Co-reporter:Benedikt Warth, Scott Spangler, Mingliang Fang, Caroline H. Johnson, Erica M. Forsberg, Ana Granados, Richard L. Martin, Xavier Domingo-Almenara, Tao Huan, Duane Rinehart, J. Rafael Montenegro-Burke, Brian Hilmers, Aries Aisporna, Linh T. Hoang, Winnie Uritboonthai, H. Paul Benton, Susan D. Richardson, Antony J. Williams, and Gary Siuzdak
Analytical Chemistry November 7, 2017 Volume 89(Issue 21) pp:11505-11505
Publication Date(Web):September 25, 2017
DOI:10.1021/acs.analchem.7b02759
Concurrent exposure to a wide variety of xenobiotics and their combined toxic effects can play a pivotal role in health and disease, yet are largely unexplored. Investigating the totality of these exposures, i.e., the “exposome”, and their specific biological effects constitutes a new paradigm for environmental health but still lacks high-throughput, user-friendly technology. We demonstrate the utility of mass spectrometry-based global exposure metabolomics combined with tailored database queries and cognitive computing for comprehensive exposure assessment and the straightforward elucidation of biological effects. The METLIN Exposome database has been redesigned to help identify environmental toxicants, food contaminants and supplements, drugs, and antibiotics as well as their biotransformation products, through its expansion with over 700 000 chemical structures to now include more than 950 000 unique small molecules. More importantly, we demonstrate how the XCMS/METLIN platform now allows for the readout of the biological effect of a toxicant through metabolomic-derived pathway analysis, and further, artificial intelligence provides a means of assessing the role of a potential toxicant. The presented workflow addresses many of the methodological challenges current exposomics research is facing and will serve to gain a deeper understanding of the impact of environmental exposures and combinatory toxic effects on human health.
Co-reporter:William R. Wikoff;Ewa Kalisak;Sunia Trauger;Marianne Manchester
Journal of Proteome Research July 6, 2009 Volume 8(Issue 7) pp:3578-3587
Publication Date(Web):Publication Date (Web): June 4, 2009
DOI:10.1021/pr900275p
Lymphocytic choriomeningitis virus (LCMV) infection of mice is noncytopathic, producing well-characterized changes reflecting the host immune response. Untargeted metabolomics using mass spectrometry identified endogenous small molecule changes in blood from mice inoculated with LCMV, sampled at days 1, 3, 7, and 14 post infection. These time points correspond to well characterized events during acute LCMV infection and the immune response. Diverse pathways were altered, including TCA cycle intermediates, γ-glutamyl dipeptides, lysophosphatidyl cholines, and fatty acids. The kynurenine pathway was activated, surprising because it is stimulated by IFN-γ, which LCMV suppresses, thus, suggesting alternative activators. In contrast, biopterin/neopterin, another IFN-γ stimulated pathway, was not activated. Many metabolites followed “response and recovery” kinetics, decreasing after infection to a minimum at days 3−7, and returning to normal by day 14. The TCA pathway followed this pattern, including citrate, cis-aconitate and α-ketoglutarate, intriguing because succinate has been shown to mediate cellular immunity. This response and recovery dynamic tracks the immune response, including the rise and fall of natural killer cell populations, serum TNF receptor concentration, and viral clearance. Metabolomics can provide target pathways for molecular diagnostics or therapeutics of viral infection and immunity.Keywords: innate immunity; mass spectrometry; metabolism; systems biology; untargeted metabolomics;
Co-reporter:Erica Forsberg;Mingliang Fang
Journal of The American Society for Mass Spectrometry 2017 Volume 28( Issue 1) pp:14-20
Publication Date(Web):2017 January
DOI:10.1007/s13361-016-1440-y
Mass spectrometry has traditionally been the technology of choice for small molecule analysis, making significant inroads into metabolism, clinical diagnostics, and pharmacodynamics since the 1960s. In the mid-1980s, with the discovery of electrospray ionization (ESI) for biomolecule analysis, a new door opened for applications beyond small molecules. Initially, proteins were widely examined, followed by oligonucleotides and other nonvolatile molecules. Then in 1991, three intriguing studies reported using mass spectrometry to examine noncovalent protein complexes, results that have been expanded on for the last 25 years. Those experiments also raised the questions: How soft is ESI, and can it be used to examine even more complex interactions? Our lab addressed these questions with the analyses of viruses, which were initially tested for viability following electrospray ionization and their passage through a quadrupole mass analyzer by placing them on an active medium that would allow them to propagate. This observation has been replicated on multiple different systems, including experiments on an even bigger microbe, a spore. The question of analysis was also addressed in the early 2000s with charge detection mass spectrometry. This unique technology could simultaneously measure mass-to-charge and charge, allowing for the direct determination of the mass of a virus. More recent experiments on spores and enveloped viruses have given us insight into the range of mass spectrometry’s capabilities (reaching 100 trillion Da), beginning to answer fundamental questions regarding the complexity of these organisms beyond proteins and genes, and how small molecules are integral to these supramolecular living structures.
Co-reporter:J. Rafael Montenegro-BurkeAries E. Aisporna, H. Paul Benton, Duane Rinehart, Mingliang Fang, Tao Huan, Benedikt Warth, Erica ForsbergBrian T. Abe, Julijana IvanisevicDennis W. Wolan, Luc Teyton, Luke Lairson, Gary Siuzdak
Analytical Chemistry 2017 Volume 89(Issue 2) pp:
Publication Date(Web):December 16, 2016
DOI:10.1021/acs.analchem.6b03890
The speed and throughput of analytical platforms has been a driving force in recent years in the “omics” technologies and while great strides have been accomplished in both chromatography and mass spectrometry, data analysis times have not benefited at the same pace. Even though personal computers have become more powerful, data transfer times still represent a bottleneck in data processing because of the increasingly complex data files and studies with a greater number of samples. To meet the demand of analyzing hundreds to thousands of samples within a given experiment, we have developed a data streaming platform, XCMS Stream, which capitalizes on the acquisition time to compress and stream recently acquired data files to data processing servers, mimicking just-in-time production strategies from the manufacturing industry. The utility of this XCMS Online-based technology is demonstrated here in the analysis of T cell metabolism and other large-scale metabolomic studies. A large scale example on a 1000 sample data set demonstrated a 10 000-fold time savings, reducing data analysis time from days to minutes. Further, XCMS Stream has the capability to increase the efficiency of downstream biochemical dependent data acquisition (BDDA) analysis by initiating data conversion and data processing on subsets of data acquired, expanding its application beyond data transfer to smart preliminary data decision-making prior to full acquisition.
Co-reporter:Caroline H. Johnson;Antonio F. Santidrian;Sarah E. LeBoeuf
Cancer & Metabolism 2017 Volume 5( Issue 1) pp:9
Publication Date(Web):31 October 2017
DOI:10.1186/s40170-017-0171-2
Cancer cells that enter the metastatic cascade require traits that allow them to survive within the circulation and colonize distant organ sites. As disseminating cancer cells adapt to their changing microenvironments, they also modify their metabolism and metabolite production.A mouse xenograft model of spontaneous tumor metastasis was used to determine the metabolic rewiring that occurs between primary cancers and their metastases. An “autonomous” mass spectrometry-based untargeted metabolomic workflow with integrative metabolic pathway analysis revealed a number of differentially regulated metabolites in primary mammary fat pad (MFP) tumors compared to microdissected paired lung metastases. The study was further extended to analyze metabolites in paired normal tissues which determined the potential influence of metabolites from the microenvironment.Metabolomic analysis revealed that multiple metabolites were increased in metastases, including cholesterol sulfate and phospholipids (phosphatidylglycerols and phosphatidylethanolamine). Metabolite analysis of normal lung tissue in the mouse model also revealed increased levels of these metabolites compared to tissues from normal MFP and primary MFP tumors, indicating potential extracellular uptake by cancer cells in lung metastases. These results indicate a potential functional importance of cholesterol sulfate and phospholipids in propagating metastasis. In addition, metabolites involved in DNA/RNA synthesis and the TCA cycle were decreased in lung metastases compared to primary MFP tumors.Using an integrated metabolomic workflow, this study identified a link between cholesterol sulfate and phospholipids, metabolic characteristics of the metastatic niche, and the capacity of tumor cells to colonize distant sites.
Co-reporter:Benedikt Warth, Nadine Levin, Duane Rinehart, John Teijaro, H. Paul Benton, Gary Siuzdak
Trends in Biotechnology 2017 Volume 35, Issue 6(Issue 6) pp:
Publication Date(Web):1 June 2017
DOI:10.1016/j.tibtech.2016.12.010
Cloud-based bioinformatic platforms address the fundamental demands of creating a flexible scientific environment, facilitating data processing and general accessibility independent of a countries’ affluence. These platforms have a multitude of advantages as demonstrated by omics technologies, helping to support both government and scientific mandates of a more open environment.
Co-reporter:J. Rafael Montenegro-Burke, Thiery Phommavongsay, Aries E. Aisporna, Tao Huan, Duane Rinehart, Erica Forsberg, Farris L. Poole, Michael P. Thorgersen, Michael W. W. Adams, Gregory Krantz, Matthew W. Fields, Trent R. Northen, Paul D. Robbins, Laura J. Niedernhofer, Luke Lairson, H. Paul Benton, and Gary Siuzdak
Analytical Chemistry 2016 Volume 88(Issue 19) pp:9753
Publication Date(Web):August 25, 2016
DOI:10.1021/acs.analchem.6b02676
Active data screening is an integral part of many scientific activities, and mobile technologies have greatly facilitated this process by minimizing the reliance on large hardware instrumentation. In order to meet with the increasingly growing field of metabolomics and heavy workload of data processing, we designed the first remote metabolomic data screening platform for mobile devices. Two mobile applications (apps), XCMS Mobile and METLIN Mobile, facilitate access to XCMS and METLIN, which are the most important components in the computer-based XCMS Online platforms. These mobile apps allow for the visualization and analysis of metabolic data throughout the entire analytical process. Specifically, XCMS Mobile and METLIN Mobile provide the capabilities for remote monitoring of data processing, real time notifications for the data processing, visualization and interactive analysis of processed data (e.g., cloud plots, principle component analysis, box-plots, extracted ion chromatograms, and hierarchical cluster analysis), and database searching for metabolite identification. These apps, available on Apple iOS and Google Android operating systems, allow for the migration of metabolomic research onto mobile devices for better accessibility beyond direct instrument operation. The utility of XCMS Mobile and METLIN Mobile functionalities was developed and is demonstrated here through the metabolomic LC-MS analyses of stem cells, colon cancer, aging, and bacterial metabolism.
Co-reporter:Michael E. Kurczy, Erica M. Forsberg, Michael P. Thorgersen, Farris L. Poole II, H. Paul Benton, Julijana Ivanisevic, Minerva L. Tran, Judy D. Wall, Dwayne A. Elias, Michael W. W. Adams, and Gary Siuzdak
ACS Chemical Biology 2016 Volume 11(Issue 6) pp:1677
Publication Date(Web):April 5, 2016
DOI:10.1021/acschembio.6b00082
Nitrogen cycling is a microbial metabolic process essential for global ecological/agricultural balance. To investigate the link between the well-established ammonium and the alternative nitrate assimilation metabolic pathways, global isotope metabolomics was employed to examine three nitrate reducing bacteria using 15NO3 as a nitrogen source. In contrast to a control (Pseudomonas stutzeri RCH2), the results show that two of the isolates from Oak Ridge, Tennessee (Pseudomonas N2A2 and N2E2) utilize nitrate and ammonia for assimilation concurrently with differential labeling observed across multiple classes of metabolites including amino acids and nucleotides. The data reveal that the N2A2 and N2E2 strains conserve nitrogen-containing metabolites, indicating that the nitrate assimilation pathway is a conservation mechanism for the assimilation of nitrogen. Co-utilization of nitrate and ammonia is likely an adaption to manage higher levels of nitrite since the denitrification pathways utilized by the N2A2 and N2E2 strains from the Oak Ridge site are predisposed to the accumulation of the toxic nitrite. The use of global isotope metabolomics allowed for this adaptive strategy to be investigated, which would otherwise not have been possible to decipher.
Co-reporter:Liliana P. Paris;Caroline H. Johnson;Edith Aguilar;Yoshihiko Usui
Metabolomics 2016 Volume 12( Issue 1) pp:
Publication Date(Web):2016 January
DOI:10.1007/s11306-015-0877-5
Proliferative diabetic retinopathy (PDR) is the most severe form of diabetic retinopathy and, along with diabetic macular edema, is responsible for the majority of blindness in adults below the age of 65. Therapeutic strategies for PDR are ineffective at curtailing disease progression in all cases; however a deeper understanding of the ocular metabolic landscape in PDR through metabolomic analysis may offer new therapeutic targets. Here, global and targeted mass spectrometry-based metabolomics were used to investigate metabolism. Initial analyses on vitreous humor from patients with PDR (n = 9) and non-diabetic controls (n = 11) revealed an increase of arginine and acylcarnitine metabolism in PDR. The oxygen-induced-retinopathy (OIR) mouse model, which exhibits comparable pathological manifestations to human PDR, revealed similar increases of arginine and other metabolites in the urea cycle, as well as downregulation of purine metabolism. We validated our findings by targeted multiple reaction monitoring and through the analysis of a second set of patient samples [PDR (n = 11) and non-diabetic controls (n = 20)]. These results confirmed a predominant and consistent increase in proline in both the OIR mouse model and vitreous samples from patients with PDR, suggesting that over activity in the arginine-to-proline pathway could be used as a therapeutic target in diabetic retinopathy.
Co-reporter:Mingliang Fang, Julijana Ivanisevic, H. Paul Benton, Caroline H. Johnson, Gary J. Patti, Linh T. Hoang, Winnie Uritboonthai, Michael E. Kurczy, and Gary Siuzdak
Analytical Chemistry 2015 Volume 87(Issue 21) pp:10935
Publication Date(Web):October 4, 2015
DOI:10.1021/acs.analchem.5b03003
Thermal processes are widely used in small molecule chemical analysis and metabolomics for derivatization, vaporization, chromatography, and ionization, especially in gas chromatography mass spectrometry (GC/MS). In this study the effect of heating was examined on a set of 64 small molecule standards and, separately, on human plasma metabolite extracts. The samples, either derivatized or underivatized, were heated at three different temperatures (60, 100, and 250 °C) at different exposure times (30 s, 60 s, and 300 s). All the samples were analyzed by liquid chromatography coupled to electrospray ionization mass spectrometry (LC/MS) and the data processed by XCMS Online (xcmsonline.scripps.edu). The results showed that heating at an elevated temperature of 100 °C had an appreciable effect on both the underivatized and derivatized molecules, and heating at 250 °C created substantial changes in the profile. For example, over 40% of the molecular peaks were altered in the plasma metabolite analysis after heating (250 °C, 300s) with a significant formation of degradation and transformation products. The analysis of 64 small molecule standards validated the temperature-induced changes observed on the plasma metabolites, where most of the small molecules degraded at elevated temperatures even after minimal exposure times (30 s). For example, tri- and diorganophosphates (e.g., adenosine triphosphate and adenosine diphosphate) were readily degraded into a mono-organophosphate (e.g., adenosine monophosphate) during heating. Nucleosides and nucleotides (e.g., inosine and inosine monophosphate) were also found to be transformed into purine derivatives (e.g., hypoxanthine). A newly formed transformation product, oleoyl ethyl amide, was identified in both the underivatized and derivatized forms of the plasma extracts and small molecule standard mixture, and was likely generated from oleic acid. Overall these analyses show that small molecules and metabolites undergo significant time-sensitive alterations when exposed to elevated temperatures, especially those conditions that mimic sample preparation and analysis in GC/MS experiments.
Co-reporter:H. Paul Benton, Julijana Ivanisevic, Nathaniel G. Mahieu, Michael E. Kurczy, Caroline H. Johnson, Lauren Franco, Duane Rinehart, Elizabeth Valentine, Harsha Gowda, Baljit K. Ubhi, Ralf Tautenhahn, Andrew Gieschen, Matthew W. Fields, Gary J. Patti, and Gary Siuzdak
Analytical Chemistry 2015 Volume 87(Issue 2) pp:884
Publication Date(Web):December 12, 2014
DOI:10.1021/ac5025649
An autonomous metabolomic workflow combining mass spectrometry analysis with tandem mass spectrometry data acquisition was designed to allow for simultaneous data processing and metabolite characterization. Although previously tandem mass spectrometry data have been generated on the fly, the experiments described herein combine this technology with the bioinformatic resources of XCMS and METLIN. As a result of this unique integration, we can analyze large profiling datasets and simultaneously obtain structural identifications. Validation of the workflow on bacterial samples allowed the profiling on the order of a thousand metabolite features with simultaneous tandem mass spectra data acquisition. The tandem mass spectrometry data acquisition enabled automatic search and matching against the METLIN tandem mass spectrometry database, shortening the current workflow from days to hours. Overall, the autonomous approach to untargeted metabolomics provides an efficient means of metabolomic profiling, and will ultimately allow the more rapid integration of comparative analyses, metabolite identification, and data analysis at a systems biology level.
Co-reporter:Caroline H. Johnson, Julijana Ivanisevic, H. Paul Benton, and Gary Siuzdak
Analytical Chemistry 2015 Volume 87(Issue 1) pp:147
Publication Date(Web):November 12, 2014
DOI:10.1021/ac5040693
Co-reporter:Julijana Ivanisevic
Journal of Neuroimmune Pharmacology 2015 Volume 10( Issue 3) pp:391-395
Publication Date(Web):2015 September
DOI:10.1007/s11481-015-9621-1
This special edition of the Journal of Neuroimmune Pharmacology focuses on the leading edge of metabolomics in brain metabolism research. The topics covered include a metabolomic field overview and the challenges in neuroscience metabolomics. The workflow and utility of different analytical platforms to profile complex biological matrices that include biofluids, brain tissue and cells, are shown in several case studies. These studies demonstrate how global and targeted metabolite profiling can be applied to distinguish disease stages and to understand the effects of drug action on the central nervous system (CNS). Finally, we discuss the importance of metabolomics to advance the understanding of brain function that includes ligand-receptor interactions and new insights into the mechanisms of CNS disorders.
Co-reporter:Caroline H. Johnson;Gary J. Patti
Journal of Neuroimmune Pharmacology 2015 Volume 10( Issue 3) pp:396-401
Publication Date(Web):2015 September
DOI:10.1007/s11481-015-9624-y
Therapeutic options for neuropathic pain have improved over the last 20 years yet still only provide partial relief with numerous side effects. Recently, metabolomics revealed that the concentration of the endogenous metabolite N,N-dimethylsphingosine (DMS) is increased in the spinal cord in a model of neuropathic pain. Additionally, it was shown that introduction of DMS to the central nervous system (CNS) resulted in mechanical allodynia. Here, we have examined two compounds; pregabalin (Lyrica®), a drug used to treat neuropathic pain, and N-oleoylethanolamine (NOE), an endogenous endocannabinoid-like compound that is known to affect multiple lipid pathways. We found that the concentration of DMS in the spinal cord was not significantly altered upon pregabalin treatment of rats suffering from neuropathic pain. We further explored whether modulating lipid metabolism may impact neuropathic pain by testing NOE as a potential novel therapeutic.
Co-reporter:Julijana Ivanisevic, Adrian A. Epstein, Michael E. Kurczy, Paul H. Benton, Winnie Uritboonthai, Howard S. Fox, Michael D. Boska, Howard E. Gendelman, Gary Siuzdak
Chemistry & Biology 2014 Volume 21(Issue 11) pp:1575-1584
Publication Date(Web):20 November 2014
DOI:10.1016/j.chembiol.2014.09.016
•Regional brain metabolomics reveals metabolite patterns across brain tissue•Global LC/MS profiling provides information on a broad array of brain metabolites•NIMS imaging provides subregional information about metabolite distributionHistorically, studies of brain metabolism have been based on targeted analyses of a limited number of metabolites. Here we present an untargeted mass spectrometry-based metabolomic strategy that has successfully uncovered differences in a broad array of metabolites across anatomical regions of the mouse brain. The NSG immunodeficient mouse model was chosen because of its ability to undergo humanization leading to numerous applications in oncology and infectious disease research. Metabolic phenotyping by hydrophilic interaction liquid chromatography and nanostructure imaging mass spectrometry revealed both water-soluble and lipid metabolite patterns across brain regions. Neurochemical differences in metabolic phenotypes were mainly defined by various phospholipids and several intriguing metabolites including carnosine, cholesterol sulfate, lipoamino acids, uric acid, and sialic acid, whose physiological roles in brain metabolism are poorly understood. This study helps define regional homeostasis for the normal mouse brain to give context to the reaction to pathological events.Figure optionsDownload full-size imageDownload high-quality image (469 K)Download as PowerPoint slide
Co-reporter:Kevin Cho, Nathaniel Mahieu, Julijana Ivanisevic, Winnie Uritboonthai, Ying-Jr Chen, Gary Siuzdak, and Gary J. Patti
Analytical Chemistry 2014 Volume 86(Issue 19) pp:9358
Publication Date(Web):August 28, 2014
DOI:10.1021/ac5029177
The METLIN metabolite database has become one of the most widely used resources in metabolomics for making metabolite identifications. However, METLIN is not designed to identify metabolites that have been isotopically labeled. As a result, unbiasedly tracking the transformation of labeled metabolites with isotope-based metabolomics is a challenge. Here, we introduce a new database, called isoMETLIN (http://isometlin.scripps.edu/), that has been developed specifically to identify metabolites incorporating isotopic labels. isoMETLIN enables users to search all computed isotopologues derived from METLIN on the basis of mass-to-charge values and specified isotopes of interest, such as 13C or 15N. Additionally, isoMETLIN contains experimental MS/MS data on hundreds of isotopomers. These data assist in localizing the position of isotopic labels within a metabolite. From these experimental MS/MS isotopomer spectra, precursor atoms can be mapped to fragments. The MS/MS spectra of additional isotopomers can then be computationally generated and included within isoMETLIN. Given that isobaric isotopomers cannot be separated chromatographically or by mass but are likely to occur simultaneously in a biological system, we have also implemented a spectral-mixing function in isoMETLIN. This functionality allows users to combine MS/MS spectra from various isotopomers in different ratios to obtain a theoretical MS/MS spectrum that matches the MS/MS spectrum from a biological sample. Thus, by searching MS and MS/MS experimental data, isoMETLIN facilitates the identification of isotopologues as well as isotopomers from biological samples and provides a platform to drive the next generation of isotope-based metabolomic studies.
Co-reporter:Harsha Gowda, Julijana Ivanisevic, Caroline H. Johnson, Michael E. Kurczy, H. Paul Benton, Duane Rinehart, Thomas Nguyen, Jayashree Ray, Jennifer Kuehl, Bernardo Arevalo, Peter D. Westenskow, Junhua Wang, Adam P. Arkin, Adam M. Deutschbauer, Gary J. Patti, and Gary Siuzdak
Analytical Chemistry 2014 Volume 86(Issue 14) pp:6931
Publication Date(Web):June 16, 2014
DOI:10.1021/ac500734c
XCMS Online (xcmsonline.scripps.edu) is a cloud-based informatic platform designed to process and visualize mass-spectrometry-based, untargeted metabolomic data. Initially, the platform was developed for two-group comparisons to match the independent, “control” versus “disease” experimental design. Here, we introduce an enhanced XCMS Online interface that enables users to perform dependent (paired) two-group comparisons, meta-analysis, and multigroup comparisons, with comprehensive statistical output and interactive visualization tools. Newly incorporated statistical tests cover a wide array of univariate analyses. Multigroup comparison allows for the identification of differentially expressed metabolite features across multiple classes of data while higher order meta-analysis facilitates the identification of shared metabolic patterns across multiple two-group comparisons. Given the complexity of these data sets, we have developed an interactive platform where users can monitor the statistical output of univariate (cloud plots) and multivariate (PCA plots) data analysis in real time by adjusting the threshold and range of various parameters. On the interactive cloud plot, metabolite features can be filtered out by their significance level (p-value), fold change, mass-to-charge ratio, retention time, and intensity. The variation pattern of each feature can be visualized on both extracted-ion chromatograms and box plots. The interactive principal component analysis includes scores, loadings, and scree plots that can be adjusted depending on scaling criteria. The utility of XCMS functionalities is demonstrated through the metabolomic analysis of bacterial stress response and the comparison of lymphoblastic leukemia cell lines.
Co-reporter:Gary J. Patti;Ralf Tautenhahn;Darcy Johannsen;Ewa Kalisiak
Metabolomics 2014 Volume 10( Issue 4) pp:737-743
Publication Date(Web):2014 August
DOI:10.1007/s11306-013-0608-8
The manipulation of distinct signaling pathways and transcription factors has been shown to influence life span in a cell-non-autonomous manner in multicellular model organisms such as Caenorhabditis elegans. These data suggest that coordination of whole-organism aging involves endocrine signaling, however, the molecular identities of such signals have not yet been determined and their potential relevance in humans is unknown. Here we describe a novel metabolomic approach to identify molecules directly associated with extended life span in C. elegans that represent candidate compounds for age-related endocrine signals. To identify metabolic perturbations directly linked to longevity, we developed metabolomic software for meta-analysis that enabled intelligent comparisons of multiple different mutants. Simple pairwise comparisons of long-lived glp-1, daf-2, and isp-1 mutants to their respective controls resulted in more than 11,000 dysregulated metabolite features of statistical significance. By using meta-analysis, we were able to reduce this number to six compounds most likely to be associated with life-span extension. Mass spectrometry-based imaging studies suggested that these metabolites might be localized to C. elegans muscle. We extended the metabolomic analysis to humans by comparing quadricep muscle tissue from young and old individuals and found that two of the same compounds associated with longevity in worms were also altered in human muscle with age. These findings provide candidate compounds that may serve as age-related endocrine signals and implicate muscle as a potential tissue regulating their levels in humans.
Co-reporter:Caroline H. Johnson;Timothy S. Fisher;Linh T. Hoang;Brunhilde H. Felding
Metabolomics 2014 Volume 10( Issue 3) pp:354-360
Publication Date(Web):2014 June
DOI:10.1007/s11306-014-0622-5
Luciferase transfected cell lines are used extensively for cancer models, revealing valuable biological information about disease mechanisms. However, these genetically encoded reporters, while useful for monitoring tumor response in cancer models, can impact cell metabolism. Indeed firefly luciferase and fatty acyl-CoA synthetases differ by a single amino acid, raising the possibility that luciferase activity might alter metabolism and introduce experimental artifacts. Therefore knowledge of the metabolic response to luciferase transfection is of significant importance, especially given the thousands of research studies using luciferase as an in vivo bioluminescence imaging reporter. Untargeted metabolomics experiments were performed to examine three different types of lymphoblastic leukemia cell lines (Ramos, Raji and SUP-T1) commonly used in cancer research, each were analyzed with and without vector transduction. The Raji model was also tested under perturbed starvation conditions to examine potential luciferase-mediated stress responses. The results showed that no significant metabolic differences were observed between parental and luciferase transduced cells for each cell line, and that luciferase overexpression does not alter cell metabolism under basal or perturbed conditions.
Co-reporter:Gary J. Patti, Ralf Tautenhahn, Duane Rinehart, Kevin Cho, Leah P. Shriver, Marianne Manchester, Igor Nikolskiy, Caroline H. Johnson, Nathaniel G. Mahieu, and Gary Siuzdak
Analytical Chemistry 2013 Volume 85(Issue 2) pp:798
Publication Date(Web):December 3, 2012
DOI:10.1021/ac3029745
Global metabolomics describes the comprehensive analysis of small molecules in a biological system without bias. With mass spectrometry-based methods, global metabolomic data sets typically comprise thousands of peaks, each of which is associated with a mass-to-charge ratio, retention time, fold change, p-value, and relative intensity. Although several visualization schemes have been used for metabolomic data, most commonly used representations exclude important data dimensions and therefore limit interpretation of global data sets. Given that metabolite identification through tandem mass spectrometry data acquisition is a time-limiting step of the untargeted metabolomic workflow, simultaneous visualization of these parameters from large sets of data could facilitate compound identification and data interpretation. Here, we present such a visualization scheme of global metabolomic data using a so-called “cloud plot” to represent multidimensional data from septic mice. While much attention has been dedicated to lipid compounds as potential biomarkers for sepsis, the cloud plot shows that alterations in hydrophilic metabolites may provide an early signature of the disease prior to the onset of clinical symptoms. The cloud plot is an effective representation of global mass spectrometry-based metabolomic data, and we describe how to extract it as standard output from our XCMS metabolomic software.
Co-reporter:Julijana Ivanisevic, Zheng-Jiang Zhu, Lars Plate, Ralf Tautenhahn, Stephen Chen, Peter J. O’Brien, Caroline H. Johnson, Michael A. Marletta, Gary J. Patti, and Gary Siuzdak
Analytical Chemistry 2013 Volume 85(Issue 14) pp:6876
Publication Date(Web):June 19, 2013
DOI:10.1021/ac401140h
Although the objective of any ‘omic science is broad measurement of its constituents, such coverage has been challenging in metabolomics because the metabolome is comprised of a chemically diverse set of small molecules with variable physical properties. While extensive studies have been performed to identify metabolite isolation and separation methods, these strategies introduce bias toward lipophilic or water-soluble metabolites depending on whether reversed-phase (RP) or hydrophilic interaction liquid chromatography (HILIC) is used, respectively. Here we extend our consideration of metabolome isolation and separation procedures to integrate RPLC/MS and HILIC/MS profiling. An aminopropyl-based HILIC/MS method was optimized on the basis of mobile-phase additives and pH, followed by evaluation of reproducibility. When applied to the untargeted study of perturbed bacterial metabolomes, the HILIC method enabled the accurate assessment of key, dysregulated metabolites in central carbon pathways (e.g., amino acids, organic acids, phosphorylated sugars, energy currency metabolites), which could not be retained by RPLC. To demonstrate the value of the integrative approach, bacterial cells, human plasma, and cancer cells were analyzed by combined RPLC/HILIC separation coupled to ESI positive/negative MS detection. The combined approach resulted in the observation of metabolites associated with lipid and central carbon metabolism from a single biological extract, using 80% organic solvent (ACN:MeOH:H2O 2:2:1). It enabled the detection of more than 30,000 features from each sample type, with the highest number of uniquely detected features by RPLC in ESI positive mode and by HILIC in ESI negative mode. Therefore, we conclude that when time and sample are limited, the maximum amount of biological information related to lipid and central carbon metabolism can be acquired by combining RPLC ESI positive and HILIC ESI negative mode analysis.
Co-reporter:Gary J. Patti,
Oscar Yanes
&
Gary Siuzdak
Nature Reviews Molecular Cell Biology 2012 13(4) pp:263
Publication Date(Web):2012-03-22
DOI:10.1038/nrm3314
Metabolites, the chemical entities that are transformed during metabolism, provide a functional readout of cellular biochemistry. With emerging technologies in mass spectrometry, thousands of metabolites can now be quantitatively measured from minimal amounts of biological material, which has thereby enabled systems-level analyses. By performing global metabolite profiling, also known as untargeted metabolomics, new discoveries linking cellular pathways to biological mechanism are being revealed and are shaping our understanding of cell biology, physiology and medicine.
Co-reporter:Athanasia D Panopoulos, Oscar Yanes, Sergio Ruiz, Yasuyuki S Kida, Dinh Diep, Ralf Tautenhahn, Aída Herrerías, Erika M Batchelder, Nongluk Plongthongkum, Margaret Lutz, W Travis Berggren, Kun Zhang, Ronald M Evans, Gary Siuzdak and Juan Carlos Izpisua Belmonte
Cell Research 2012 22(1) pp:168-177
Publication Date(Web):November 8, 2011
DOI:10.1038/cr.2011.177
Metabolism is vital to every aspect of cell function, yet the metabolome of induced pluripotent stem cells (iPSCs) remains largely unexplored. Here we report, using an untargeted metabolomics approach, that human iPSCs share a pluripotent metabolomic signature with embryonic stem cells (ESCs) that is distinct from their parental cells, and that is characterized by changes in metabolites involved in cellular respiration. Examination of cellular bioenergetics corroborated with our metabolomic analysis, and demonstrated that somatic cells convert from an oxidative state to a glycolytic state in pluripotency. Interestingly, the bioenergetics of various somatic cells correlated with their reprogramming efficiencies. We further identified metabolites that differ between iPSCs and ESCs, which revealed novel metabolic pathways that play a critical role in regulating somatic cell reprogramming. Our findings are the first to globally analyze the metabolome of iPSCs, and provide mechanistic insight into a new layer of regulation involved in inducing pluripotency, and in evaluating iPSC and ESC equivalence.
Co-reporter:Oscar Yanes, Ralf Tautenhahn, Gary J. Patti, and Gary Siuzdak
Analytical Chemistry 2011 Volume 83(Issue 6) pp:2152
Publication Date(Web):February 17, 2011
DOI:10.1021/ac102981k
Mass spectrometry-based metabolomics is the comprehensive study of naturally occurring small molecules collectively known as the metabolome. Given the vast structural diversity and chemical properties of endogenous metabolites, biological extraction and chromatography methods bias the number, property, and concentration of metabolites detected by mass spectrometry and creates a challenge for global untargeted studies. In this work, we used Escherichia coli bacterial cells to explore the influence of solvent polarity, temperature, and pH in extracting polar and nonpolar metabolites simultaneously. In addition, we explored chromatographic conditions involving different stationary and mobile phases that optimize the separation and ionization of endogenous metabolite extracts as well as a mixture of synthetic standards. Our results reveal that hot polar solvents are the most efficient in extracting both hydrophilic and hydrophobic metabolites simultaneously. In addition, ammonium fluoride in the mobile phase substantially improved ionization efficiency in negative electrospray ionization mode by an average increase in signal intensity of 5.7 and over a 2-fold increase in the total number of features detected. The improvement in sensitivity with ammonium fluoride resulted in 3.5 times as many metabolite hits in databases compared to ammonium acetate or formic acid enriched mobile phases and allowed for the identification of unique metabolites involved in fundamental cellular pathways.
Co-reporter:Ralf Tautenhahn, Gary J. Patti, Ewa Kalisiak, Takashi Miyamoto, Manuela Schmidt, Fang Yin Lo, Joshua McBee, Nitin S. Baliga, and Gary Siuzdak
Analytical Chemistry 2011 Volume 83(Issue 3) pp:696
Publication Date(Web):December 21, 2010
DOI:10.1021/ac102980g
Mass spectrometry-based untargeted metabolomics often results in the observation of hundreds to thousands of features that are differentially regulated between sample classes. A major challenge in interpreting the data is distinguishing metabolites that are causally associated with the phenotype of interest from those that are unrelated but altered in downstream pathways as an effect. To facilitate this distinction, here we describe new software called metaXCMS for performing second-order (“meta”) analysis of untargeted metabolomics data from multiple sample groups representing different models of the same phenotype. While the original version of XCMS was designed for the direct comparison of two sample groups, metaXCMS enables meta-analysis of an unlimited number of sample classes to facilitate prioritization of the data and increase the probability of identifying metabolites causally related to the phenotype of interest. metaXCMS is used to import XCMS results that are subsequently filtered, realigned, and ultimately compared to identify shared metabolites that are up- or down-regulated across all sample groups. We demonstrate the software’s utility by identifying histamine as a metabolite that is commonly altered in three different models of pain. metaXCMS is freely available at http://metlin.scripps.edu/metaxcms/.
Co-reporter:Gary J. Patti, Hin-Koon Woo, Oscar Yanes, Leah Shriver, Diane Thomas, Wilasinee Uritboonthai, Junefredo V. Apon, Rick Steenwyk, Marianne Manchester and Gary Siuzdak
Analytical Chemistry 2010 Volume 82(Issue 1) pp:121
Publication Date(Web):December 4, 2009
DOI:10.1021/ac9014353
Nanostructure-initiator mass spectrometry (NIMS) is a highly sensitive, matrix-free technique that is well suited for biofluid analysis and imaging of biological tissues. Here we provide a new technical variation of NIMS to analyze carbohydrates and steroids, molecules that are challenging to detect with traditional mass spectrometric approaches. Analysis of carbohydrates and steroids was accomplished by spray depositing NaCl or AgNO3 on the NIMS porous silicon surface to provide a uniform environment rich with cationization agents prior to desporption of the fluorinated polymer initiator. Laser desorption/ionization of the ion-coated NIMS surface allowed for Na+ cationization of carbohydrates and Ag+ cationization of steroids. The reliability of the approach is quantitatively demonstrated with a calibration curve over the physiological range of glucose and cholesterol concentrations in human serum (1−200 μM). Additionally, we illustrate the sensitivity of the method by showing its ability to detect carbohydrates and steroids down to the 800-amol and 100-fmol levels, respectively. The technique developed is well suited for tissue imaging of biologically significant metabolites such as sucrose and cholesterol. To highlight its applicability, we used cation-enhanced NIMS to image the distribution of sucrose in a Gerbera jamesonii flower stem and the distribution of cholesterol in a mouse brain. The flower stem and brain sections were placed directly on the ion-coated NIMS surface without further preparation and analyzed directly. The overall results reported underscore the potential of NIMS to analyze and image chemically diverse compounds that have been traditionally challenging to observe with mass spectrometry-based techniques.
Co-reporter:Oscar Yanes, Hin-Koon Woo, Trent R. Northen, Stacey R. Oppenheimer, Leah Shriver, Jon Apon, Mayra N. Estrada, Michael J. Potchoiba, Rick Steenwyk, Marianne Manchester and Gary Siuzdak
Analytical Chemistry 2009 Volume 81(Issue 8) pp:2969
Publication Date(Web):March 20, 2009
DOI:10.1021/ac802576q
Nanostructure initiator mass spectrometry (NIMS) is a recently introduced matrix-free desorption/ionization platform that requires minimal sample preparation. Its application to xenobiotics and endogenous metabolites in tissues is demonstrated, where clozapine and N-desmethylclozapine were observed from mouse and rat brain sections. It has also been applied to direct biofluid analysis where ketamine and norketamine were observed from plasma and urine. Detection of xenobiotics from biofluids was made even more effective using a novel NIMS on-surface extraction method taking advantage of the hydrophobic nature of the initiator. Linear response and limit of detection were also evaluated for xenobiotics such as methamphetamine, codeine, alprazolam, and morphine, revealing that NIMS can be used for quantitative analysis. Overall, our results demonstrate the capacity of NIMS to perform sensitive, simple, and rapid analyses from highly complex biological tissues and fluids.
Co-reporter:Bridgit Crews, William R. Wikoff, Gary J. Patti, Hin-Koon Woo, Ewa Kalisiak, Johanna Heideker and Gary Siuzdak
Analytical Chemistry 2009 Volume 81(Issue 20) pp:8538
Publication Date(Web):September 18, 2009
DOI:10.1021/ac9014947
Analytical and biological variability are issues of central importance to human metabolomics studies. Here both types of variation are examined in human plasma and cerebrospinal fluid (CSF) using a global liquid chromatography/mass spectrometry (LC/MS) metabolomics strategy. The platform shows small analytical variation with a median coefficient of variation (CV) of 15−16% for both plasma and CSF sample matrixes when the integrated area of each peak in the mass spectra is considered. Analysis of biological variation shows that human CSF has a median CV of 35% and plasma has a median CV of 46%. To understand the difference in CV between the biofluids, we compared plasma and CSF independently obtained from different healthy humans. Additionally, we analyzed another group of patients from whom we compared matched CSF and plasma (plasma and CSF obtained from the same human subject). A similar number of features was observed in both biofluids, although the majority of features appeared with greater intensity in plasma. More than a dozen metabolites shared between the human CSF and plasma metabolomes were identified based on accurate mass measurements, retention times, and MS/MS spectra. The fold change in these metabolites was consistent with the median biological CV determined for all peaks. The measured median biological CV together with analysis of intragroup variation of healthy individuals suggests that fold changes above 2 in metabolomics studies investigating plasma or CSF are statistically relevant with respect to the inherent variability of a healthy control group. These data demonstrate the reproducibility of the global metabolomics platform using LC/MS and reveal the robustness of the approach for biomarker discovery.
Co-reporter:William R. Wikoff;Andrew T. Anfora;Jun Liu;Peter G. Schultz;Scott A. Lesley;Eric C. Peters
PNAS 2009 Volume 106 (Issue 10 ) pp:3698-3703
Publication Date(Web):2009-03-10
DOI:10.1073/pnas.0812874106
Although it has long been recognized that the enteric community of bacteria that inhabit the human distal intestinal track
broadly impacts human health, the biochemical details that underlie these effects remain largely undefined. Here, we report
a broad MS-based metabolomics study that demonstrates a surprisingly large effect of the gut “microbiome” on mammalian blood
metabolites. Plasma extracts from germ-free mice were compared with samples from conventional (conv) animals by using various
MS-based methods. Hundreds of features were detected in only 1 sample set, with the majority of these being unique to the
conv animals, whereas ≈10% of all features observed in both sample sets showed significant changes in their relative signal
intensity. Amino acid metabolites were particularly affected. For example, the bacterial-mediated production of bioactive
indole-containing metabolites derived from tryptophan such as indoxyl sulfate and the antioxidant indole-3-propionic acid
(IPA) was impacted. Production of IPA was shown to be completely dependent on the presence of gut microflora and could be
established by colonization with the bacterium Clostridium sporogenes. Multiple organic acids containing phenyl groups were also greatly increased in the presence of gut microbes. A broad, drug-like
phase II metabolic response of the host to metabolites generated by the microbiome was observed, suggesting that the gut microflora
has a direct impact on the drug metabolism capacity of the host. Together, these results suggest a significant interplay between
bacterial and mammalian metabolism.
Co-reporter:H. P. Benton, D. M. Wong, S. A. Trauger and G. Siuzdak
Analytical Chemistry 2008 Volume 80(Issue 16) pp:6382
Publication Date(Web):July 16, 2008
DOI:10.1021/ac800795f
Mass spectrometry based metabolomics represents a new area for bioinformatics technology development. While the computational tools currently available such as XCMS statistically assess and rank LC−MS features, they do not provide information about their structural identity. XCMS2 is an open source software package which has been developed to automatically search tandem mass spectrometry (MS/MS) data against high quality experimental MS/MS data from known metabolites contained in a reference library (METLIN). Scoring of hits is based on a “shared peak count” method that identifies masses of fragment ions shared between the analytical and reference MS/MS spectra. Another functional component of XCMS2 is the capability of providing structural information for unknown metabolites, which are not in the METLIN database. This “similarity search” algorithm has been developed to detect possible structural motifs in the unknown metabolite which may produce characteristic fragment ions and neutral losses to related reference compounds contained in METLIN, even if the precursor masses are not the same.
Co-reporter:Sunia A. Trauger, Ewa Kalisak, Jaroslaw Kalisiak, Hirotoshi Morita, Michael V. Weinberg, Angeli Lal Menon, Farris L. Poole II, Michael W. W. Adams and Gary Siuzdak
Journal of Proteome Research 2008 Volume 7(Issue 3) pp:1027-1035
Publication Date(Web):2017-2-22
DOI:10.1021/pr700609j
We have performed a comprehensive characterization of global molecular changes for a model organism Pyrococcus furiosus using transcriptomic (DNA microarray), proteomic, and metabolomic analysis as it undergoes a cold adaptation response from its optimal 95 to 72 °C. Metabolic profiling on the same set of samples shows the down-regulation of many metabolites. However, some metabolites are found to be strongly up-regulated. An approach using accurate mass, isotopic pattern, database searching, and retention time is used to putatively identify several metabolites of interest. Many of the up-regulated metabolites are part of an alternative polyamine biosynthesis pathway previously established in a thermophilic bacterium Thermus thermophilus. Arginine, agmatine, spermidine, and branched polyamines N 4-aminopropylspermidine and N 4-( N-acetylaminopropyl)spermidine were unambiguously identified based on their accurate mass, isotopic pattern, and matching of MS/MS data acquired under identical conditions for the natural metabolite and a high purity standard. Both DNA microarray and semiquantitative proteomic analysis using a label-free spectral counting approach indicate the down-regulation of a large majority of genes with diverse predicted functions related to growth such as transcription, amino acid biosynthesis, and translation. Some genes are, however, found to be up-regulated through the measurement of their relative mRNA and protein levels. The complimentary information obtained by the various “omics” techniques is used to catalogue and correlate the overall molecular changes.
Co-reporter:Jinq-Chyi Lee;Steven M. Yannone;Jason Raymond;Chi-Huey Wong;Trent R. Northen;Linh Hoang;Der-Ren Hwang
PNAS 2008 Volume 105 (Issue 10 ) pp:3678-3683
Publication Date(Web):2008-03-11
DOI:10.1073/pnas.0712332105
We describe a Nanostructure-Initiator Mass Spectrometry (NIMS) enzymatic (Nimzyme) assay in which enzyme substrates are immobilized
on the mass spectrometry surface by using fluorous-phase interactions. This “soft” immobilization allows efficient desorption/ionization
while also enabling the use of surface-washing steps to reduce signal suppression from complex biological samples, which results
from the preferential retention of the tagged products and reactants. The Nimzyme assay is sensitive to subpicogram levels
of enzyme, detects both addition and cleavage reactions (sialyltransferase and galactosidase), is applicable over a wide range
of pHs and temperatures, and can measure activity directly from crude cell lysates. The ability of the Nimzyme assay to analyze
complex mixtures is illustrated by identifying and directly characterizing β-1,4-galactosidase activity from a thermophilic
microbial community lysate. The optimal enzyme temperature and pH were found to be 65°C and 5.5, respectively, and the activity
was inhibited by both phenylethyl-β-d-thiogalactopyranoside and deoxygalactonojirimycin. Metagenomic analysis of the community suggests that the activity is from
an uncultured, unsequenced γ-proteobacterium. In general, this assay provides an efficient method for detection and characterization
of enzymatic activities in complex biological mixtures prior to sequencing or cloning efforts. More generally, this approach
may have important applications for screening both enzymatic and inhibitor libraries, constructing and screening glycan microarrays,
and complementing fluorous-phase organic synthesis.
Co-reporter:Trent R. Northen;Hin-Koon Woo
Journal of The American Society for Mass Spectrometry 2007 Volume 18( Issue 11) pp:1945-1949
Publication Date(Web):2007 November
DOI:10.1016/j.jasms.2007.08.009
The surface structure of porous silicon used in desorption/ionization on porous silicon (DIOS) mass analysis is known to play a primary role in the desorption/ionization (D/I) process. In this study, mass spectrometry and scanning electron microscopy (SEM) are used to examine the correlation between intact ion generation with surface ablation and surface morphology. The DIOS process is found to be highly laser energy dependent and correlates directly with the appearance of surface ions (Sin+ and OSiH+). A threshold laser energy for DIOS is observed (10 mJ/cm2), which supports that DIOS is driven by surface restructuring and is not a strictly thermal process. In addition, three DIOS regimes are observed that correspond to surface restructuring and melting. These results suggest that higher surface area silicon substrates may enhance DIOS performance. A recent example that fits into this mechanism is the surface of silicon nanowires, which has a high surface energy and concomitantly requires lower laser energy for analyte desorption.
Co-reporter:David M Mutch;Grace O'Maille;William R Wikoff;Therese Wiedmer
Genome Biology 2007 Volume 8( Issue 3) pp:
Publication Date(Web):2007 March
DOI:10.1186/gb-2007-8-3-r38
The obesity epidemic has prompted the search for candidate genes capable of influencing adipose function. One such candidate, that encoding phospholipid scramblase 3 (PLSCR3), was recently identified, as genetic deletion of it led to lipid accumulation in abdominal fat pads and changes characteristic of metabolic syndrome. Because adipose tissue is increasingly recognized as an endocrine organ, capable of releasing small molecules that modulate disparate physiological processes, we examined the plasma from wild-type, Plscr1-/-, Plscr3-/- and Plscr1&3-/- mice. Using an untargeted comprehensive metabolite profiling approach coupled with targeted gene expression analyses, the perturbed biochemistry and functional redundancy of PLSCR proteins was assessed.Nineteen metabolites were differentially and similarly regulated in both Plscr3-/- and Plscr1&3-/- animals, of which five were characterized from accurate mass, tandem mass spectrometry data and their correlation to the Metlin database as lysophosphatidylcholine (LPC) species enriched with C16:1, C18:1, C20:3, C20:5 and C22:5 fatty acids. No significant changes in the plasma metabolome were detected upon elimination of PLSCR1, indicating that increases in pro-inflammatory lipids are specifically associated with the obese state of Plscr3-deficient animals. Correspondingly, increases in white adipose lipogenic gene expression confirm a role for PLSCR3 in adipose lipid metabolism.The untargeted profiling of circulating metabolites suggests no detectable functional redundancies between PLSCR proteins; however, this approach simultaneously identified previously unrecognized lipid metabolites that suggest a novel molecular link between obesity, inflammation and the downstream consequences associated with PLSCR3-deficiency.
Co-reporter:Trent R. Northen,
Oscar Yanes,
Michael T. Northen,
Dena Marrinucci,
Winnie Uritboonthai,
Junefredo Apon,
Stephen L. Golledge,
Anders Nordström
&
Gary Siuzdak
Nature 2007 449(7165) pp:1033
Publication Date(Web):2007-10-25
DOI:10.1038/nature06195
The ability of mass spectrometry to generate intact biomolecular ions efficiently in the gas phase has led to its widespread application in metabolomics1, proteomics2, biological imaging3, biomarker discovery4 and clinical assays (namely neonatal screens5). Matrix-assisted laser desorption/ionization6, 7 (MALDI) and electrospray ionization8 have been at the forefront of these developments. However, matrix application complicates the use of MALDI for cellular, tissue, biofluid and microarray analysis and can limit the spatial resolution because of the matrix crystal size9 (typically more than 10 μm), sensitivity and detection of small compounds (less than 500 Da). Secondary-ion mass spectrometry10 has extremely high lateral resolution (100 nm) and has found biological applications11, 12 although the energetic desorption/ionization is a limitation owing to molecular fragmentation. Here we introduce nanostructure-initiator mass spectrometry (NIMS), a tool for spatially defined mass analysis. NIMS uses ‘initiator’ molecules trapped in nanostructured surfaces or ‘clathrates’ to release and ionize intact molecules adsorbed on the surface. This surface responds to both ion and laser irradiation. The lateral resolution (ion-NIMS about 150 nm), sensitivity, matrix-free and reduced fragmentation of NIMS allows direct characterization of peptide microarrays, direct mass analysis of single cells, tissue imaging, and direct characterization of blood and urine.
Co-reporter:Elizabeth J. Want Dr.;Benjamin F. Cravatt
ChemBioChem 2005 Volume 6(Issue 11) pp:
Publication Date(Web):4 OCT 2005
DOI:10.1002/cbic.200500151
Mass spectrometry has a strong history in drug-metabolite analysis and has recently emerged as the foremost technology in endogenous metabolite research. The advantages of mass spectrometry include a wide dynamic range, the ability to observe a diverse number of molecular species, and reproducible quantitative analysis. These attributes are important in addressing the issue of metabolite profiling, as the dynamic range easily exceeds nine orders of magnitude in biofluids, and the diversity of species ranges from simple amino acids to lipids to complex carbohydrates. The goals of the application of mass spectrometry range from basic biochemistry to clinical biomarker discovery with challenges in generating a comprehensive profile, data analysis, and structurally characterizing physiologically important metabolites. The precedent for this work has already been set in neonatal screening, as blood samples from millions of neonates are tested routinely by mass spectrometry as a diagnostic tool for inborn errors of metabolism. In this review, we will discuss the background from which contemporary metabolite research emerged, the techniques involved in this exciting area, and the current and future applications of this field.
Co-reporter:Brian Bothner Dr. Dr.
ChemBioChem 2004 Volume 5(Issue 3) pp:
Publication Date(Web):26 FEB 2004
DOI:10.1002/cbic.200300754
Staying together: Electrospray ionization has been shown to generate intact gas-phase viral ions that can be used to examine mass and structure of whole viruses such as the tabac mosaic virus. That these particles are viable can be demonstrated by the infection of tobacco plants, which then develop characteristic lesions (see picture).
Co-reporter:Sunia A. Trauger Dr.;Eugene Wu Dr.;Steve J. Bark Dr.;Glen R. Nemerow Dr.
ChemBioChem 2004 Volume 5(Issue 8) pp:
Publication Date(Web):2 AUG 2004
DOI:10.1002/cbic.200400037
A tandem mass spectrometry-based approach is demonstrated for detecting a receptor for Ad37, one of the causative agents for epidemic keratoconjunctivitis. Partial purification of membrane glycoproteins was performed by using lectin-affinity chromatography and SDS-PAGE. Gel bands that were shown to bind Ad37 by using Viral Overlay Protein Blot Assay (VOPBA) were excised, proteolyzed and analyzed by using nanoLC-MS/MS to identify putative receptors contained in a mixture of proteins. Four candidate receptors were identified among approximately 50 proteins based on a search against a protein database. Inhibition of gene delivery mediated by an Ad37 vector, with antibodies against the glycoproteins identified by tandem mass spectrometry, strongly indicated that Membrane Cofactor Protein (MCP), a member of the complement regulatory family of proteins, is the receptor. This rapid and sensitive MS/MS-based strategy is perceived to have wide potential applications for the detection of viral receptors.
Co-reporter:Zhouxin Shen Dr.;Eden P. Go Dr.;Alejra Gamez Dr.;Junefredo V. Apon;Valery Fokin Dr.;Mike Greig;Manuel Ventura Dr.;John E. Crowell Dr.;Ola Blixt Dr.;James C. Paulson ;Raymond C. Stevens ;M. G. Finn Dr. Dr.
ChemBioChem 2004 Volume 5(Issue 7) pp:
Publication Date(Web):1 JUL 2004
DOI:10.1002/cbic.200400008
A surface-based laser desorption/ionization mass spectrometry assay that makes use of Desorption/Ionization on Silicon Mass Spectrometry (DIOS-MS) has been developed to monitor enzyme activity and enzyme inhibition. DIOS-MS has been used to characterize inhibitors from a library and then to monitor their activity against selected enzyme targets, including proteases, glycotransferase, and acetylcholinesterase. An automated DIOS-MS system was also used as a high-throughput screen for the activity of novel enzymes and enzyme inhibitors. On two different commercially available instruments, a sampling rate of up to 38 inhibitors per minute was accomplished, with thousands of inhibitors being monitored. The ease of applying mass spectrometry toward developing enzyme assays and the speed of surface-based assays such as DIOS for monitoring inhibitor effectiveness and enzyme activity makes it attractive for a broad range of screening applications.
Co-reporter:Zhouxin Shen Dr.;Eden P. Go Dr.;Alejra Gamez Dr.;Junefredo V. Apon;Valery Fokin Dr.;Mike Greig;Manuel Ventura Dr.;John E. Crowell Dr.;Ola Blixt Dr.;James C. Paulson ;Raymond C. Stevens ;M. G. Finn Dr. Dr.
ChemBioChem 2004 Volume 5(Issue 7) pp:
Publication Date(Web):1 JUL 2004
DOI:10.1002/cbic.200490028
Co-reporter:Warren G. Lewis, Zhouxin Shen, M.G. Finn, Gary Siuzdak
International Journal of Mass Spectrometry 2003 Volume 226(Issue 1) pp:107-116
Publication Date(Web):15 March 2003
DOI:10.1016/S1387-3806(02)00973-9
Desorption/ionization on silicon mass spectrometry (DIOS-MS) is a matrix-less laser vaporization method for generating gas-phase ions. The physical properties of the silicon surfaces are crucial to DIOS-MS performance and are controlled by the selection of silicon type and the silicon etching conditions. DIOS-MS has been examined for its applicability to small-molecule analysis, quantitative studies, reaction monitoring, chromatography, protein identification, and protein functional characterization. In organic chemistry, DIOS has been applied to the analysis of reactions directed toward development of new catalysts and transformations. Because DIOS offers a chip-based format, it is capable of being used to raster the silicon surface for biological and chemical screening applications, such as enzymatic activity assays. DIOS-MS extends the mass analysis capabilities of laser desorption to small biomolecules and thus offers a platform on which multiple experiments can be performed on a wide variety of molecules.
Co-reporter:John J. Thomas, Zhouxin Shen, Robert Blackledge, Gary Siuzdak
Analytica Chimica Acta 2001 Volume 442(Issue 2) pp:183-190
Publication Date(Web):5 September 2001
DOI:10.1016/S0003-2670(01)01107-2
Desorption–ionization on silicon (DIOS) is a new, matrix-free laser desorption mass spectrometry approach that allows for the direct identification of low molecular weight compounds in the presence of potentially interfering compounds. The porous silicon surfaces provide a scaffold for trapping analyte molecules, and are readily adaptable to commercial time-of-flight instruments. As an example of its utility in forensic cases, DIOS mass spectrometry was used to distinguish between similar synthetic polymers and identify specific polymers from complex biological media. Despite the absence of matrix, specific low molecular weight polymers were rapidly identified without any fragmentation. This method was applied to the rapid identification of ethoxylate polymers during a criminal investigation.
Co-reporter:Stephen D. Fuerstenau Dr.;W. Henry Benner Dr.;John J. Thomas Dr.;Christophe Brugidou Dr.;Brian Bothner Dr.
Angewandte Chemie 2001 Volume 113(Issue 6) pp:
Publication Date(Web):14 MAR 2001
DOI:10.1002/1521-3757(20010316)113:6<1011::AID-ANGE10112>3.0.CO;2-C
Co-reporter:Stephen D. Fuerstenau Dr.;W. Henry Benner Dr.;John J. Thomas Dr.;Christophe Brugidou Dr.;Brian Bothner Dr.
Angewandte Chemie 2001 Volume 113(Issue 3) pp:
Publication Date(Web):30 JAN 2001
DOI:10.1002/1521-3757(20010202)113:3<559::AID-ANGE559>3.0.CO;2-#
Co-reporter:John J. Thomas;Zhouxin Shen;John E. Crowell;M. G. Finn
PNAS 2001 Volume 98 (Issue 9 ) pp:4932-4937
Publication Date(Web):2001-04-24
DOI:10.1073/pnas.081069298
Since the advent of matrix-assisted laser desorption/ionization
and electrospray ionization, mass spectrometry has played an
increasingly important role in protein functional characterization,
identification, and structural analysis. Expanding this role,
desorption/ionization on silicon (DIOS) is a new approach that allows
for the analysis of proteins and related small molecules. Despite the
absence of matrix, DIOS-MS yields little or no fragmentation and is
relatively tolerant of moderate amounts of contaminants commonly found
in biological samples. Here, functional assays were performed on an
esterase, a glycosidase, a lipase, as well as exo- and endoproteases by
using enzyme-specific substrates. Enzyme activity also was monitored in
the presence of inhibitors, successfully demonstrating the ability of
DIOS to be used as an inhibitor screen. Because DIOS is a matrix-free
desorption technique, it also can be used as a platform for multiple
analyses to be performed on the same protein. This unique advantage was
demonstrated with acetylcholine esterase for qualitative and
quantitative characterization and also by its subsequent identification
directly from the DIOS platform.
Co-reporter:Klas Broo;Jing Wei;Dawn Marshall;Fred Brown;Thomas J. Smith;John E. Johnson;Anette Schneemann
PNAS 2001 Volume 98 (Issue 5 ) pp:2274-2277
Publication Date(Web):2001-02-27
DOI:10.1073/pnas.051598298
Mass spectrometry and fluorescent probes have provided direct
evidence that alkylating agents permeate the protein capsid of naked
viruses and chemically inactivate the nucleic acid.
N-acetyl-aziridine and a fluorescent alkylating agent,
dansyl sulfonate aziridine, inactivated three different viruses, flock
house virus, human rhinovirus-14, and foot and mouth disease virus.
Mass spectral studies as well as fluorescent probes showed that
alkylation of the genome was the mechanism of inactivation. Because
particle integrity was not affected by selective alkylation (as shown
by electron microscopy and sucrose gradient experiments), it was
reasoned that the dynamic nature of the viral capsid acts as a conduit
to the interior of the particle. Potential applications include
fluorescent labeling for imaging viral genomes in living cells, the
sterilization of blood products, vaccine development, and viral
inactivation in vivo.
Co-reporter:Jing Wei,
Jillian M. Buriak
and
Gary Siuzdak
Nature 1999 399(6733) pp:243
Publication Date(Web):
DOI:10.1038/20400
Desorption mass spectrometry has undergone significant improvements since the original experiments were performed more than 90 years ago1. The most dramatic change occurred in the early1980s with the introduction of an organic matrix2,3,4 to transfer energy to the analyte. This reduces ion fragmentation but also introduces background ions from the matrix. Here we describe a matrix-free strategy forbiomolecular mass spectrometry based on pulsed-laser desorption–ionization from a porous silicon5 surface. Our method uses porous silicon to trap analytes deposited on the surface, and laser irradiation to vaporize and ionize them. We show that the method works at femtomole and attomole levels of analyte, and induces little or no fragmentation, in contrast to what is typically observed with other such approaches6,7,8,9,10,11. The ability to perform these measurements without a matrix3,4,12,13 also makes itmore amenable to small-molecule analysis. Chemical14 and structural15 modification of the porous silicon has enabled optimization of the ionization characteristics of the surface. Our technique offers good sensitivity as well as compatibility with silicon-based microfluidics and microchip technologies.
Co-reporter:Trent R. Northen, Hin-Koon Woo, Michael T. Northen, Anders Nordström, Winnie Uritboonthail, Kimberly L. Turner, Gary Siuzdak
Journal of the American Society for Mass Spectrometry (November 2007) Volume 18(Issue 11) pp:1945-1949
Publication Date(Web):1 November 2007
DOI:10.1016/j.jasms.2007.08.009
The surface structure of porous silicon used in desorption/ionization on porous silicon (DIOS) mass analysis is known to play a primary role in the desorption/ionization (D/I) process. In this study, mass spectrometry and scanning electron microscopy (SEM) are used to examine the correlation between intact ion generation with surface ablation and surface morphology. The DIOS process is found to be highly laser energy dependent and correlates directly with the appearance of surface ions (Sin+ and OSiH+). A threshold laser energy for DIOS is observed (10 mJ/cm2), which supports that DIOS is driven by surface restructuring and is not a strictly thermal process. In addition, three DIOS regimes are observed that correspond to surface restructuring and melting. These results suggest that higher surface area silicon substrates may enhance DIOS performance. A recent example that fits into this mechanism is the surface of silicon nanowires, which has a high surface energy and concomitantly requires lower laser energy for analyte desorption.
.jpg)
![L-Aspartic acid,N-[[[(4S)-4-amino-4-carboxybutyl]amino]iminomethyl]-](http://img.cochemist.com/ccimg/2400/2387-71-5.png)
![L-Aspartic acid,N-[[[(4S)-4-amino-4-carboxybutyl]amino]iminomethyl]-](http://img.cochemist.com/ccimg/2400/2387-71-5_b.png)
![1-Propanaminium,3-carboxy-N,N,N-trimethyl-2-[(1-oxooctadecyl)oxy]-, inner salt](http://img.cochemist.com/ccimg/2000/1976-27-8.png)
![1-Propanaminium,3-carboxy-N,N,N-trimethyl-2-[(1-oxooctadecyl)oxy]-, inner salt](http://img.cochemist.com/ccimg/2000/1976-27-8_b.png)










































![4,6,10-Trioxa-5-phosphatetracosanoicacid, 2-amino-5-hydroxy-11-oxo-8-[(1-oxotetradecyl)oxy]-, 5-oxide, (2S,8R)-](http://img.cochemist.com/ccimg/64100/64023-32-1.png)
![4,6,10-Trioxa-5-phosphatetracosanoicacid, 2-amino-5-hydroxy-11-oxo-8-[(1-oxotetradecyl)oxy]-, 5-oxide, (2S,8R)-](http://img.cochemist.com/ccimg/64100/64023-32-1_b.png)
![4H-Imidazo[1,5-a][1,4]benzodiazepin-4-ol,8-chloro-6-(2-fluorophenyl)-1-methyl-](http://img.cochemist.com/ccimg/59500/59468-85-8.png)
![4H-Imidazo[1,5-a][1,4]benzodiazepin-4-ol,8-chloro-6-(2-fluorophenyl)-1-methyl-](http://img.cochemist.com/ccimg/59500/59468-85-8_b.png)




![L-Ornithine,N5-[imino(nitroamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/51000/50903-99-6.png)
![L-Ornithine,N5-[imino(nitroamino)methyl]-, methyl ester](http://img.cochemist.com/ccimg/51000/50903-99-6_b.png)




![1-Propanaminium, 3-carboxy-N,N,N-trimethyl-2-[(1-oxotetradecyl)oxy]-, inner salt, (2R)-](/data/chemimg/3784600/25597-07-3.png)
![1-Propanaminium, 3-carboxy-N,N,N-trimethyl-2-[(1-oxotetradecyl)oxy]-, inner salt, (2R)-](/data/chemimg/3784600/25597-07-3_b.png)

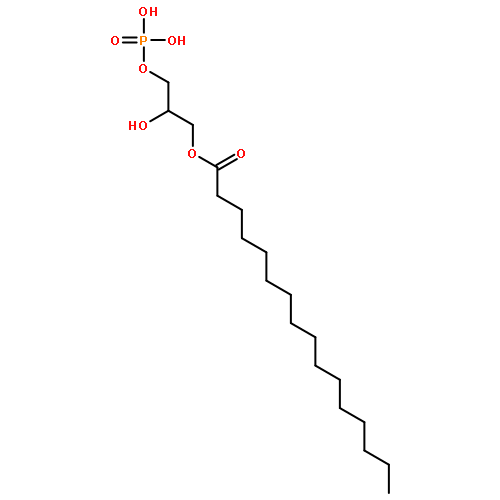
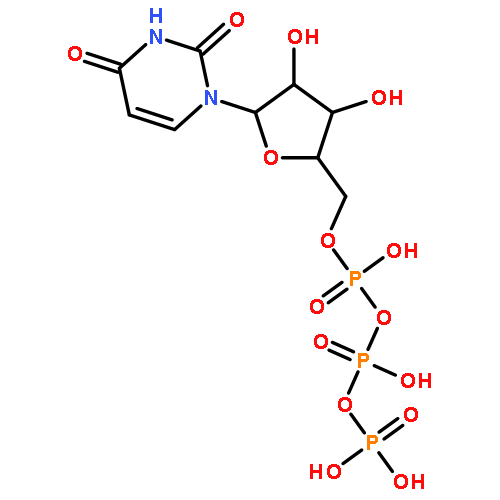
![[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[(3,4,5-trihydroxyoxolan-2-yl)methoxy]phosphoryl] Hydrogen Phosphate](http://img.cochemist.com/ccimg/20800/20762-30-5.png)
![[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[(3,4,5-trihydroxyoxolan-2-yl)methoxy]phosphoryl] Hydrogen Phosphate](http://img.cochemist.com/ccimg/20800/20762-30-5_b.png)
![3,5,9-Trioxa-4-phosphaheptacosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxotetradecyl)oxy]-, inner salt,4-oxide, (7R)-](http://img.cochemist.com/ccimg/20700/20664-02-2.png)
![3,5,9-Trioxa-4-phosphaheptacosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxotetradecyl)oxy]-, inner salt,4-oxide, (7R)-](http://img.cochemist.com/ccimg/20700/20664-02-2_b.png)



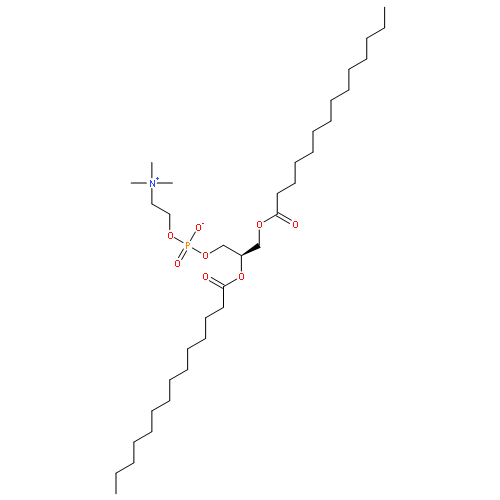
![3,5,9-Trioxa-4-phosphaheneicosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxododecyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/18200/18194-25-7.png)
![3,5,9-Trioxa-4-phosphaheneicosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxododecyl)oxy]-, inner salt, 4-oxide,(7R)-](http://img.cochemist.com/ccimg/18200/18194-25-7_b.png)







![2-[[2-[[2-[[2-[[2-[[2-amino-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoic Acid](http://img.cochemist.com/ccimg/7000/6934-38-9.png)
![2-[[2-[[2-[[2-[[2-[[2-amino-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-(4-hydroxyphenyl)propanoic Acid](http://img.cochemist.com/ccimg/7000/6934-38-9_b.png)
![Glycine, L-g-glutamyl-S-[(4-nitrophenyl)methyl]-L-cysteinyl-](http://img.cochemist.com/ccimg/6900/6803-19-6.png)
![Glycine, L-g-glutamyl-S-[(4-nitrophenyl)methyl]-L-cysteinyl-](http://img.cochemist.com/ccimg/6900/6803-19-6_b.png)
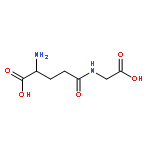


![2-AMINO-5-[[1-(CARBOXYMETHYLAMINO)-1-OXO-3-SULFOPROPAN-2-YL]AMINO]-5-OXOPENTANOIC ACID](http://img.cochemist.com/ccimg/3800/3773-07-7.png)
![2-AMINO-5-[[1-(CARBOXYMETHYLAMINO)-1-OXO-3-SULFOPROPAN-2-YL]AMINO]-5-OXOPENTANOIC ACID](http://img.cochemist.com/ccimg/3800/3773-07-7_b.png)
![2-METHYL-2-PROPANYL 4-[(3-AMINOPHENYL)CARBAMOYL]-1-PIPERIDINECARB<WBR />OXYLATE](http://img.cochemist.com/ccimg/3600/3545-76-4.png)
![2-METHYL-2-PROPANYL 4-[(3-AMINOPHENYL)CARBAMOYL]-1-PIPERIDINECARB<WBR />OXYLATE](http://img.cochemist.com/ccimg/3600/3545-76-4_b.png)
![3,5,9-Trioxa-4-phosphatetracosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxopentadecyl)oxy]-, inner salt,4-oxide, (7R)-](http://img.cochemist.com/ccimg/3400/3355-27-9.png)
![3,5,9-Trioxa-4-phosphatetracosan-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[(1-oxopentadecyl)oxy]-, inner salt,4-oxide, (7R)-](http://img.cochemist.com/ccimg/3400/3355-27-9_b.png)





![Benzo[g]pteridine-2,4(1H,3H)-dione,7,8-dimethyl-](http://img.cochemist.com/ccimg/1100/1086-80-2.png)
![Benzo[g]pteridine-2,4(1H,3H)-dione,7,8-dimethyl-](http://img.cochemist.com/ccimg/1100/1086-80-2_b.png)




![2,5-Cyclohexadiene-1,4-dione,2-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-5,6-dimethoxy-3-methyl-](http://img.cochemist.com/ccimg/700/606-06-4.png)
![2,5-Cyclohexadiene-1,4-dione,2-[(2E)-3,7-dimethyl-2,6-octadien-1-yl]-5,6-dimethoxy-3-methyl-](http://img.cochemist.com/ccimg/700/606-06-4_b.png)























![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3.png)
![2-[[(4r)-4-[(3r,5s,7r,8r,9s,10s,12s,13r,14s,17r)-3,7,12-trihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]pentanoyl]amino]ethanesulfonic Acid](http://img.cochemist.com/ccimg/100/81-24-3_b.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)






![b-D-Glucopyranosiduronic acid,6-bromo-3-[[(2-methoxyphenyl)amino]carbonyl]-2-naphthalenyl](http://img.cochemist.com/ccimg/100/37-87-6.png)
![b-D-Glucopyranosiduronic acid,6-bromo-3-[[(2-methoxyphenyl)amino]carbonyl]-2-naphthalenyl](http://img.cochemist.com/ccimg/100/37-87-6_b.png)