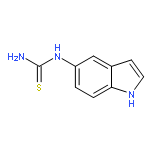
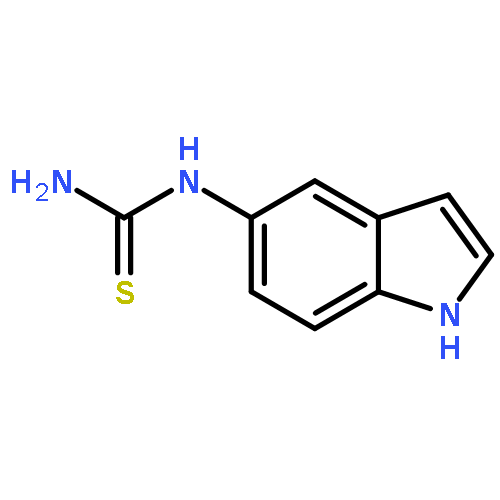
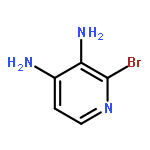
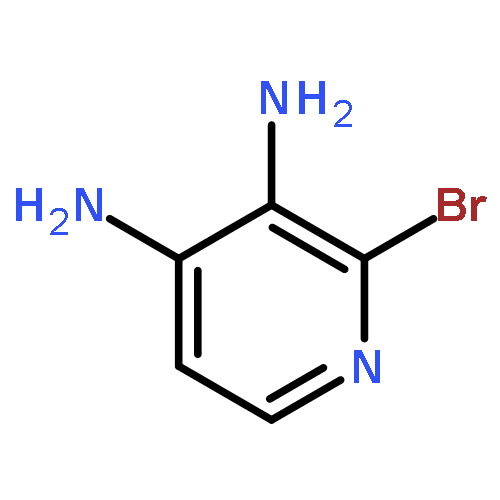
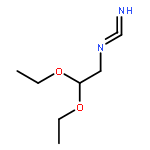
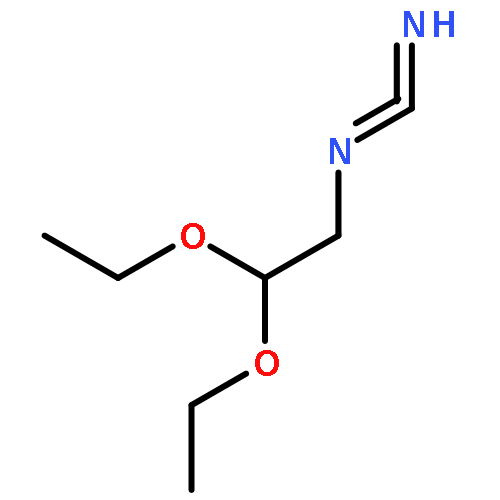
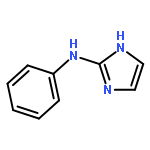
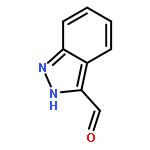
Co-reporter:Zongtao Lin; Srinivasa R. Marepally; Dejian Ma; Tae-Kang Kim; Allen SW. Oak; Linda K. Myers; Robert C. Tuckey; Andrzej T. Slominski; Duane D. Miller;Wei Li
Journal of Medicinal Chemistry 2016 Volume 59(Issue 10) pp:5102-5108
Publication Date(Web):April 12, 2016
DOI:10.1021/acs.jmedchem.6b00182
The vitamin D3 metabolite, 20S,23S-dihydroxyvitamin D3, was chemically synthesized for the first time and identified to be the same as the enzymatically produced metabolite. The C23 absolute configurations of both 20S,23S/R-dihydroxyvitamin D3 epimers were unambiguously assigned by NMR and Mosher ester analysis. Their kinetics of CYP27B1 metabolism were investigated during the production of their 1α-hydroxylated derivatives. Bioactivities of these products were compared in terms of vitamin D3 receptor activation, anti-inflammatory, and antiproliferative activities.
Co-reporter:Zongtao Lin, Srinivasa Reddy Marepally, Dejian Ma, Linda K. Myers, Arnold E. Postlethwaite, Robert C. Tuckey, Chloe Y. S. Cheng, Tae-Kang Kim, Junming Yue, Andrzej T. Slominski, Duane D. Miller, and Wei Li
Journal of Medicinal Chemistry 2015 Volume 58(Issue 19) pp:7881-7887
Publication Date(Web):September 14, 2015
DOI:10.1021/acs.jmedchem.5b00881
Bioactive vitamin D3 metabolites 20S,24S-dihydroxyvitamin D3 [20S,24S(OH)2D3] and 20S,24R-dihydroxyvitamin D3 [20S,24R(OH)2D3] were chemically synthesized and confirmed to be identical to their enzymatically generated counterparts. The absolute configurations at C24 and its influence on the kinetics of 1α-hydroxylation by CYP27B1 were determined. Their corresponding 1α-hydroxyl derivatives were subsequently produced. Biological comparisons of these products showed different properties with respect to vitamin D3 receptor activation, anti-inflammatory activity, and antiproliferative activity, with 1α,20S,24R(OH)2D3 being the most potent compound.
Co-reporter:Dong-Jin Hwang, Jin Wang, Wei Li, and Duane D. Miller
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 9) pp:993
Publication Date(Web):August 6, 2015
DOI:10.1021/acsmedchemlett.5b00208
A new series of indole analogues based on our earlier lead compound, 2-(1H-indol-5-yl)-4-(3,4,5-trimethoxyphenyl)-1H-imidazo[4,5-c]pyridine (42), was prepared as tubulin inhibitors in an effort to find a molecule with improved cytotoxic potency and metabolic stability. A series of indolyl-imidazopyridines (IIP) were synthesized and exhibited potent tubulin polymerization inhibitory activity with potent IC50 values ranging from 3 to 175 nM against a panel of human melanoma and prostate cancer cell lines. Among these compounds, the 6-indolyl compound 43 showed improved cytotoxic potency (average IC50 of 9.75 nM vs 55.75 nM) and metabolic stability in human liver microsomes (half-life time was 56.3 min vs. 45.4 min) as compared to previously reported 42. It was also shown to be effective against P-glycoprotein (P-gp) mediated multiple drug resistance (MDR) and taxol resistance.Keywords: antiproliferative activity; Colchicine-binding site; liver microsomal stability; melanoma; multiple drug resistance; prostate cancer; structure−activity relationship (SAR); tubulin polymerization inhibitor;
Co-reporter:Yan Lu ; Jianjun Chen ; Jin Wang ; Chien-Ming Li ; Sunjoo Ahn ; Christina M. Barrett ; James T. Dalton ; Wei Li ;Duane D. Miller
Journal of Medicinal Chemistry 2014 Volume 57(Issue 17) pp:7355-7366
Publication Date(Web):August 14, 2014
DOI:10.1021/jm500764v
To block the metabolically labile sites of novel tubulin inhibitors targeting the colchicine binding site based on SMART, ABI, and PAT templates, we have designed, synthesized, and biologically tested three focused sets of new derivatives with modifications at the carbonyl linker, the para-position in the C ring of SMART template, and modification of A ring of the PAT template. Structure–activity relationships of these compounds led to the identification of new benzimidazole and imidazo[4,5-c]pyridine-fused ring templates, represented by compounds 4 and 7, respectively, which showed enhanced antitumor activity and substantially improved the metabolic stability in liver microsomes compared to SMART. MOM group replaced TMP C ring and generated a potent analogue 15, which showed comparable potency to the parent SMART compound. Further modification of PAT template yielded another potent analogue 33 with 5-indolyl substituent at A ring.
Co-reporter:Zongtao Lin;Ruinan Yang;Zheng Guan;Ailiang Chen;Wei Li
Phytochemical Analysis 2014 Volume 25( Issue 6) pp:485-494
Publication Date(Web):
DOI:10.1002/pca.2517
ABSTRACT
Introduction
As an essential medicine and tea source in many countries, Plumula Nelumbinis potentially exerts its major biological activities through its alkaloids. However, the activities of Plumula Nelumbinis are not fully understood due to the lack of studies on its chemical components.
Objective
To establish an ultra-performance liquid chromatography combined with diode-array detector (UPLC/DAD) method, coupled to an electrospray ionisation with quadrupole time-of-flight mass spectrometry (ESI/QTOF/MS) method, for the separation and identification of Plumula Nelumbinis alkaloids.
Methods
The eluant from an UPLC separation of an ethanol extract of Plumula Nelumbinis was directly infused into an ESI/QTOF/MS system. Both positive and negative ion modes of ESI with low and high collision energy (CE) were used to obtain sufficient MS information.
Results
Twenty-one alkaloids were tentatively identified based on their chromatographic characteristics, UV spectra, exact mass, MS fragments and literature reports. They consist of six bis-1-benzyltetrahydroisoquinoline, eleven benzyltetrahydroisoquinoline (including two glycoalkaloids and two quaternary ammoniums), two aporphine, one proaporphine and one indole alkaloids. Eleven were identified in Plumula Nelumbinis for the first time and seven were first reported in Nelumbo nucifera Gaertn. Five compounds, namely norcoclaurine-4′-O-glucoside, norcoclaurine-6-O-glucoside, isolotusine, 6-demethyl-4-demethylN-methylcoclaurine and N-norisoliensinine, were characterised and proposed as new compounds.
Conclusion
The established UPLC/DAD − ESI/QTOF/MS method is efficient for systematic identification of the alkaloids in Plumula Nelumbinis extract. Copyright © 2014 John Wiley & Sons, Ltd.
Co-reporter:Min Xiao ; Sunjoo Ahn ; Jin Wang ; Jianjun Chen ; Duane. D. Miller ; James T Dalton ;Wei Li
Journal of Medicinal Chemistry 2013 Volume 56(Issue 8) pp:3318-3329
Publication Date(Web):April 2, 2013
DOI:10.1021/jm4001117
A series of 4-aryl-2-benzoyl-imidazoles were designed and synthesized based on our previously reported 2-aryl-4-benzoyl-imidazole (ABI) derivatives. The new structures reversed the aryl group and the benzoyl group of previous ABI structures and were named as reverse ABI (RABI) analogues. RABIs were evaluated for biological activity against eight cancer cell lines including multidrug-resistant cancer cell lines. In vitro assays indicated that several RABI compounds had excellent antiproliferative properties, with IC50 values in the low nanomolar range. The average IC50 of the most active compound 12a is 14 nM. In addition, the mechanism of action of these new analogues was investigated by cell cycle analysis, tubulin polymerization assay, competitive mass spectrometry binding assay, and molecular docking studies. These studies confirmed that these new RABI analogues maintain their mechanisms of action by disrupting tubulin polymerization, similar to their parental ABI analogues.
Co-reporter:Yan Lu ; Jianjun Chen ; Zorica Janjetovic ; Phillip Michaels ; Edith K. Y. Tang ; Jin Wang ; Robert C. Tuckey ; Andrzej T. Slominski ; Wei Li ;Duane D. Miller
Journal of Medicinal Chemistry 2012 Volume 55(Issue 7) pp:3573-3577
Publication Date(Web):March 9, 2012
DOI:10.1021/jm201478e
The non-naturally occurring 20R epimer of 20-hydroxyvitamin D3 is synthesized based on chemical design and hypothesis. The 20R isomer is separated by semipreparative HPLC, and its structure is characterized. A comparison of 20R isomer to its 20S counterpart in biological evaluation demonstrates that they have different behaviors in antiproliferative and metabolic studies.
Co-reporter:Jianjun Chen ; Sunjoo Ahn ; Jin Wang ; Yan Lu ; James T. Dalton ; Duane D. Miller ;Wei Li
Journal of Medicinal Chemistry 2012 Volume 55(Issue 16) pp:7285-7289
Publication Date(Web):July 11, 2012
DOI:10.1021/jm300564b
Novel ABI-III compounds were designed and synthesized based on our previously reported ABI-I and ABI-II analogues. ABI-III compounds are highly potent against a panel of melanoma and prostate cancer cell lines, with the best compound having an average IC50 value of 3.8 nM. They are not substrate of Pgp and thus may effectively overcome Pgp-mediated multidrug resistance. ABI-III analogues maintain their mechanisms of action by inhibition of tubulin polymerization.
Co-reporter:Shivaputra A. Patil, Jin Wang, Xiaochen S. Li, Jianjun Chen, Terreia S. Jones, Amira Hosni-Ahmed, Renukadevi Patil, William L. Seibel, Wei Li, Duane D. Miller
Bioorganic & Medicinal Chemistry Letters 2012 Volume 22(Issue 13) pp:4458-4461
Publication Date(Web):1 July 2012
DOI:10.1016/j.bmcl.2012.04.074
As a continuation of our efforts to discover and develop small molecules as anticancer agents, we identified GRI-394837 as an initial hit from similarity search on RGD and its analogs. Based on GRI-394837, we designed and synthesized a focused set of novel chromenes (4a–e) in a single step using microwave method. All five compounds showed activity in the nanomolar range (IC50: 7.4–640 nM) in two melanoma, three prostate and four glioma cancer cell lines. The chromene 4e is active against all the cell lines and particularly against the A172 human glioma cell line (IC50: 7.4 nM). Interestingly, in vitro tubulin polymerization assay shows 4e to be a weak tubulin polymerization inhibitor but it shows very strong cytotoxicity in cellular assays, therefore there must be additional unknown mechanism(s) for the anticancer activity. Additionally, the strong antiproliferative activity was verified by one of the selected chromene (4a) by the NCI 60 cell line screen. These results strongly suggest that the novel chromenes could be further developed as a potential therapeutic agent for a variety of aggressive cancers.
Co-reporter:Wei Li
Pharmaceutical Research 2012 Volume 29( Issue 11) pp:2939-2942
Publication Date(Web):2012 November
DOI:10.1007/s11095-012-0886-2
Co-reporter:Zhao Wang;Jianjun Chen;Jin Wang;Sunjoo Ahn
Pharmaceutical Research 2012 Volume 29( Issue 11) pp:3040-3052
Publication Date(Web):2012 November
DOI:10.1007/s11095-012-0726-4
To evaluate abilities of 2-aryl-4-benzoyl-imidazoles (ABI) to overcome multidrug resistance (MDR), define their cellular target, and assess in vivo antimelanoma efficacy.MDR cell lines that overexpressed P-glycoprotein, MDR-associated proteins, and breast cancer resistance protein were used to evaluate ABI ability to overcome MDR. Cell cycle analysis, molecular modeling, and microtubule imaging were used to define ABI cellular target. SHO mice bearing A375 human melanoma xenograft were used to evaluate ABI in vivo antitumor activity. B16-F10/C57BL mouse melanoma lung metastasis model was used to test ABI efficacy to inhibit tumor lung metastasis.ABIs showed similar potency to MDR cells compared to matching parent cells. ABIs were identified to target tubulin on the colchicine binding site. After 31 days of treatment, ABI-288 dosed at 25 mg/kg inhibited melanoma tumor growth by 69%; dacarbazine at 60 mg/kg inhibited growth by 52%. ABI-274 dosed at 25 mg/kg showed better lung metastasis inhibition than dacarbazine at 60 mg/kg.This new class of antimitotic compounds can overcome several clinically important drug resistant mechanisms in vitro and are effective in inhibiting melanoma lung metastasis in vivo, supporting their further development.
Co-reporter:Jianjun Chen, Chien-Ming Li, Jin Wang, Sunjoo Ahn, Zhao Wang, Yan Lu, James T. Dalton, Duane D. Miller, Wei Li
Bioorganic & Medicinal Chemistry 2011 19(16) pp: 4782-4795
Publication Date(Web):
DOI:10.1016/j.bmc.2011.06.084
Co-reporter:Jianjun Chen ; Zhao Wang ; Chien-Ming Li ; Yan Lu ; Pavan K. Vaddady ; Bernd Meibohm ; James T. Dalton ; Duane D. Miller ;Wei Li
Journal of Medicinal Chemistry 2010 Volume 53(Issue 20) pp:7414-7427
Publication Date(Web):October 4, 2010
DOI:10.1021/jm100884b
A series of 2-aryl-4-benzoyl-imidazoles (ABI) was synthesized as a result of structural modifications based on the previous set of 2-aryl-imidazole-4-carboxylic amide (AICA) derivatives and 4-substituted methoxylbenzoyl-aryl-thiazoles (SMART). The average IC50 of the most active compound (5da) was 15.7 nM. ABI analogues have substantially improved aqueous solubility (48.9 μg/mL for 5ga vs 0.909 μg/mL for SMART-1, 0.137 μg/mL for paclitaxel, and 1.04 μg/mL for combretastatin A4). Mechanism of action studies indicate that the anticancer activity of ABI analogues is through inhibition of tubulin polymerization by interacting with the colchicine binding site. Unlike paclitaxel and colchicine, the ABI compounds were equally potent against multidrug resistant cancer cells and the sensitive parental melanoma cancer cells. In vivo results indicated that 5cb was more effective than DTIC in inhibiting melanoma xenograph tumor growth. Our results suggest that the novel ABI compounds may be developed to effectively treat drug-resistant tumors.
Co-reporter:Georgina N. Masoud, Wei Li
Acta Pharmaceutica Sinica B (September 2015) Volume 5(Issue 5) pp:378-389
Publication Date(Web):September 2015
DOI:10.1016/j.apsb.2015.05.007
Co-reporter:Wei Li, Jianjun Chen, Zorica Janjetovic, Tae-Kang Kim, Trevor Sweatman, Yan Lu, Jordan Zjawiony, Robert C. Tuckey, Duane Miller, Andrzej Slominski
Steroids (July 2013) Volume 78(Issue 7) pp:731
Publication Date(Web):1 July 2013
DOI:10.1016/j.steroids.2013.04.001
Co-reporter:Qinghui Wang, Zongtao Lin, Tae-Kang Kim, Andrzej T. Slominski, Duane D. Miller, Wei Li
Steroids (December 2015) Volume 104() pp:153-162
Publication Date(Web):1 December 2015
DOI:10.1016/j.steroids.2015.09.009
•Key intermediates 7 and 12 were generated in parallel via modified synthetic ways.•Simultaneous removal of EOM and TBS was completed using camphorsulfonic acid.•20S-Hydroxyvitamin D3 was synthesized in 16 steps with an overall yield of 0.4%.•Synthetic 20S-(OH)D3 activated VDR and initiated expression of downstream genes.•20S-(OH)D3 was potent in inhibition of melanoma cells proliferation.A total synthetic strategy of 20S-hydroxyvitamin D3 [20S-(OH)D3] involving modified synthesis of key intermediates 7 and 12, Grignard reaction to stereoselectively generate 20S-OH and Wittig–Horner coupling to establish D3 framework, was completed in 16 steps with an overall yield of 0.4%. The synthetic 20S-(OH)D3 activated vitamin D receptor (VDR) and initiated the expression of downstream genes. In addition, 20S-(OH)D3 showed similar inhibitory potency as calcitriol [1,25(OH)2D3] on proliferation of melanoma cells.Download full-size image
Co-reporter:Wei Li, Jianjun Chen, Zorica Janjetovic, Tae-Kang Kim, Trevor Sweatman, Yan Lu, Jordan Zjawiony, Robert C. Tuckey, Duane Miller, Andrzej Slominski
Steroids (December 2010) Volume 75(Issue 12) pp:926-935
Publication Date(Web):1 December 2010
DOI:10.1016/j.steroids.2010.05.021
20S-hydroxyvitamin D3 (20S-(OH)D3), an in vitro product of vitamin D3 metabolism by the cytochrome P450scc, was recently isolated, identified and shown to possess antiproliferative activity without inducing hypercalcemia. The enzymatic production of 20S-(OH)D3 is tedious, expensive, and cannot meet the requirements for extensive chemical and biological studies. Here we report for the first time the chemical synthesis of 20S-(OH)D3 which exhibited biological properties characteristic of the P450scc-generated compound. Specifically, it was hydroxylated to 20,23-dihydroxyvitamin D3 and 17,20-dihydroxyvitamin D3 by P450scc and was converted to 1α,20-dihydroxyvitamin D3 by CYP27B1. It inhibited proliferation of human epidermal keratinocytes with lower potency than 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3) in normal epidermal human keratinocytes, but with equal potency in immortalized HaCaT keratinocytes. It also stimulated VDR gene expression with similar potency to 1,25(OH)2D3, and stimulated involucrin (a marker of differentiation) and CYP24 gene expression, showing a lower potency for the latter gene than 1,25(OH)2D3. Testing performed with hamster melanoma cells demonstrated a dose-dependent inhibition of cell proliferation and colony forming capabilities similar or more pronounced than those of 1,25(OH)2D3. Thus, we have developed a chemical method for the synthesis of 20S-(OH)D3, which will allow the preparation of a series of 20S-(OH)D3 analogs to study structure–activity relationships to further optimize this class of compound for therapeutic use.
Co-reporter:Tae-Kang Kim, Jin Wang, Zorica Janjetovic, Jianjun Chen, Robert C. Tuckey, Minh N. Nguyen, Edith K.Y. Tang, Duane Miller, Wei Li, Andrzej T. Slominski
Molecular and Cellular Endocrinology (25 September 2012) Volume 361(Issues 1–2) pp:143-152
Publication Date(Web):25 September 2012
DOI:10.1016/j.mce.2012.04.001
To define the interaction of novel secosteroids produced by the action of cytochrome P450scc with vitamin D receptor (VDR), we used a human melanoma line overexpressing VDR fused with enhanced green fluorescent protein (EGFP) and tested the ligand induced translocation of VDR from the cytoplasm to the nucleus. Hydroxyderivatives of vitamin D3 with a full length (D3) side chain and hydroxy-secosteroids with a shortened side chain (pD) stimulated VDR translocation and inhibited proliferation, however, with different potencies. In general the D3 were more potent than pD analogues. Molecular modeling of the binding of the secosteroids to the VDR genomic binding pocket (G-pocket) correlated well with the experimental data for VDR translocation. In contrast, docking scores for the non-genomic binding site of the VDR were poor. In conclusion, both the length of the side chain and the number and position of hydroxyl groups affect the activation of VDR by novel secosteroids.Highlights► We tested ligand induced vitamin D receptor translocation by novel secosteroids. ► We modeled secosteroids bindings to the genomic and non-genomic pockets. ► Good correlation between binding to the genomic pocket and translocation exists. ► Novel vitamin D hydroxyderivatives inhibit cell proliferation. ► Both chain length and the number or positions of hydroxyl groups are important.