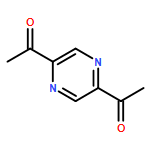
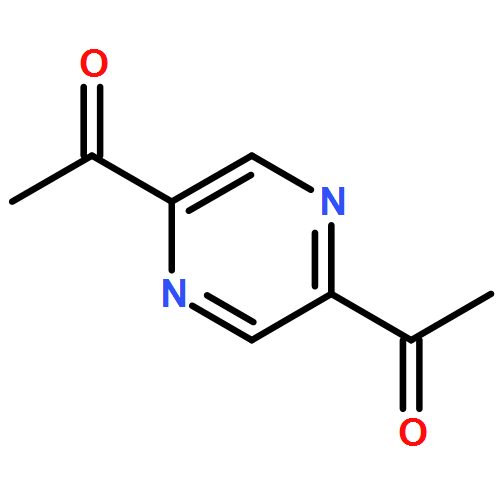
Co-reporter:Xavier Lucas, Antonio Bauzá, Antonio Frontera and David Quiñonero
Chemical Science 2016 vol. 7(Issue 2) pp:1038-1050
Publication Date(Web):05 Jun 2015
DOI:10.1039/C5SC01386K
Noncovalent interactions have a constitutive role in the science of intermolecular relationships, particularly those involving aromatic rings such as π–π and cation–π. In recent years, anion–π contact has also been recognized as a noncovalent bonding interaction with important implications in chemical processes. Yet, its involvement in biological processes has been scarcely reported. Herein we present a large-scale PDB analysis of the occurrence of anion–π interactions in proteins and nucleic acids. In addition we have gone a step further by considering the existence of cooperativity effects through the inclusion of a second noncovalent interaction, i.e. π-stacking, T-shaped, or cation–π interactions to form anion–π–π and anion–π–cation triads. The statistical analysis of the thousands of identified interactions reveals striking selectivities and subtle cooperativity effects among the anions, π-systems, and cations in a biological context. The reported results stress the importance of anion–π interactions and the cooperativity that arises from ternary contacts in key biological processes, such as protein folding and function and nucleic acids–protein and protein–protein recognition. We include examples of anion–π interactions and triads putatively involved in enzymatic catalysis, epigenetic gene regulation, antigen–antibody recognition, and protein dimerization.
Co-reporter:A. Bauzá, P. M. Deyà, A. Frontera and D. Quiñonero
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 4) pp:1322-1326
Publication Date(Web):19 Nov 2013
DOI:10.1039/C3CP54147A
The controversial proposal that substituent effects in cation–π interactions can be attributed mainly to electrostatic effects between the ion and local dipoles has been theoretically studied by analyzing 171 aromatics interacting with Na+. Our results stress the importance of both electrostatic and π-polarization effects to properly describe cation–π interactions.
Co-reporter:María García-Valverde, Ignacio Alfonso, David Quiñonero, and Roberto Quesada
The Journal of Organic Chemistry 2012 Volume 77(Issue 15) pp:6538-6544
Publication Date(Web):July 19, 2012
DOI:10.1021/jo301008c
A conformational analysis of a synthetic model prodiginine was carried out. In solution this compound showed a strong preference for the β conformation, in which all the heterocycles are mutually cis. This conformation provided an ideal alignment of the three N–H groups for interacting with anions when the molecule is protonated. A different conformation was also detected in d6-DMSO for the mesylate salt, assigned to the α conformation, in which the C ring is engaged in an intramolecular hydrogen bond with the OMe group. The formation of a homodimer was observed in concentrated CDCl3 solutions of the neutral free base form of this prodiginine derivative. DFT calculations and the solid state structures of the hydrochloric and methanesulfonic acid salts were in good agreement with the results observed in solution. A complete study of the relative energies of different tautomers, isomers, and supramolecular complexes supported the preference for the β conformation both in water and in the gas phase.
Co-reporter:David Quiñonero, Antonio Frontera, and Pere M. Deyà
The Journal of Physical Chemistry C 2012 Volume 116(Issue 39) pp:21083-21092
Publication Date(Web):September 13, 2012
DOI:10.1021/jp306586f
The adsorption of CO2 by zigzag and armchair single-walled carbon nanotubes (SWNTs) of different diameters (4.70–10.85 Å) has been studied using DFT with empirical dispersion correction (B97-D/SVP). Different binding sites have been considered, namely, in the interior (side-on and end-on binding modes) and on the surface (parallel or perpendicular) of the nanotube. Our calculations predict larger interaction energies for interior than exterior adsorption, with the strongest interactions observed for the (9,0) and (5,5) SWNTs (−12.8 and −12.5 kcal·mol–1, respectively). Therefore, these SWNTs can be considered to be very good potential candidates for carbon capture and storage in reducing CO2 emissions, as corroborated by the computed ΔH and ΔG adsorption energies. Moreover, we predict that interior adsorption would be more favorable than interstitial adsorption in bundles for (9,0), (10,0), (11,0), and (5,5) nanotubes. Furthemore, the diffusion of CO2 from the outside to the interior of the (5,5) SWNT is an energetically barrierless and favorable process. We have also analyzed the interplay between CO2·SWNT and CO2·CO2 interactions when more than one CO2 molecule is inside the tube, showing interesting cooperativity effects for SWNTs with large diameters. Finally, the symmetry-adapted perturbation theory partition scheme was used to investigate the physical nature of the interactions and to analyze the different energy contributions to the binding energy.
Co-reporter:David Quiñonero, Carolina Estarellas, Antonio Frontera, Pere M. Deyà
Chemical Physics Letters 2011 Volume 508(1–3) pp:144-148
Publication Date(Web):18 May 2011
DOI:10.1016/j.cplett.2011.04.004
Abstract
High-level ab initio calculations were carried out to estimate non-covalent interactions of systems based on π interactions, namely, ion–π, π–π, lone pair– or X–H⋯π interactions. It is important to obtain accurate geometric and energetic values since these interactions are present in relevant biological and chemical systems. The binding energies were calculated by means of the RI-MP2, SCS-RI-MP2 and CCSD(T) methods using the aug-cc-pVTZ and aug-cc-pVQZ basis sets. In principle, the most accurate methodology should give the most precise answer. However, the results obtained in this work indicate that the use of the most expensive computational method is not strictly necessary to achieve a good description of the above-mentioned non-covalent interactions.
Co-reporter:David Quiñonero, Pere M. Deyà, M. Pilar Carranza, Ana M. Rodríguez, Félix A. Jalón and Blanca R. Manzano
Dalton Transactions 2010 vol. 39(Issue 3) pp:794-806
Publication Date(Web):18 Nov 2009
DOI:10.1039/B915794H
The synthesis of octahedral copper and zinc coordination complexes containing ligands of the type 6R,2,4-bis(3,5-dimethylpyrazol-1-yl)triazine is described. They exhibit the simultaneous presence of C–H/π and anion–π interactions on both sides of the same triazine ring. When the pyrazolyl groups are not methylated, lone pair–π and anion–π interactions coexist on the same triazine ring. In addition, the interplay between C–H/π and anion–π interactions is studied by means of high level correlation ab initio calculations. They demonstrate that synergistic effects are present when both interactions coexist. These synergistic effects have been evaluated using the genuine non-additivity energies and symmetry adapted perturbation theory (SAPT).
Co-reporter:Xavier Lucas;Carolina Estarellas;Daniel Escudero;Antonio Frontera Dr.;David Quiñonero Dr. ;Pere M. Deyà
ChemPhysChem 2009 Volume 10( Issue 13) pp:2256-2264
Publication Date(Web):
DOI:10.1002/cphc.200900157
Abstract
The interplay between two important non-covalent interactions involving aromatic rings (namely anion–π and hydrogen bonding) is investigated. Very interesting cooperativity effects are present in complexes where anion–π and hydrogen bonding interactions coexist. These effects are found in systems where the distance between the anion and the hydrogen-bond donor/acceptor molecule is as long as ∼11 Å. These effects are studied theoretically using the energetic and geometric features of the complexes, which were computed using ab initio calculations. We use and discuss several criteria to analyze the mutual influence of the non-covalent interactions studied herein. In addition we use Bader’s theory of atoms-in-molecules to characterize the interactions and to analyze the strengthening or weakening of the interactions depending upon the variation of the charge density at the critical points.
Co-reporter:Antonio Frontera ;David Quiñonero Dr.;Daniel Escudero;Pablo Ballester ;Antonio Costa Dr.;Pere M. Deyà Dr.
ChemPhysChem 2007 Volume 8(Issue 8) pp:1182-1187
Publication Date(Web):10 MAY 2007
DOI:10.1002/cphc.200700100
Several complexes of tropylium (1) with anions are optimized at the RI-MP2(full)/6-31++G** level of theory. This binding unit can interact very favorably with anions, and it combines the strength of the electrostatic interaction with the directionality of the anion–π interaction. The complexes of 1 with anions are characterized by means of the Bader theory of “atoms-in-molecules,” and the physical nature of the interaction has been analyzed by means of the molecular interaction potential with polarization tool. Experimental evidence of anion–π interactions involving seven-membered rings has been found in the solid state.
Co-reporter:A. Bauzá, P. M. Deyà, A. Frontera and D. Quiñonero
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 4) pp:NaN1326-1326
Publication Date(Web):2013/11/19
DOI:10.1039/C3CP54147A
The controversial proposal that substituent effects in cation–π interactions can be attributed mainly to electrostatic effects between the ion and local dipoles has been theoretically studied by analyzing 171 aromatics interacting with Na+. Our results stress the importance of both electrostatic and π-polarization effects to properly describe cation–π interactions.
Co-reporter:Xavier Lucas, Antonio Bauzá, Antonio Frontera and David Quiñonero
Chemical Science (2010-Present) 2016 - vol. 7(Issue 2) pp:NaN1050-1050
Publication Date(Web):2015/06/05
DOI:10.1039/C5SC01386K
Noncovalent interactions have a constitutive role in the science of intermolecular relationships, particularly those involving aromatic rings such as π–π and cation–π. In recent years, anion–π contact has also been recognized as a noncovalent bonding interaction with important implications in chemical processes. Yet, its involvement in biological processes has been scarcely reported. Herein we present a large-scale PDB analysis of the occurrence of anion–π interactions in proteins and nucleic acids. In addition we have gone a step further by considering the existence of cooperativity effects through the inclusion of a second noncovalent interaction, i.e. π-stacking, T-shaped, or cation–π interactions to form anion–π–π and anion–π–cation triads. The statistical analysis of the thousands of identified interactions reveals striking selectivities and subtle cooperativity effects among the anions, π-systems, and cations in a biological context. The reported results stress the importance of anion–π interactions and the cooperativity that arises from ternary contacts in key biological processes, such as protein folding and function and nucleic acids–protein and protein–protein recognition. We include examples of anion–π interactions and triads putatively involved in enzymatic catalysis, epigenetic gene regulation, antigen–antibody recognition, and protein dimerization.
Co-reporter:David Quiñonero, Pere M. Deyà, M. Pilar Carranza, Ana M. Rodríguez, Félix A. Jalón and Blanca R. Manzano
Dalton Transactions 2010 - vol. 39(Issue 3) pp:NaN806-806
Publication Date(Web):2009/11/18
DOI:10.1039/B915794H
The synthesis of octahedral copper and zinc coordination complexes containing ligands of the type 6R,2,4-bis(3,5-dimethylpyrazol-1-yl)triazine is described. They exhibit the simultaneous presence of C–H/π and anion–π interactions on both sides of the same triazine ring. When the pyrazolyl groups are not methylated, lone pair–π and anion–π interactions coexist on the same triazine ring. In addition, the interplay between C–H/π and anion–π interactions is studied by means of high level correlation ab initio calculations. They demonstrate that synergistic effects are present when both interactions coexist. These synergistic effects have been evaluated using the genuine non-additivity energies and symmetry adapted perturbation theory (SAPT).
Co-reporter:David Quiñonero, Ibon Alkorta and José Elguero
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 40) pp:NaN27950-27950
Publication Date(Web):2016/09/09
DOI:10.1039/C6CP03662G
Stable minima showing halogen bonds between charged molecules with the same sign have been explored by means of theoretical calculations. The dissociation transition states and their corresponding barriers have also been characterized. In all cases, the results indicate that the complexes are thermodynamically unstable but kinetically stable with respect to the isolated monomers in gas phase. A corrected binding energy profile by removing the charge–charge repulsion of the monomers shows a profile similar to the one observed for the dissociation of analogous neutral systems. The nature of the interaction in the minima and TSs has been analyzed using the symmetry adapted perturbation theory (SAPT) method. The results indicate the presence of local favorable electrostatic interactions in the minima that vanish in the TSs. Natural bond orbital (NBO) and “atoms-in-molecules” (AIM) theories were used to analyze the complexes, obtaining good correlations between Laplacian and electron density values with both bond distances and charge-transfer energy contributions E(2). The largest E(2) orbital interaction energies for cation–cation and anion–anion complexes are 561.2 and 197.9 kJ mol−1, respectively.