K. We then used the theory to predict the effect of doping Li into the hexagonal MOFs, which leads to substantial H2 density even at ambient temperatures. For example, IRMOF-2-96-Li leads to 6.0 wt % H2 storage at 273 K and 100 bar, the first material to attain the 2010 DOE target.
Co-reporter:Yuki Matsuda ; Wei-Qiao Deng ; III
The Journal of Physical Chemistry C 2008 Volume 112(Issue 29) pp:11042-11049
Publication Date(Web):June 27, 2008
DOI:10.1021/jp8021776
It is anticipated that future nanoelectronic devices will utilize carbon nanotubes (CNT) and/or single graphene sheets (SGS) as the low-level on-chip interconnects or functional elements. Here we address the contact resistance of Cu for higher level on-chip interconnects with CNT or SGS elements. We use first-principles quantum mechanical (QM) density functional and matrix Green’s function methods to show that perfect Cu−SGS contact has a contact resistance of 16.3 MΩ for a one square nanometer contact. Then we analyzed possible improvements in contact resistance through incorporation of simple functional groups such as aryl (−C6H4−), acetylene (−CC−), carboxyl (−COO−), and amide (−CONH−), on CNT. We find that all four anchors enhance the interfacial mechanical stabilities and electrical conductivity. The best scenario is −COOH functionalized CNT which reduces the contact resistance to the Cu by a factor of 275 and increases the mechanical stability by 26 times.
Co-reporter:Yun Hee Jang ; III
The Journal of Physical Chemistry C 2008 Volume 112(Issue 23) pp:8715-8720
Publication Date(Web):May 14, 2008
DOI:10.1021/jp800201z
1,2-Dithiolane is a promising anchor group for attaching molecules to metal electrodes in molecular junction devices. This five-membered cyclic disulfide adsorbs on Au surfaces either in a cyclic fashion (with its disulfide bond intact, via molecular adsorption) or in an acyclic fashion (with its disulfide bond broken, via dissociative adsorption). Our density functional theory calculations show that the dissociative adsorption is slightly preferred, but both are stable. We also report nonequilibrium Green’s function calculations showing that molecular junctions of cyclic and acyclic 1,2-dithiolanes sandwiched between two gold electrodes exhibit essentially the same insulating current–voltage characteristics at moderate bias voltages, despite the significant difference in their states of adsorption.
Co-reporter:Daniel H. Ess, Steven M. Bischof, Jonas Oxgaard, Roy A. Periana and William A. Goddard III.
Organometallics 2008 Volume 27(Issue 24) pp:6440-6445
Publication Date(Web):November 13, 2008
DOI:10.1021/om8006568
Chelate-assisted and internal electrophilic substitution type transition states were studied using a DFT-based energy decomposition method. Interaction energies for benzene and methane C−H bond activation by (acac-O,O)2Ir(X) complexes (X = CH3COO and OH) were evaluated using the absolutely localized molecular orbital energy decomposition analysis (ALMO-EDA). A ratio of ∼1.5:1 for forward to reverse charge-transfer between (acac-O,O)2Ir(X) and benzene or methane transition state fragments confirms “ambiphilic” bonding, the result of an interplay between the electrophilic iridium center and the internal base component. This analysis also revealed that polarization effects account for a significant amount of transition state stabilization. The energy penalty to deform reactants into their transition state geometry, distortion energy, was also used to understand the large activation energy difference between six-membered and four-membered acetate-assisted transition states and help explain why these complexes do not activate the methane C−H bond.
Co-reporter:Zhitao Xu ; Jonas Oxgaard ;William Goddard
Organometallics 2008 Volume 27(Issue 15) pp:3770-3773
Publication Date(Web):July 16, 2008
DOI:10.1021/om800274f
Quantum mechanical studies on methane CH activation catalyzed by PtCl2 in concentrated H2SO4 and ionic liquid solution show that the effect of the ionic liquid is to enable Shilov-like chemistry in an oxidizing medium, by solvating the otherwise insoluble PtCl2(s) in H2SO4. Other possible mechanisms have been investigated and discarded.
Co-reporter:William A. Goddard III;Kimberly Chenoweth;Sanja Pudar
Topics in Catalysis 2008 Volume 50( Issue 1-4) pp:2-18
Publication Date(Web):2008 November
DOI:10.1007/s11244-008-9096-x
In order to determine the chemical mechanism for the (amm)oxidation of propane and propene on multi-metal oxide (MMO) catalysts, we have carried out quantum mechanical (QM) calculations for model reactions on small clusters that we have used to train the parameters for the ReaxFF reactive force field, which enables molecular dynamics (MD) simulations for reactions on the complex reconstructed surfaces of MMO. We report here insights from the QM on the reaction mechanisms of selective (amm)oxidation of propene on bismuth molybdate catalysts and the oxidative dehydrogenation of propane on vanadium oxide catalysts. We also report the application of ReaxFF to predict the stable surfaces of the M1 phases of the MoVTeNbO catalysts.
Co-reporter:Amos G. Anderson, William A. Goddard III, Peter Schröder
Computer Physics Communications 2007 Volume 177(Issue 3) pp:298-306
Publication Date(Web):1 August 2007
DOI:10.1016/j.cpc.2007.03.004
Quantum Monte Carlo (QMC) is among the most accurate methods for solving the time independent Schrödinger equation. Unfortunately, the method is very expensive and requires a vast array of computing resources in order to obtain results of a reasonable convergence level. On the other hand, the method is not only easily parallelizable across CPU clusters, but as we report here, it also has a high degree of data parallelism. This facilitates the use of recent technological advances in Graphical Processing Units (GPUs), a powerful type of processor well known to computer gamers. In this paper we report on an end-to-end QMC application with core elements of the algorithm running on a GPU. With individual kernels achieving as much as 30× speed up, the overall application performs at up to 6× faster relative to an optimized CPU implementation, yet requires only a modest increase in hardware cost. This demonstrates the speedup improvements possible for QMC in running on advanced hardware, thus exploring a path toward providing QMC level accuracy as a more standard tool. The major current challenge in running codes of this type on the GPU arises from the lack of fully compliant IEEE floating point implementations. To achieve better accuracy we propose the use of the Kahan summation formula in matrix multiplications. While this drops overall performance, we demonstrate that the proposed new algorithm can match CPU single precision.
Co-reporter:Sang Soo Han Dr.;Wei-Qiao Deng ;William A. Goddard III
Angewandte Chemie International Edition 2007 Volume 46(Issue 33) pp:
Publication Date(Web):18 JUL 2007
DOI:10.1002/anie.200700303
Quick on the uptake: The metal–organic framework Mg-MOF-C30 (see picture) contains Mg4O(CO2)6 building units (pink) and aromatic organic linkers containing 30 carbon atoms (teal). This material leads to 8.08 wt % H2 uptake at 77 K and 20 bar, the highest among investigated MOF structures.
Co-reporter:Sang Soo Han Dr.;Wei-Qiao Deng ;William A. Goddard III
Angewandte Chemie 2007 Volume 119(Issue 33) pp:
Publication Date(Web):18 JUL 2007
DOI:10.1002/ange.200700303
Gierig nach Wasserstoff: In dem metall-organischen Gerüst (MOF) Mg-MOF-C30 (siehe Bild) sind {Mg4O(CO2)6}-Bausteine (rosa) durch aromatische organische Brücken (cyan) mit 30 Kohlenstoffatomen verbunden. Mit 8.08 Gew.-% (bei 77 K und 20 bar H2) nimmt das Material mehr Wasserstoff auf als alle übrigen untersuchten MOF-Strukturen.
Co-reporter:Jiyoung Heo Dr.;Sang-Kyou Han Dr.;Nagarajan Vaidehi Dr.;John Wendel;Peter Kekenes-Huskey III
ChemBioChem 2007 Volume 8(Issue 13) pp:
Publication Date(Web):23 JUL 2007
DOI:10.1002/cbic.200700188
We report the 3D structure predicted for the mouse MrgC11 (mMrgC11) receptor by using the MembStruk computational protocol, and the predicted binding site for the F-M-R-F-NH2 neuropeptide together with four singly chirally modified ligands. We predicted that the R-F-NH2 part of the tetrapeptide sticks down into the protein between the transmembrane (TM) domains 3, 4, 5, and 6. The Phe (F-NH2) interacted favorably with Tyr110 (TM3), while the Arg makes salt bridges to Asp161 (TM4) and Asp179 (TM5). We predicted that the Met extends from the binding site, but the terminal Phe residue sticks back into an aromatic/hydrophobic site flanked by Tyr237, Leu238, Leu240, and Tyr256 (TM6), and Trp162 (TM4). We carried out subsequent mutagenesis experiments followed by intracellular calcium-release assays that demonstrated the dramatic decrease in activity for the Tyr110Ala, Asp161Ala, and Asp179Ala substitutions, which was predicted by our model. These experiments provide strong evidence that our predicted G protein-coupled receptor (GPCR) structure is sufficiently accurate to identify binding sites for selective ligands. Similar studies were made with the mMrgA1 receptor, which did not bind the R-F-NH2 dipeptide; we explain this to be due to the increased hydrophobic character of the binding pocket in mMrgA1.
Co-reporter:Jiyoung Heo Dr.;Sang-Kyou Han Dr.;Nagarajan Vaidehi Dr.;John Wendel;Peter Kekenes-Huskey III
ChemBioChem 2007 Volume 8(Issue 13) pp:
Publication Date(Web):27 AUG 2007
DOI:10.1002/cbic.200790042
The cover picture shows the predicted structure of the mouse MrgC11 G protein-coupled receptor (GPCR) with the predicted binding site for a signaling tetrapeptide (Phe-(D)Met-Arg-Phe-NH2, a member of the FMRFa family of signal peptides). MrgC11 belongs to the MAS-related gene family and is thought to be involved in pain sensation or modulation. These predictions led to the identification of several residues critical for ligand binding (see inset), while mutagenesis and intracellular-calcium-release experiments confirm the dramatic decrease in activity predicted for the Y110A, D161A, and D179A mutants. This validates the accuracy of these predicted structures, which can now be used for structure-based drug design to find small nonpeptide agonists and antagonists for mMrgC11 and perhaps to identify the endogenous ligand. This also validates the MembStruk methodology used in these predictions, which is equally applicable to all GPCR proteins. Details of the modeling, docking, and experimental verification are in the article by W. A. Goddart III et al. on p. 1527 ff.
Co-reporter:Joyce Yao-chun Peng;Nagarajan Vaidehi;Spencer E. Hall;William A. Goddard III
ChemMedChem 2006 Volume 1(Issue 8) pp:
Publication Date(Web):31 JUL 2006
DOI:10.1002/cmdc.200600047
The muscarinic acetylcholine G-protein-coupled receptors are implicated in diseases ranging from cognitive dysfunctions to smooth-muscle disorders. To provide a structural basis for drug design, we used the MembStruk computational method to predict the 3D structure of the human M1 muscarinic receptor. We validated this structure by using the HierDock method to predict the binding sites for three agonists and four antagonists. The intermolecular ligand–receptor contacts at the predicted binding sites agree well with deductions from available mutagenesis experiments, and the calculated relative binding energies correlate with measured binding affinities. The predicted binding site of all four antagonists is located between transmembrane (TM) helices 3, 4, 5, 6, and 7, whereas the three agonists prefer a site involving residues from TM3, TM6, and TM7. We find that Trp 157(4) contributes directly to antagonist binding, whereas Pro 159(4) provides an indirect conformational switch to position Trp 157(4) in the binding site (the number in parentheses indicates the TM helix). This explains the large decrease in ligand binding affinity and signaling efficacy by mutations of Trp 157(4) and Pro 159(4) not previously explained by homology models. We also found that Asp 105(3) and aromatic residues Tyr 381(6), Tyr 404(7), and Tyr 408(7) are critical for binding the quaternary ammonium head group of the ligand through cation–π interactions. For ligands with a charged tertiary amine head group, we suggest that proton transfer from the ligand to Asp 105(3) occurs upon binding. Furthermore, we found that an extensive aromatic network involving Tyr 106(3), Trp 157(4), Phe 197(5), Trp 378(6), and Tyr 381(6) is important in stabilizing antagonist binding. For antagonists with two terminal phenyl rings, this aromatic network extends to Trp 164(4), Tyr 179(extracellular loop 2), and Phe 390(6) located at the extracellular end of the TMs. We find that Asn 382(6) forms hydrogen bonds with selected antagonists. Tyr381(6) and Ser 109(3) form hydrogen bonds with the ester moiety of acetylcholine, which binds in the gauche conformation.
Co-reporter:Qingsong Zhang;Tahir Cagin III;
Proceedings of the National Academy of Sciences 2006 103(40) pp:14695-14700
Publication Date(Web):September 25, 2006
DOI:10.1073/pnas.0606612103
Using quantum mechanics (QM, Density Functional Theory) we show that all four phases of barium titanate (BaTiO3) have local Ti distortions toward 〈111〉 (an octahedral face). The stable rhombohedral phase has all distortions in phase
(ferroelectric, FE), whereas higher temperature phases have antiferroelectric coupling (AFE) in one, two, or three dimensions
(orthorhombic, tetragonal, cubic). This FE–AFE model from QM explains such puzzling aspects of these systems as the allowed
Raman excitation observed for the cubic phase, the distortions toward 〈111〉 observed in the cubic phase using x-ray fine structure,
the small transition entropies, the heavily damped soft phonon modes, and the strong diffuse x-ray scattering in all but the
rhombohedral phase. In addition, we expect to see additional weak Bragg peaks at the face centers of the reciprocal lattice
for the cubic phase. Similar FE–AFE descriptions are expected to occur for other FE materials. Accounting for this FE–AFE
nature of these phases is expected to be important in accurately simulating the domain wall structures, energetics, and dynamics,
which in turn may lead to the design of improved materials.
Co-reporter:Xin Xu III;
Proceedings of the National Academy of Sciences 2004 101(9) pp:2673-2677
Publication Date(Web):February 23, 2004
DOI:10.1073/pnas.0308730100
We derive the form for an exact exchange energy density for a density decaying with Gaussian-like behavior at long range.
Based on this, we develop the X3LYP (extended hybrid functional combined with Lee–Yang–Parr correlation functional) extended
functional for density functional theory to significantly improve the accuracy for hydrogen-bonded and van der Waals complexes
while also improving the accuracy in heats of formation, ionization potentials, electron affinities, and total atomic energies
[over the most popular and accurate method, B3LYP (Becke three-parameter hybrid functional combined with Lee–Yang–Parr correlation
functional)]. X3LYP also leads to a good description of dipole moments, polarizabilities, and accurate excitation energies
from s to d orbitals for transition metal atoms and ions. We suggest that X3LYP will be useful for predicting ligand binding
in proteins and DNA.
Co-reporter:M. Yashar S. Kalani;Nagarajan Vaidehi;Peter L. Freddolino;Victor Wai Tak Kam;Spencer E. Hall;Rene J. Trabanino;Maziyar A. Kalani III;Wely B. Floriano
PNAS 2004 Volume 101 (Issue 11 ) pp:3815-3820
Publication Date(Web):2004-03-16
DOI:10.1073/pnas.0400100101
Dopamine neurotransmitter and its receptors play a critical role in the cell signaling process responsible for information
transfer in neurons functioning in the nervous system. Development of improved therapeutics for such disorders as Parkinson's
disease and schizophrenia would be significantly enhanced with the availability of the 3D structure for the dopamine receptors
and of the binding site for dopamine and other agonists and antagonists. We report here the 3D structure of the long isoform
of the human D2 dopamine receptor, predicted from primary sequence using first-principles theoretical and computational techniques
(i.e., we did not use bioinformatic or experimental 3D structural information in predicting structures). The predicted 3D
structure is validated by comparison of the predicted binding site and the relative binding affinities of dopamine, three
known dopamine agonists (antiparkinsonian), and seven known antagonists (antipsychotic) in the D2 receptor to experimentally
determined values. These structures correctly predict the critical residues for binding dopamine and several antagonists,
identified by mutation studies, and give relative binding affinities that correlate well with experiments. The predicted binding
site for dopamine and agonists is located between transmembrane (TM) helices 3, 4, 5, and 6, whereas the best antagonists
bind to a site involving TM helices 2, 3, 4, 6, and 7 with minimal contacts to TM helix 5. We identify characteristic differences
between the binding sites of agonists and antagonists.
Co-reporter:Peter L. Freddolino;M. Yashar S. Kalani;Nagarajan Vaidehi;Wely B. Floriano;Spencer E. Hall;Rene J. Trabanino;Victor Wai Tak Kam III;
Proceedings of the National Academy of Sciences 2004 101(9) pp:2736-2741
Publication Date(Web):February 23, 2004
DOI:10.1073/pnas.0308751101
We report the 3D structure of human β2 adrenergic receptor (AR) predicted by using the MembStruk first principles method.
To validate this structure, we use the HierDock first principles method to predict the ligand-binding sites for epinephrine
and norepinephrine and for eight other ligands, including agonists and antagonists to β2 AR and ligands not observed to bind
to β2 AR. The binding sites agree well with available mutagenesis data, and the calculated relative binding energies correlate
reasonably with measured binding affinities. In addition, we find characteristic differences in the predicted binding sites
of known agonists and antagonists that allow us to infer the likely activity of other ligands. The predicted ligand-binding
properties validate the methods used to predict the 3D structure and function. This validation is a successful step toward
applying these procedures to predict the 3D structures and function of the other eight subtypes of ARs, which should enable
the development of subtype-specific antagonists and agonists with reduced side effects.
Co-reporter:Nagarajan Vaidehi;Wely B. Floriano;Rene Trabanino;Spencer E. Hall;Peter Freddolino;Eun Jung Choi;Georgios Zamanakos III;
Proceedings of the National Academy of Sciences 2002 99(20) pp:12622-12627
Publication Date(Web):September 26, 2002
DOI:10.1073/pnas.122357199
G protein-coupled receptors (GPCRs) mediate our sense of vision, smell, taste, and pain. They are also involved in cell recognition
and communication processes, and hence have emerged as a prominent superfamily for drug targets. Unfortunately, the atomic-level
structure is available for only one GPCR (bovine rhodopsin), making it difficult to use structure-based methods to design
drugs and mutation experiments. We have recently developed first principles methods (MembStruk and HierDock) for predicting
structure of GPCRs, and for predicting the ligand binding sites and relative binding affinities. Comparing to the one case
with structural data, bovine rhodopsin, we find good accuracy in both the structure of the protein and of the bound ligand.
We report here the application of MembStruk and HierDock to β1-adrenergic receptor, endothelial differential gene 6, mouse
and rat I7 olfactory receptors, and human sweet receptor. We find that the predicted structure of β1-adrenergic receptor leads
to a binding site for epinephrine that agrees well with the mutation experiments. Similarly the predicted binding sites and
affinities for endothelial differential gene 6, mouse and rat I7 olfactory receptors, and human sweet receptor are consistent
with the available experimental data. These predicted structures and binding sites allow the design of mutation experiments
to validate and improve the structure and function prediction methods. As these structures are validated they can be used
as targets for the design of new receptor-selective antagonists or agonists for GPCRs.
Co-reporter:Xin Xu III
PNAS 2002 99 (24 ) pp:15308-15312
Publication Date(Web):2002-11-26
DOI:10.1073/pnas.202596799
The recent observation [Wentworth, P., Jones, L. H., Wentworth, A. D., Zhu, X. Y., Larsen, N. A., Wilson, I. A., Xu, X., Goddard,
W. A., Janda, K. D., Eschenmoser, A. & Lerner, R. A. (2001) Science 293, 1806–1811] that antibodies form H2O2 from 1O2 plus H2O was explained in terms of the formation of the H2O3 species that in the antibody reacts with a second H2O3 to form H2O2. There have been few reports of the chemistry for forming H2O3, but recently Engdahl and Nelander [Engdahl, A. & Nelander, B. (2002) Science 295, 482–483] reported that photolysis of the ozone–hydrogen peroxide complex in argon matrices leads to significant concentrations
of H2O3. We report here the chemical mechanism for this process, determined by using first-principles quantum mechanics. We show
that in an argon matrix it is favorable (3.5 kcal/mol barrier) for H2O2 and O3 to form a [(HO2)(HO3)] hydrogen-bonded complex [head-to-tail seven-membered ring (7r)]. In this complex, the barrier for forming H2O3 plus 3O2 is only 4.8 kcal/mol, which should be observable by means of thermal processes (not yet reported). Irradiation of the [(HO2)(HO3)-7r] complex should break the HO–OO bond of the HO3 moiety, eliminating 3O2 and leading to [(HO2)(HO)]. This [(HO2)(HO)] confined in the matrix cage is expected to rearrange to also form H2O3 (observed experimentally). We show that these two processes can be distinguished isotopically. These results (including the
predicted vibrational frequencies) suggest strategies for synthesizing H2O3 and characterizing its chemistry. We suggest that the [(HO2)(HO3)-7r] complex and H2O3 are involved in biological, atmospheric, and environmental oxidative processes.
Co-reporter:Deqiang Zhang;Nagarajan Vaidehi III;Joseph F. Danzer;Derek Debe
PNAS 2002 Volume 99 (Issue 10 ) pp:6579-6584
Publication Date(Web):2002-05-14
DOI:10.1073/pnas.052150499
Although incorporation of amino acid analogs provides a powerful means of producing new protein structures with interesting
functions, many amino acid analogs cannot be incorporated easily by using the wild-type aminoacyl-tRNA synthetase (aaRS).
To be able to incorporate specific amino acid analogs site-specifically, it is useful to build a mutant aaRS that preferentially
activates the analog compared with the natural amino acids. Experimental combinatorial studies to find such mutant aaRSs have
been successful but can easily become costly and time-consuming. In this article, we describe the clash opportunity progressive
(COP) computational method for designing a mutant aaRS to preferentially take up the analog compared with the natural amino
acids. To illustrate this COP procedure, we apply it to the design of mutant Methanococcus jannaschii tyrosyl-tRNA synthetase (M.jann-TyrRS). Because the three-dimensional structure for M.jann-TyrRS was not available, we used the STRUCTFAST homology modeling procedure plus molecular dynamics with continuum solvent
forces to predict the structure of wild-type M.jann-TyrRS. We validate this structure by predicting the binding site for tyrosine and calculating the binding energies of the
20 natural amino acids, which shows that tyrosine binds the strongest. With the COP design algorithm we then designed a mutant
tyrosyl tRNA synthetase to activate O-methyl-l-tyrosine preferentially compared with l-tyrosine. This mutant [Y32Q, D158A] is similar to the mutant designed with combinatorial experiments, [Y32Q, D158A, E107T,
L162P], by Wang et al. [Wang, L., Brock, A., Herberich, B. & Schultz, P. G. (2001) Science 292, 498–500]. We predict that the new one will have much greater activity while retaining significant discrimination between
O-methyl-l-tyrosine and tyrosine.
Co-reporter:P.P. Knops-Gerrits, W.A. Goddard III
Journal of Molecular Catalysis A: Chemical 2001 Volume 166(Issue 1) pp:135-145
Publication Date(Web):22 January 2001
DOI:10.1016/S1381-1169(00)00460-X
The conversion of methane to methanol over zeolitic α-oxygen sites has been demonstrated using Fe-ZSM-5. To discriminate between mono- and poly-nuclear active sites, we prepared the [Fe]-ZEO with iron in the ZEOlite lattice via direct synthesis and Fex-ZEO, by dispersion of x wt.% iron on the ZEOlite. Shape-selective formation of nano-clusters of iron oxides with various sizes is realized inside the pore-sizes varying from 10.0 to 8.0 and 6.3 to 4.3 Å of the CFI, MOR, MFI, and CHA zeolites. The Fe–K edge X-ray absorption data were obtained for the Fe-CIT-5, Fe-ZSM-5, Fe-MOR and Fe-CHA zeolites containing iron clusters. In Mossbauer spectroscopy the absence and presence of a hyperfine magnetic field (HMF) for [Fe]-CIT-5 and Fe-CIT-5 are seen. The quantum mechanics calculations analyze the different environments of iron, e.g. the tetrahedral lattice occluded species, the di-nuclear sites attached to the zeolite, the nano-phase hematite sites. The molecular mechanics calculations involve a new molecular mechanics force field, the universal force field (UFF). α-Oxygen can be formed on di-nuclear iron sites in zeolites by N2O decomposition at elevated temperatures and is dependent on the zeolite structure utilized. Fe-chabazite (CHA), Fe-mordenite (MOR) and Fe-CIT-5 (CFI) were found to be less active than Fe-ZSM-5. A range of preparative and activation conditions were studied preceding methane conversion. Proper activation is essential to maximize catalyst actvity, e.g. pretreatment under vacuum at 800–900°C, activation with N2O at 250°C and reaction with methane at 20°C. Extraction of methanol from the catalyst is performed with H2O. Structure–activity effects are discussed.
Co-reporter:Malgorzata Witko, Peter-Paul Knops-Gerrits, Roberto Millini, William A. Goddard III
Journal of Molecular Catalysis A: Chemical 2001 Volume 166(Issue 1) pp:1-2
Publication Date(Web):22 January 2001
DOI:10.1016/S1381-1169(00)00454-4
Co-reporter:Wely B. Floriano;Michael S. Singer;Nagarajan Vaidehi;Gordon M. Shepherd III
PNAS 2000 Volume 97 (Issue 20 ) pp:10712-10716
Publication Date(Web):2000-09-26
DOI:10.1073/pnas.97.20.10712
The prevailing paradigm for G protein-coupled receptors is that
each receptor is narrowly tuned to its ligand and closely related
agonists. An outstanding problem is whether this paradigm applies to
olfactory receptor (ORs), which is the largest gene family in the
genome, in which each of 1,000 different G protein-coupled receptors is
believed to interact with a range of different odor molecules from the
many thousands that comprise “odor space.” Insights into how
these interactions occur are essential for understanding the sense of
smell. Key questions are: (i) Is there a binding pocket?
(ii) Which amino acid residues in the binding pocket
contribute to peak affinities? (iii) How do affinities
change with changes in agonist structure? To approach these questions,
we have combined single-cell PCR results [Malnic, B., Hirono, J.,
Sato, T. & Buck, L. B. (1999) Cell 96,
713–723] and well-established molecular dynamics methods to model the
structure of a specific OR (OR S25) and its interactions with 24 odor
compounds. This receptor structure not only points to a likely
odor-binding site but also independently predicts the two compounds
that experimentally best activate OR S25. The results provide a
mechanistic model for olfactory transduction at the molecular level and
show how the basic G protein-coupled receptor template is adapted for
encoding the enormous odor space. This combined approach can
significantly enhance the identification of ligands for the many
members of the OR family and also may shed light on other protein
families that exhibit broad specificities, such as chemokine receptors
and P450 oxidases.
Co-reporter:Hai Xiao, Andrés Jaramillo-Botero, Patrick L. Theofanis, William A. Goddard III
Mechanics of Materials (November 2015) Volume 90() pp:243-252
Publication Date(Web):1 November 2015
DOI:10.1016/j.mechmat.2015.02.008
•Introduce a unique method for modeling the excited dynamics of large-scale systems with explicit electrons.•Provide effective core pseudo potentials (ECP) in a non-adiabatic modeling framework for high-Z materials (including C, Si, O, Al, and others).•Extends the electron force field method (eFF), originally introduced by Su and Goddard (2007) for Z < 6, to Z > 6 and call it eFF–ECP.•Provide multiple validation of eFF–ECP against first principles quantum mechanics and experiments (e.g. hypervelocity impact, brittle fracture, among others).•The eFF–ECP is the only method available for modeling the dynamics of non-adiabatic system with millions of atoms over nanoseconds.Modeling non-adiabatic phenomena and materials at extremes has been a long-standing challenge for computational chemistry and materials science, particularly for systems that undergo irreversible phase transformations due to significant electronic excitations. Ab initio and existing quantum mechanics approximations to the Schrödinger equation have been limited to ground-state descriptions or few excited electronic states, less than 100 atoms, and sub-picosecond timescales of dynamics evolution. Recently, the electron force field (eFF) introduced by Su and Goddard (2007) presented a cost-efficient alternative to describe the dynamics of electronic and nuclear degrees of freedom. eFF describes an N-electron wave function as a Hartree product of one-electron floating spherical Gaussian wave packets propagating via the time-dependent Schrödinger equation under a mixed quantum–classical Hamiltonian evaluated as sum of self- and pairwise potential interactions. Local Pauli potential corrections replace the need for explicit anti-symmetrization of total electronic wavefunction, a wavefunction kinetic energy term accounts for Heisenberg’s uncertainty, and classical electrostatics complete the total eFF energy expression. However, due to the spherical symmetry of the underlying Gaussian basis functions, the original eFF formulation is limited to low-Z numbers with electrons of predominant s-character. To overcome this, we introduce here a formal set of potential form extensions that enable accurate description of p-block elements in the periodic table. The extensions consist of a model representing the core electrons of an atom together with the nucleus as a single pseudo particle with wave-like behavior, interacting with valence electrons, nuclei, and other cores through effective core pseudopotentials (ECPs). We demonstrate and validate the ECP extensions for complex bonding structures, geometries and energetics of systems with p-block character (containing silicon, oxygen, carbon, or aluminum atoms and combination thereof) and apply them to study materials under extreme mechanical loading conditions.
Co-reporter:William A. Goddard III, Soo-Kyung Kim, Youyong Li, Bartosz Trzaskowski, Adam R. Griffith, Ravinder Abrol
Journal of Structural Biology (April 2010) Volume 170(Issue 1) pp:10-20
Publication Date(Web):1 April 2010
DOI:10.1016/j.jsb.2010.01.001
G protein-coupled receptors (GPCRs) are therapeutic targets for many diseases, but progress in developing active and selective therapeutics has been severely hampered by the difficulty in obtaining accurate structures. We have been developing methods for predicting the structures for GPCR ligand complexes, but validation has been hampered by a lack of experimental structures with which to compare our predictions. We report here the predicted structures of the human adenosine GPCR subtypes (A1, A2A, A2B, and A3) and the binding sites for adenosine agonist and eight antagonists to this predicted structure, making no use of structural data, and compare with recent experimental crystal structure for ZM241385 bound human A2A receptor. The predicted structure correctly identifies 9 of the 12 crystal binding site residues. Moreover, the predicted binding energies of eight antagonists to the predicted structure of A2A correlate quite well with experiment. These excellent predictions resulted when we used Monte Carlo techniques to optimize the loop structures, particularly the cysteine linkages. Ignoring these linkages led to a much worse predicted binding site (identifying only 3 of the 12 important residues).These results indicate that computational methods can predict the three-dimensional structure of GPCR membrane proteins sufficiently accurately for use in designing subtype selective ligands for important GPCR therapeutics targets.
Co-reporter:Sijia S. Dong, William A. Goddard III, Ravinder Abrol
Biophysical Journal (21 June 2016) Volume 110(Issue 12) pp:
Publication Date(Web):21 June 2016
DOI:10.1016/j.bpj.2016.04.028
We present a hybrid computational methodology to predict multiple energetically accessible conformations for G protein-coupled receptors (GPCRs) that might play a role in binding to ligands and different signaling partners. To our knowledge, this method, termed ActiveGEnSeMBLE, enables the first quantitative energy profile for GPCR activation that is consistent with the qualitative profile deduced from experiments. ActiveGEnSeMBLE starts with a systematic coarse grid sampling of helix tilts/rotations (∼13 trillion transmembrane-domain conformations) and selects the conformational landscape based on energy. This profile identifies multiple potential active-state energy wells, with the TM3–TM6 intracellular distance as an approximate activation coordinate. These energy wells are then sampled locally using a finer grid to find locally minimized conformation in each energy well. We validate this strategy using the inactive and active experimental structures of β2 adrenergic receptor (hβ2AR) and M2 muscarinic acetylcholine receptor. Structures of membrane-embedded hβ2AR along its activation coordinate are subjected to molecular-dynamics simulations for relaxation and interaction energy analysis to generate a quantitative energy landscape for hβ2AR activation. This landscape reveals several metastable states along this coordinate, indicating that for hβ2AR, the agonist alone is not enough to stabilize the active state and that the G protein is necessary, consistent with experimental observations. The method’s application to somatostatin receptor SSTR5 (no experimental structure available) shows that to predict an active conformation it is better to start from an inactive structure template based on a close homolog than to start from an active template based on a distant homolog. The energy landscape for hSSTR5 activation is consistent with hβ2AR in the role of the G protein. These results demonstrate the utility of the ActiveGEnSeMBLE method for predicting multiple conformations along the pathways for activating GPCRs and the corresponding energy landscapes, thereby providing detailed structural insights into the initial molecular events of GPCR function that are not easily accessible by experiments.
Co-reporter:Michael S. Webster-Gardiner, Ross Fu, George C. Fortman, Robert J. Nielsen, T. Brent Gunnoe and William A. Goddard III
Catalysis Science & Technology (2011-Present) 2015 - vol. 5(Issue 1) pp:NaN100-100
Publication Date(Web):2014/10/20
DOI:10.1039/C4CY00972J
The Rh(I) complexes [(FlDAB)Rh(coe)(TFA)] (1) and [(BOZO)Rh(coe)(TFA)] (2) [FlDAB = N,N-bis-(pentafluorophenyl)-2,3-dimethyl-1,4-diaza-1,3-butadiene, coe = cyclooctene, TFA = trifluoroacetate, BOZO = bis(2-oxazolin-2-yl)] are efficient catalyst precursors for H/D exchange between arenes and DTFA. Catalyst precursor 1 exhibits a TOF of 0.06 s−1 at 150 °C for benzene H/D exchange. DFT calculations revealed that H/D exchange through reversible oxidative addition or internal electrophilic substitution of benzene is a viable pathway.
Co-reporter:Mu-Jeng Cheng, Ross Fu and William A. Goddard, III
Chemical Communications 2014 - vol. 50(Issue 14) pp:NaN1750-1750
Publication Date(Web):2013/12/18
DOI:10.1039/C3CC47502F
We use our recent discovery of the reduction-coupled oxo activation (ROA) principle to design a series of organometallic molecules that activate C–H bonds through this unique proton/electron-decoupled hydrogen abstraction mechanism, in which the main group oxo moiety binds to the proton while the electron is transferred to the transition metal. Here we illustrate this general class of catalyst clusters with several examples that are validated through quantum mechanics calculations.
Co-reporter:Mårten Ahlquist, Roy A. Periana and William A. Goddard III
Chemical Communications 2009(Issue 17) pp:
Publication Date(Web):
DOI:10.1039/B821854D
Co-reporter:Kenneth J. H. Young, Jonas Oxgaard, Daniel H. Ess, Steven K. Meier, Timothy Stewart, William A. Goddard, III and Roy A. Periana
Chemical Communications 2009(Issue 22) pp:NaN3272-3272
Publication Date(Web):2009/04/23
DOI:10.1039/B823303A
A discrete, air, protic, and thermally stable (NNC)Ir(III) pincer complex was synthesized that catalytically activates the CH bond of methane in trifluoroacetic acid; functionalization using NaIO4 and KIO3 gives the oxy-ester.
Co-reporter:Diego Benitez, Ekaterina Tkatchouk and William A. Goddard III
Chemical Communications 2008(Issue 46) pp:NaN6196-6196
Publication Date(Web):2008/10/23
DOI:10.1039/B815665D
Using density functional theory with the B3LYP and M06 functionals, we show conclusively that the (H2IMes)(Cl)2Ru olefin metathesis mechanism is bottom-bound with the chlorides remaining trans throughout the reaction, thus attempts to effect diastereo- and enantioselectivity should focus on manipulations that maintain the trans-dichloro Ru geometry.
Co-reporter:Alessandro Fortunelli, William A. Goddard III, Luca Sementa, Giovanni Barcaro, Fabio R. Negreiros and Andrés Jaramillo-Botero
Chemical Science (2010-Present) 2015 - vol. 6(Issue 7) pp:NaN3925-3925
Publication Date(Web):2015/04/29
DOI:10.1039/C5SC00840A
Recently Debe et al. reported that Pt3Ni7 leads to extraordinary Oxygen Reduction Reaction (ORR) activity. However, several reports show that hardly any Ni remains in the layers of the catalysts close to the surface (“Pt-skin effect”). This paradox that Ni is essential to the high catalytic activity with the peak ORR activity at Pt3Ni7 while little or no Ni remains close to the surface is explained here using large-scale first-principles-based simulations. We make the radical assumption that processing Pt–Ni catalysts under ORR conditions would leach out all Ni accessible to the solvent. To simulate this process we use the ReaxFF reactive force field, starting with random alloy particles ranging from 50% Ni to 90% Ni and containing up to ∼300000 atoms, deleting the Ni atoms, and equilibrating the resulting structures. We find that the Pt3Ni7 case and a final particle radius around 7.5 nm lead to internal voids in communication with the exterior, doubling the external surface footprint, in fair agreement with experiment. Then we examine the surface character of these nanoporous systems and find that a prominent feature in the surface of the de-alloyed particles is a rhombic structure involving 4 surface atoms which is crystalline-like but under-coordinated. Using density-functional theory, we calculate the energy barriers of ORR steps on Pt nanoporous catalysts, focusing on the Oad-hydration reaction (Oad + H2Oad → OHad + OHad) but including the barriers of O2 dissociation (O2ad → Oad + Oad) and water formation (OHad + Had → H2Oad). We find that the reaction barrier for the Oad-hydration rate-determining-step is reduced significantly on the de-alloyed surface sites compared to Pt(111). Moreover we find that these active sites are prevalent on the surface of particles de-alloyed from a Pt–Ni 30:70 initial composition. These simulations explain the peak in surface reactivity at Pt3Ni7, and provide a rational guide to use for further optimization of improved catalytic and nanoporous materials.
Co-reporter:Xiang Y. Liu, Kapil S. Lokare, Somesh K. Ganesh, Jason M. Gonzales, Jonas Oxgaard, William A. Goddard III and Roy A. Periana
Dalton Transactions 2011 - vol. 40(Issue 1) pp:NaN304-304
Publication Date(Web):2010/11/11
DOI:10.1039/C0DT00997K
Using tetradentate, dianionic ligands, several new rhodium complexes have been prepared. Some of these diamine-bis(phenolate) compounds, are active for C–H activation of benzene. These complexes are air and thermally stable. All four complexes were characterized by X-ray diffraction.
Co-reporter:Cai-Chao Ye, Qi An, William A. Goddard III, Tao Cheng, Wei-Guang Liu, Sergey V. Zybin and Xue-Hai Ju
Journal of Materials Chemistry A 2015 - vol. 3(Issue 5) pp:NaN1978-1978
Publication Date(Web):2014/11/25
DOI:10.1039/C4TA05676K
Di-tetrazine-tetroxide (DTTO) was predicted in 2001 to have a density (up to 2.3 g cm−3) and heat of detonation (up to 421.0 kcal mol−1) better than other explosives, making it the “holy grail” of energetic materials (EMs), but all attempts at synthesis have failed. We report Density Functional Theory (DFT) molecular dynamics simulations (DFT-MD) on DTTO crystal for the two most stable monomers. We predict that the most stable isomer (c1) has a density of ρ = 1.96 g cm−3 with an estimated detonation velocity (Dv) of 9.70 km s−1 and a detonation pressure (Dp) of 43.0 GPa, making it comparable to RDX (ρ = 1.82 g cm−3, Dv = 8.75 km s−1, Dp = 35.0 GPa), HMX (ρ = 1.91 g cm−3, Dv = 9.10 km s−1, Dp = 39.3 GPa) and CL-20 (ρ = 2.04 g cm−3, Dv = 9.38 km s−1, Dp = 44.1 GPa). The DFT-MD studies find that the initial reaction at lower pressure is unimolecular decomposition to form two N2O molecules (barrier 45.9 kcal mol−1), while at higher pressure it is intermolecular oxygen-transfer with a barrier of 40.1 kcal mol−1. For the c2 isomer (less stable by 1.2 kcal mol−1) the initial reaction involves two DTTO molecules reacting to form a dimer which then releases N2 as a direct product (barrier 48.1 kcal mol−1), a unique initial reaction among EMs. These results suggest that DTTO may have a higher thermal stability (barrier >7.0 kcal mol−1 higher) than RDX, HMX, and CL-20.
Co-reporter:Qi An, Jin Qian, Robert R. Nielsen, Luca Sementa, Giovanni Barcaro, Fabio R. Negreiros, Alessandro Fortunelli and William A. Goddard III
Journal of Materials Chemistry A 2016 - vol. 4(Issue 31) pp:NaN12045-12045
Publication Date(Web):2016/06/14
DOI:10.1039/C6TA03669D
Experimental evidence that surface acoustic waves (SAW) can significantly enhance the rate of catalytic oxidation of CO to CO2 over the Pt(110) catalyst surface [S. Kelling et al., Faraday Disc., 1997, 107, 435–444] is examined using quantum mechanics (QM) simulations. First we determined the QM based mechanism for the O2-rich régime of the reaction, and the energy landscape of CO interacting with an O-covered reconstructed Pt(110) surface at both static and dynamic levels, but in the absence of SAW. We then utilized ab initio molecular dynamic (AIMD) simulations to determine how SAW might modify the kinetics. We focus here on the short (picosecond time scale) shock spikes induced by switching of domains in the piezoelectric driver on which the catalyst is deposited. We find that SAW-induced spikes promote dynamic changes in the diffusion and desorption, from which we estimate the influence of SAW on CO oxidation rate over Pt(110). We find good agreement with the experimentally observed catalytic enhancement by SAW. With an atomistic mechanism in place one can now consider how to use SAW to enhance other catalytic reactions.
Co-reporter:Dezhou Guo, Sergey V. Zybin, Qi An, William A. Goddard III and Fenglei Huang
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 3) pp:NaN2022-2022
Publication Date(Web):2015/11/17
DOI:10.1039/C5CP04516A
The combustion or detonation of reacting materials at high temperature and pressure can be characterized by the Chapman–Jouguet (CJ) state that describes the chemical equilibrium of the products at the end of the reaction zone of the detonation wave for sustained detonation. This provides the critical properties and product kinetics for input to macroscale continuum simulations of energetic materials. We propose the ReaxFF Reactive Dynamics to CJ point protocol (Rx2CJ) for predicting the CJ state parameters, providing the means to predict the performance of new materials prior to synthesis and characterization, allowing the simulation based design to be done in silico. Our Rx2CJ method is based on atomistic reactive molecular dynamics (RMD) using the QM-derived ReaxFF force field. We validate this method here by predicting the CJ point and detonation products for three typical energetic materials. We find good agreement between the predicted and experimental detonation velocities, indicating that this method can reliably predict the CJ state using modest levels of computation.
Co-reporter:Kimberly Chenoweth, David Chenoweth and William A. Goddard III
Organic & Biomolecular Chemistry 2009 - vol. 7(Issue 24) pp:NaN5258-5258
Publication Date(Web):2009/11/09
DOI:10.1039/B911482C
With the goal of identifying alkyne-like reagents for use in click chemistry, but without Cu catalysts, we used B3LYP density function theory (DFT) to investigate the trends in activation barriers for the 1,3-dipolar cycloadditions of azides with various cyclooctyne, dibenzocyclooctyne, and azacyclooctyne compounds. Based on these trends, we find monobenzocyclooctyne-based reagents that are predicted to have dramatically improved reactivity over currently employed reagents.
Co-reporter:Tingting Zhou, Lianchi Liu, William A. Goddard III, Sergey V. Zybin and Fenglei Huang
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 43) pp:NaN23791-23791
Publication Date(Web):2014/09/17
DOI:10.1039/C4CP03781B
Recently quantum mechanical (QM) calculations on a single Si-PETN (silicon-pentaerythritol tetranitrate) molecule were used to explain its colossal sensitivity observed experimentally in terms of a unique Liu carbon-silyl nitro-ester rearrangement (R3Si–CH2–O–R2 → R3Si–O–CH2–R2). In this paper we expanded the study of Si-PETN from a single molecule to a bulk system by extending the ReaxFF reactive force field to describe similar Si–C–H–O–N systems with parameters optimized to reproduce QM results. The reaction mechanisms and kinetics of thermal decomposition of solid Si-PETN were investigated using ReaxFF reactive molecular dynamics (ReaxFF-RMD) simulations at various temperatures to explore the origin of the high sensitivity. We find that at lower temperatures, the decomposition of Si-PETN is initiated by the Liu carbon-silyl nitro-ester rearrangement forming Si–O bonds which is not observed in PETN. As the reaction proceeds, the exothermicity of Si–O bond formation promotes the onset of NO2 formation from N–OC bond cleavage which does not occur in PETN. At higher temperatures PETN starts to react by the usual mechanisms of NO2 dissociation and HONO elimination; however, Si-PETN remains far more reactive. These results validate the predictions from QM that the significantly increased sensitivity of Si-PETN arises from a unimolecular process involving the unusual Liu rearrangement but not from multi-molecular collisions. It is the very low energy barrier and the high exothermicity of the Si–O bond formation providing energy early in the decomposition process that is responsible.
Co-reporter:Tod A. Pascal, Shiang-Tai Lin and William A. Goddard III
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 1) pp:NaN181-181
Publication Date(Web):2010/11/23
DOI:10.1039/C0CP01549K
We validate here the Two-Phase Thermodynamics (2PT) method for calculating the standard molar entropies and heat capacities of common liquids. In 2PT, the thermodynamics of the system is related to the total density of states (DoS), obtained from the Fourier Transform of the velocity autocorrelation function. For liquids this DoS is partitioned into a diffusional component modeled as diffusion of a hard sphere gas plus a solid component for which the DoS(υ) → 0 as υ → 0 as for a Debye solid. Thermodynamic observables are obtained by integrating the DoS with the appropriate weighting functions. In the 2PT method, two parameters are extracted from the DoS self-consistently to describe diffusional contributions: the fraction of diffusional modes, f, and DoS(0). This allows 2PT to be applied consistently and without re-parameterization to simulations of arbitrary liquids. We find that the absolute entropy of the liquid can be determined accurately from a single short MD trajectory (20 ps) after the system is equilibrated, making it orders of magnitude more efficient than commonly used perturbation and umbrella sampling methods. Here, we present the predicted standard molar entropies for fifteen common solvents evaluated from molecular dynamics simulations using the AMBER, GAFF, OPLS AA/L and Dreiding II forcefields. Overall, we find that all forcefields lead to good agreement with experimental and previous theoretical values for the entropy and very good agreement in the heat capacities. These results validate 2PT as a robust and efficient method for evaluating the thermodynamics of liquid phase systems. Indeed 2PT might provide a practical scheme to improve the intermolecular terms in forcefields by comparing directly to thermodynamic properties.
Co-reporter:Mu-Jeng Cheng, Robert J. Nielsen, Jamil Tahir-Kheli and William A. Goddard III
Physical Chemistry Chemical Physics 2011 - vol. 13(Issue 20) pp:NaN9838-9838
Publication Date(Web):2011/04/18
DOI:10.1039/C0CP02777D
We have studied the magnetic structure of the high symmetry vanadyl pyrophosphate ((VO)2P2O7, VOPO), focusing on the spin exchange couplings, using density functional theory (B3LYP) with the full three-dimensional periodicity. VOPO involves four distinct spin couplings: two larger couplings exist along the chain direction (a-axis), which we predict to be antiferromagnetic, JOPO = −156.8 K and JO = −68.6 K, and two weaker couplings appear along the c (between two layers) and b directions (between two chains in the same layer), which we calculate to be ferromagnetic, Jlayer = 19.2 K and Jchain = 2.8 K. Based on the local density of states and the response of spin couplings to varying the cell parameter a, we found that JOPO originates from a super-exchange interaction through the bridging –O–P–O– unit. In contrast, JO results from a direct overlap of 3dx2 − y2 orbitals on two vanadium atoms in the same V2O8 motif, making it very sensitive to structural fluctuations. Based on the variations in V–O bond length as a function of strain along a, we found that the V–O bonds of V–(OPO)2–V are covalent and rigid, whereas the bonds of V–(O)2–V are fragile and dative. These distinctions suggest that compression along the a-axis would have a dramatic impact on JO, changing the magnetic structure and spin gap of VOPO. This result also suggests that assuming JO to be a constant over the range of 2–300 K whilst fitting couplings to the experimental magnetic susceptibility is an invalid method. Regarding its role as a catalyst, the bonding pattern suggests that O2 can penetrate beyond the top layers of the VOPO surface, converting multiple V atoms from the +4 to +5 oxidation state, which seems crucial to explain the deep oxidation of n-butane to maleic anhydride.
Co-reporter:Tao Cheng, William A Goddard, Qi An, Hai Xiao, Boris Merinov and Sergey Morozov
Physical Chemistry Chemical Physics 2017 - vol. 19(Issue 4) pp:NaN2673-2673
Publication Date(Web):2016/12/21
DOI:10.1039/C6CP08055C
The sluggish oxygen reduction reaction (ORR) is a major impediment to the economic use of hydrogen fuel cells in transportation. In this work, we report the full ORR reaction mechanism for Pt(111) based on Quantum Mechanics (QM) based Reactive metadynamics (RμD) simulations including explicit water to obtain free energy reaction barriers at 298 K. The lowest energy pathway for 4 e− water formation is: first, *OOH formation; second, *OOH reduction to H2O and O*; third, O* hydrolysis using surface water to produce two *OH and finally *OH hydration to water. Water formation is the rate-determining step (RDS) for potentials above 0.87 Volt, the normal operating range. Considering the Eley–Rideal (ER) mechanism involving protons from the solvent, we predict the free energy reaction barrier at 298 K for water formation to be 0.25 eV for an external potential below U = 0.87 V and 0.41 eV at U = 1.23 V, in good agreement with experimental values of 0.22 eV and 0.44 eV, respectively. With the mechanism now fully understood, we can use this now validated methodology to examine the changes upon alloying and surface modifications to increase the rate by reducing the barrier for water formation.
Co-reporter:Yuan Ping, Ravishankar Sundararaman and William A. Goddard III
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 45) pp:NaN30509-30509
Publication Date(Web):2015/10/26
DOI:10.1039/C5CP05740J
The band edge positions of photocatalysts relative to the redox potentials of water play an important role in determining the efficiency of photoelectrochemical cells. These band positions depend on the structure of the solid–liquid interface, but direct ab initio molecular dynamics calculations of these interfaces, while expected to be accurate, are too computationally demanding for high-throughput materials screening. Thus rapid theoretical screening of new photocatalyst materials requires simplified continuum solvation models that are suitable for treating solid–liquid interfaces. In this paper, we evaluate the accuracy of the recently developed CANDLE and SaLSA continuum solvation models for predicting solvation effects on the band positions of several well-studied surfaces [Si(111), TiO2(110), IrO2(110) and WO3(001)] in water. We find that the solvation effects vary considerably, ranging from <0.5 eV for hydrophobic surfaces, 0.5–1 eV for many hydrophilic oxide surfaces, to ∼2 eV for oxygen-deficient surfaces. The solvation model predictions are in excellent agreement (within ∼0.1 eV) with ab initio molecular dynamics results where available, and in good agreement (within ∼0.2–0.3 eV) with experimental measurements. We also predict the energetics for surface oxygen vacancies and their effect on the band positions of the hydrated WO3(001) surface, leading to an explanation for why the solvation shift observed experimentally is substantially larger than predicted for the ideal surface. Based on the correlation between solvation shift and the type of surface and solvent, we suggest approaches to engineer the band positions of surfaces in aqueous and non-aqueous solutions.
Co-reporter:Dezhou Guo, Qi An, Sergey V. Zybin, William A. Goddard III, Fenglei Huang and Bin Tang
Journal of Materials Chemistry A 2015 - vol. 3(Issue 10) pp:NaN5419-5419
Publication Date(Web):2015/01/26
DOI:10.1039/C4TA06858K
To gain an atomistic-level understanding of the experimental observation that the cocrystal TNT/CL-20 leads to decreased sensitivity, we carried out reactive molecular dynamics (RMD) simulations using the ReaxFF reactive force field. We compared the thermal decomposition of the TNT/CL-20 cocrystal with that of pure crystals of TNT and CL-20 and with a simple physical mixture of TNT and CL-20. We find that cocrystal has a lower decomposition rate than CL-20 but higher than TNT, which is consistent with experimental observation. We find that the formation of carbon clusters arising from TNT, a carbon-rich molecule, plays an important role in the thermal decomposition process, explaining the decrease in sensitivity for the cocrystal. At low temperature and in the early stage of chemical reactions under high temperature, the cocrystal releases energy more slowly than the simple mixture of CL-20–TNT. These results confirm the expectation that co-crystallization is an effective way to decrease the sensitivity for energetic materials while retaining high performance.
Reaction mechanism from quantum molecular dynamics for the initial thermal decomposition of 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N), promising green energetic materials
Co-reporter:Cai-Chao Ye, Qi An, Tao Cheng, Sergey Zybin, Saber Naserifar, Xue-Hai Ju and William A. Goddard III
Journal of Materials Chemistry A 2015 - vol. 3(Issue 22) pp:NaN12050-12050
Publication Date(Web):2015/04/24
DOI:10.1039/C5TA02486B
Klapötke and co-workers recently designed two new materials, 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N), envisioned as candidates for green high-energy materials. However, all attempts at synthesis have failed. In order to validate the expected properties for these systems and to determine why these materials are too unstable to synthesize, we used the PBE flavor of Density Functional Theory (DFT) to predict the crystal structures for MTO and MTO3N and then we carried out DFT molecular dynamics simulations (DFT-MD) to determine the initial reaction mechanisms for decomposition. Klapötke estimated that MTO would have a density of ρ = 1.859 g cm−3 with an estimated detonation velocity (Dv) of 8.979 km s−1, making it comparable to RDX (ρ = 1.82 g cm−3, Dv = 8.75 km s−1) and β-HMX (ρ = 1.91 g cm−3, Dv = 9.10 km s−1). His estimated impact sensitivity >30 J, make it much better than HMX (7 J) and RDX (7.5 J). Our predicted crystal structure for MTO (P2(1) space group) leads to ρ = 1.859 g cm−3, in good agreement with expectations. Our DFT-MD studies find that the first step in the decomposition of MTO is intermolecular hydrogen-transfer reaction (barrier 3.0 kcal mol−1) which is followed quickly by H2O and NO release with reaction barriers of 46.5 and 35.5 kcal mol−1. In contrast for MTO3N (P2(1)/c predicted space group), we find that the first steps are a bimolecular decomposition to release NO2 (ΔH = 44.1 kcal mol−1, ΔG = 54.7 kcal mol−1) simultaneous with unimolecular NO2 cleavage (ΔH = 59.9 and ΔG = 58.2 kcal mol−1) a unique initial reaction among EMs. These results suggest that MTO3N would be significantly more thermally stabile (barrier > 6.0 kcal mol−1 higher) than RDX and HMX, making it an excellent candidate to be insensitive new green energetic materials. However we find that MTO leads to very favorable hydrogen transfer reactions that may complicate synthesis and crystallization, making MTO3N the more promising system.
Prediction of structures and properties of 2,4,6-triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N) green energetic materials from DFT and ReaxFF molecular modeling
Co-reporter:Saber Naserifar, Sergey Zybin, Cai-Chao Ye and William A. Goddard III
Journal of Materials Chemistry A 2016 - vol. 4(Issue 4) pp:NaN1276-1276
Publication Date(Web):2015/11/09
DOI:10.1039/C5TA06426K
2,4,6-Triamino-1,3,5-triazine-1,3,5-trioxide (MTO) and 2,4,6-trinitro-1,3,5-triazine-1,3,5-trioxide (MTO3N) were suggested by Klapötke et al. as candidates for green high energy density materials (HEDM), but a successful synthesis has not yet been reported. In order to predict the properties of these systems, we used quantum mechanics (PBE flavor of density functional theory) to predict the most stable conformations of MTO and MTO3N and their optimum packing into the most stable crystal structures. We found that MTO has the P21 space-group with a density of ρ = 1.92 g cm−3 while MTO3N has the P21/c space-group with a density of ρ = 2.10 g cm−3. The heats of reaction (ΔHrxn) were computed to be 1036 kcal kg−1 for MTO, 1412 kcal kg−1 for MTO3N, and 1653 kcal kg−1 for a mixture of them. These properties are comparable to those of such other useful energetic materials as RDX (ρ = 1.80 g cm−3, ΔHrxn = 1266 kcal kg−1), HMX, and PETN, making MTO and MTO3N excellent candidates for environmentally friendly HEDMs. In addition, we predicted the stability of –NH2, –NO, and –NO2 groups in water solution. We also show that the ReaxFF-lg reactive FF leads to an accurate description of the structural properties of MTO and MTO3N crystals making it practical to carry out large-scale reactive molecular dynamics simulations practical for these systems to determine the sensitivity and performance (CJ point calculation and velocity) under shear, shock, and thermal loads.
Co-reporter:Sang Soo Han, José L. Mendoza-Cortés and William A. Goddard III
Chemical Society Reviews 2009 - vol. 38(Issue 5) pp:NaN1476-1476
Publication Date(Web):2009/03/24
DOI:10.1039/B802430H
This critical review covers the application of computer simulations, including quantum calculations (ab initio and DFT), grand canonical Monte-Carlo simulations, and molecular dynamics simulations, to the burgeoning area of the hydrogen storage by metal–organic frameworks and covalent-organic frameworks. This review begins with an overview of the theoretical methods obtained from previous studies. Then strategies for the improvement of hydrogen storage in the porous materials are discussed in detail. The strategies include appropriate pore size, impregnation, catenation, open metal sites in metal oxide parts and within organic linker parts, doping of alkali elements onto organic linkers, substitution of metal oxide with lighter metals, functionalized organic linkers, and hydrogen spillover (186 references).
.jpg)




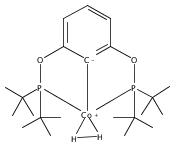
![2-Benzoxazolamine,N-[1-[[3-ethoxy-4-(1-methylethoxy)phenyl]methyl]-4-piperidinyl]-](http://img.cochemist.com/ccimg/909900/909851-03-2.png)
![2-Benzoxazolamine,N-[1-[[3-ethoxy-4-(1-methylethoxy)phenyl]methyl]-4-piperidinyl]-](http://img.cochemist.com/ccimg/909900/909851-03-2_b.png)
![2-Benzoxazolamine,N-[1-[[3-ethoxy-4-(trifluoromethyl)phenyl]methyl]-4-piperidinyl]-](http://img.cochemist.com/ccimg/909900/909850-99-3.png)
![2-Benzoxazolamine,N-[1-[[3-ethoxy-4-(trifluoromethyl)phenyl]methyl]-4-piperidinyl]-](http://img.cochemist.com/ccimg/909900/909850-99-3_b.png)
![2-Benzoxazolamine,N-[1-[(4-chloro-3-ethoxyphenyl)methyl]-4-piperidinyl]-](http://img.cochemist.com/ccimg/909900/909850-97-1.png)
![2-Benzoxazolamine,N-[1-[(4-chloro-3-ethoxyphenyl)methyl]-4-piperidinyl]-](http://img.cochemist.com/ccimg/909900/909850-97-1_b.png)














![6H-Oxireno[e][2]benzoxacyclotetradecin-6,12(7H)-dione,8-chloro-1a,14,15,15a-tetrahydro-9,11-dihydroxy-14-methyl-,(1aR,2Z,4E,14R,15aR)-](http://img.cochemist.com/ccimg/12800/12772-57-5.png)
![6H-Oxireno[e][2]benzoxacyclotetradecin-6,12(7H)-dione,8-chloro-1a,14,15,15a-tetrahydro-9,11-dihydroxy-14-methyl-,(1aR,2Z,4E,14R,15aR)-](http://img.cochemist.com/ccimg/12800/12772-57-5_b.png)