Co-reporter:Md. Saifuzzaman, Rick Morrison, Zhaohua Zheng, Stephanie Orive, Justin Hamilton, Philip E. Thompson, Jasim M.A. Al-rawi
Bioorganic & Medicinal Chemistry 2017 Volume 25, Issue 20(Issue 20) pp:
Publication Date(Web):15 October 2017
DOI:10.1016/j.bmc.2017.08.022
•Synthesis of 40 8-aryl-2-morpholino-7-O-substituted benzo[e][1,3]oxazin-4-ones.•Atropisomers of O-substituted-8-aryl-copounds resulted from hindered rotation.•Bulky substitutions impeded binding except in the case of PI3Kδ and PDE3.•Piperazinyl 26a showed ADP-induced platelet-rich plasma aggregation (IC50 of 10 μM).A series of 40 7-(O-substituted)-2-morpholino-8-aryl-4H-benzo[e][1,3]oxazin-4-one derivatives was synthesized. They were prepared via synthesis of a key precursor, 8-bromo-7-hydroxy-2-morpholino-4H-benzo[e][1,3]oxazin-4-one 13 which was amenable to ether synthesis at the 7-position and Suzuki coupling at the 8-position. The 2 protons of 7-OCH2 in compounds 18g, 18h, 18i, 18l and 18m prove to be magnetically non-equivalent, atropisomerism (axial chirality), as result of sterically hindered rotation of the bulky 8-aryl-substituent.The products were evaluated for their activities against PI3K isoforms, DNA-PK and PDE3. The results showed that this substitution pattern has a deleterious effect on PI3K activities, which may arise from steric hindrance in the active site. PI3Kδ was somewhat more tolerant of this substitution particularly where 8-(4-methoxylphenyl) substituents were present (IC50s ∼ 2–3 μM). Good activities against PDE3 were also obtained for compounds, with particular members of the 7-(2-pyridinyl) methoxy series 19 showing good inhibition (IC50s ∼ 2–3 μM), comparable to previously described analogues. A piperazinyl derivative 26a effectively inhibited ADP-induced platelet aggregation with an IC50 of 8 μM.Download high-res image (129KB)Download full-size image
Co-reporter:Simon J. Mountford;Biswaranjan Mohanty;Kade D. Roberts;Heidi H. Yu;Martin J. Scanlon;Roger L. Nation;Tony Velkov;Jian Li;Philp E. Thompson
Organic & Biomolecular Chemistry 2017 vol. 15(Issue 34) pp:7173-7180
Publication Date(Web):2017/08/30
DOI:10.1039/C7OB01493G
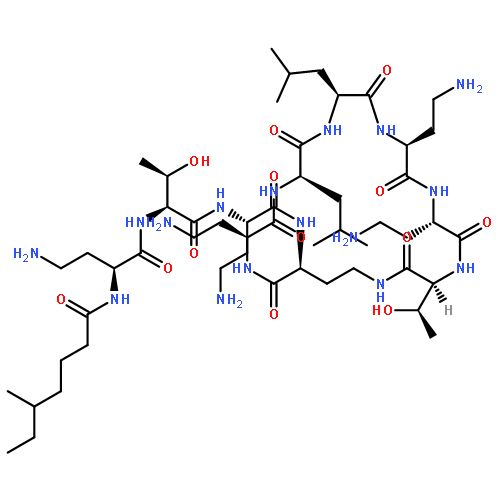
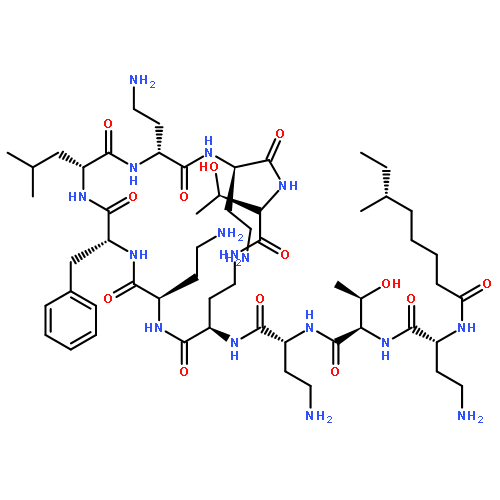
The first total synthesis of a polypeptin, PE2, as well as its solution structure is reported. Synthesis in optically pure form confirms the proposed stereochemistry of the polypeptins at the 3-position on the 3-hydroxy depsipeptide moiety. We have also determined the NMR structure of PE2 in aqueous solution, showing it to form a stable ring conformation. The synthetic peptide shows anti-bacterial activity consistent with reports for naturally derived counterparts.
Co-reporter:Mengjie Liu; Simon J. Mountford; Rachel R. Richardson; Marleen Groenen; Nicholas D. Holliday;Philip E. Thompson
Journal of Medicinal Chemistry 2016 Volume 59(Issue 13) pp:6059-6069
Publication Date(Web):June 13, 2016
DOI:10.1021/acs.jmedchem.6b00310
The dimeric peptide 1 (BVD-74D, as a diastereomeric mixture) is a potent and selective neuropeptide Y Y4 receptor agonist. It represents a valuable candidate in developing traceable ligands for pharmacological studies of Y4 receptors and as a lead compound for antiobesity drugs. Its optically pure stereoisomers along with analogues and fluorescently labeled variants were prepared by exploiting alkene metathesis reactions. The (2R,7R)-diaminosuberoyl containing peptide, (R,R)-1, had markedly higher affinity and agonist efficacy than its (S,S)-counterpart. Furthermore, the sulfo-Cy5 labeled (R,R)-14 retained high agonist potency as a novel fluorescent ligand for imaging Y4 receptors.
Co-reporter:Mengjie Liu, Rachel R. Richardson, Simon J. Mountford, Lei Zhang, Matheus H. Tempone, Herbert Herzog, Nicholas D. Holliday, and Philip E. Thompson
Bioconjugate Chemistry 2016 Volume 27(Issue 9) pp:2166
Publication Date(Web):August 11, 2016
DOI:10.1021/acs.bioconjchem.6b00376
Traceable truncated Neuropeptide Y (NPY) analogues with Y1 receptor (Y1R) affinity and selectivity are highly desirable tools in studying receptor location, regulation, and biological functions. A range of fluorescently labeled analogues of a reported Y1R/Y4R preferring ligand BVD-15 have been prepared and evaluated using high content imaging techniques. One peptide, [Lys2(sCy5), Arg4]BVD-15, was characterized as an Y1R antagonist with a pKD of 7.2 measured by saturation analysis using fluorescent imaging. The peptide showed 8-fold lower affinity for Y4R (pKD = 6.2) and was a partial agonist at this receptor. The suitability of [Lys2(sCy5), Arg4]BVD-15 for Y1R and Y4R competition binding experiments was also demonstrated in intact cells. The nature of the label was shown to be critical with replacement of sCy5 by the more hydrophobic Cy5.5 resulting in a switch from Y1R antagonist to Y1R partial agonist.
Co-reporter:Zhaohua Zheng, Jo-Anne Pinson, Simon J. Mountford, Stephanie Orive, Simone M. Schoenwaelder, David Shackleford, Andrew Powell, Erin M. Nelson, Justin R. Hamilton, Shaun P. Jackson, Ian G. Jennings, Philip E. Thompson
European Journal of Medicinal Chemistry 2016 Volume 122() pp:339-351
Publication Date(Web):21 October 2016
DOI:10.1016/j.ejmech.2016.06.010
•Potent and selective inhibitors of PI3Kβ.•Distinct mechanisms underpinning selectivity compared to known inhibitors.•Anti-platelet activity shown in platelet aggregation, activation and adhesion in vitro.•Anti-thrombotic activity in vivo in mouse electrolytic injury model without effects on haemostasis.A series of amino-substituted triazines were developed and examined for PI3Kβ inhibition and anti-platelet function. Structural adaptations of a morpholine ring of the prototype pan-PI3K inhibitor ZSTK474 yielded PI3Kβ selective compounds, where the selectivity largely derives from an interaction with the non-conserved Asp862 residue, as shown by site directed mutagenesis. The most PI3Kβ selective inhibitor from the series was studied in detail through a series of in vitro and in vivo functional studies. MIPS-9922, 10 potently inhibited ADP-induced washed platelet aggregation. It also inhibited integrin αIIbβ3 activation and αIIbβ3 dependent platelet adhesion to immobilized vWF under high shear. It prevented arterial thrombus formation in the in vivo electrolytic mouse model of thrombosis without inducing prolonged bleeding or excess blood loss.
Co-reporter:Rick Morrison, Zhaohua Zheng, Ian G. Jennings, Philip E. Thompson, Jasim M.A. Al-Rawi
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 22) pp:5534-5538
Publication Date(Web):15 November 2016
DOI:10.1016/j.bmcl.2016.10.003
•Suzuki coupling novel linear and angular fused-aryl-morpholino-naphth-oxazines.•Linear 6.7-fused 13a and 13b selective PI3Kδ isoform inhibitors (IC50 = 7.7 and 5.61 μM).•Antiplatelet activity for the angular 7,8-fused 22a, b, k and l with IC50 = 3.0,14.0, 2.0 and 5.0 μM.To continue our study of 2-morpholino-benzoxazine based compounds, which show useful activity against PI3K family enzymes or antiplatelet activity, we designed and synthesized a series of linear 6.7-fused, 5,6-angular fused and 7,8-angular fused-aryl-morpholino-naphth-oxazines. The compounds were prepared from substituted 2-hydroxynaphthoic acid to give the corresponding thioxo analogues 8, 9, 15 and 19. The thioxo products were then converted to the morpholino substituted analogue. The aryl group was introduced by Suzuki coupling of bromo precursors. The products were evaluated for activity at PI3K family enzymes and as platelet aggregation inhibitors and compared to reported unsubstituted analogues. The linear 6.7-fused product 13a and 13b were moderated potent but selective PI3Kδ isoform inhibitors (IC50 = 7.7 and 5.61 μM). Good antiplatelet activity was noticed for the angular 7,8-fused compounds 22a, b, k and l with IC50 = 3.0,14.0, 2.0 and 5.0 μM respectively. The antiplatelet activity is independent of PDE3.
Co-reporter:Brittany L. Howard, Katherine L. Harvey, Rebecca J. Stewart, Mauro F. Azevedo, Brendan S. Crabb, Ian G. Jennings, Paul R. Sanders, David T. Manallack, Philip E. Thompson, Christopher J. Tonkin, and Paul R. Gilson
ACS Chemical Biology 2015 Volume 10(Issue 4) pp:1145
Publication Date(Web):January 2, 2015
DOI:10.1021/cb501004q
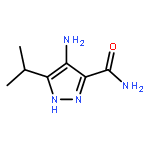
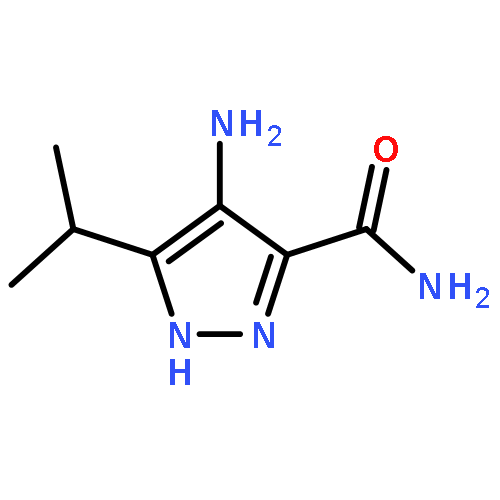
Apicomplexan parasites, including Plasmodium falciparum and Toxoplasma gondii, the causative agents of severe malaria and toxoplasmosis, respectively, undergo several critical developmental transitions during their lifecycle. Most important for human pathogenesis is the asexual cycle, in which parasites undergo rounds of host cell invasion, replication, and egress (exit), destroying host cell tissue in the process. Previous work has identified important roles for Protein Kinase G (PKG) and Protein Kinase A (PKA) in parasite egress and invasion, yet little is understood about the regulation of cyclic nucleotides, cGMP and cAMP, that activate these enzymes. To address this, we have focused upon the development of inhibitors of 3′,5′-cyclic nucleotide phosphodiesterases (PDEs) to block the breakdown of cyclic nucleotides. This was done by repurposing human PDE inhibitors noting various similarities of the human and apicomplexan PDE binding sites. The most potent inhibitors blocked the in vitro proliferation of P. falciparum and T. gondii more potently than the benchmark compound zaprinast. 5-Benzyl-3-isopropyl-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-one (BIPPO) was found to be a potent inhibitor of recombinant P. falciparum PfPDEα and activated PKG-dependent egress of T. gondii and P. falciparum, likely by promoting the exocytosis of micronemes, an activity that was reversed by a specific Protein Kinase G inhibitor. BIPPO also promotes cAMP-dependent phosphorylation of a P. falciparum ligand critical for host cell invasion, suggesting that the compound inhibits single or multiple PDE isoforms that regulate both cGMP and cAMP levels. BIPPO is therefore a useful tool for the dissection of signal transduction pathways in apicomplexan parasites.
Co-reporter:Simon J. Mountford, Zhaohua Zheng, Krithika Sundaram, Ian G. Jennings, Justin R. Hamilton, and Philip E. Thompson
ACS Medicinal Chemistry Letters 2015 Volume 6(Issue 1) pp:3
Publication Date(Web):September 24, 2014
DOI:10.1021/ml500354e
The Class II PI3 kinases are emerging from the shadows of their Class I cousins. The data emerging from PIK3C2 genetic modification studies and from siRNA knockdown suggest important roles in physiology and pathology. With some well-studied Class I isoform inhibitors showing strong Class II activity and a wealth of crystallographic information available, the structural similarity of these isoforms to Class I provides both the opportunity and the challenge in design of selective pharmacological inhibitors.Keywords: Class II; isoform selectivity; PI3 kinase
Co-reporter:Simon J. Mountford ; Anthony L. Albiston ; William N. Charman ; Leelee Ng ; Jessica K. Holien ; Michael W. Parker ; Joseph A. Nicolazzo ; Philip E. Thompson ;Siew Yeen Chai
Journal of Medicinal Chemistry 2014 Volume 57(Issue 4) pp:1368-1377
Publication Date(Web):January 28, 2014
DOI:10.1021/jm401540f
Peptide inhibitors of insulin-regulated aminopeptidase (IRAP) enhance fear avoidance and spatial memory and accelerate spatial learning in a number of memory paradigms. Using a virtual screening approach, a series of benzopyran compounds was identified that inhibited the catalytic activity of IRAP, ultimately resulting in the identification of potent and specific inhibitors. The present study describes the medicinal chemistry campaign that led to the development of the lead candidate, 3, highlighting the key structural features considered as critical for binding. Furthermore, the in vivo pharmacokinetics and brain uptake of compounds (1 and 3) were assessed in rats and were complemented with in vitro human and rat microsomal stability studies. Following intravenous administration to rodents, 3 exhibits brain exposure, albeit it is rapidly converted to 1, a compound which also exhibits potent inhibition of IRAP.
Co-reporter:Simon J. Mountford, Mengjie Liu, Lei Zhang, Marleen Groenen, Herbert Herzog, Nicholas D. Holliday and Philip E. Thompson
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 20) pp:3271-3281
Publication Date(Web):02 Apr 2014
DOI:10.1039/C4OB00176A
The potent Y1 receptor antagonist, 1229U91 has an unusual cyclic dimer structure that makes syntheses of analogue series quite challenging. We have examined three new routes to the synthesis of such peptides that has given access to novel structural variants including heterodimeric compounds, ring size variants and labelled conjugates. These compounds, including a fluorescently labelled analogue VIII show potent antagonism that can be utilised in studying Y1 receptor pharmacology.
Co-reporter:Zhaohua Zheng, Michelle S. Miller, Ian G. Jennings, and Philip E. Thompson
ACS Chemical Biology 2013 Volume 8(Issue 4) pp:679
Publication Date(Web):January 29, 2013
DOI:10.1021/cb300666s
The p110β isoform of PI3 kinase (PI3Kβ) has been implicated in pathological disorders such as thrombosis and cancer and a number of PI3Kβ-selective inhibitors have recently progressed into clinical studies. Although crystallography studies identify a binding site conformation favored by the inhibitors, no specific interaction explains the observed selectivity. Using site-directed mutagenesis we have identified a specific tyrosine residue of the binding site Y778 that dictates the ability of the PI3Kβ isoform to bind these inhibitors. When mutated to isoleucine, PI3Kβ has reduced ability to present a specific cryptic binding site into which a range of reported PI3Kβ inhibitors can bind, and conversely when tyrosine is introduced into the same position in PI3Kα, the same inhibitors gain potency. The results provide a cogent explanation for the selectivity profiles displayed by these PI3K inhibitors and maybe others as well.
Co-reporter:Michelle A. Camerino, David K. Chalmers and Philip E. Thompson
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 16) pp:2571-2573
Publication Date(Web):27 Feb 2013
DOI:10.1039/C3OB40218E
An efficient synthesis of the enantiomers of fluorenylethylchloroformate (FLEC) has been achieved that allows the routine application of the reagent for the resolution of chiral amines including unusual amino acids. The utility of the fluorenylethoxycarbonyl (Feoc) group as a chiral Fmoc equivalent, for combined resolution and protection of amino acids, in solid phase peptide synthesis is also shown.
Co-reporter:Jo-Anne Pinson, Zhaohua Zheng, Michelle S. Miller, David K. Chalmers, Ian G. Jennings, and Philip E. Thompson
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 2) pp:206
Publication Date(Web):December 20, 2012
DOI:10.1021/ml300336j
A series of aminoacyl-triazine derivatives based upon the pan-PI3K inhibitor ZSTK474 were identified as potent and isoform-selective inhibitors of PI3Kβ. The compounds showed selectivity based upon stereochemistry with l-amino acyl derivatives preferring PI3Kβ, while their d-congeners favored PI3Kδ. The mechanistic basis of this inhibition was studied using site-directed mutants. One Asp residue, D862, was identified as a critical participant in binding to the PI3Kβ-selective inhibitors, distinguishing this class from other reported PI3Kβ-selective inhibitors. The compounds show strong inhibition of cellular Akt phosphorylation and growth of PTEN-deficient MD-MBA-468 cells.Keywords: cancer; p110β; PI3 kinase; ZSTK474
Co-reporter:Michelle S. Miller, Jo-Anne Pinson, Zhaohua Zheng, Ian G. Jennings, Philip E. Thompson
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 3) pp:802-805
Publication Date(Web):1 February 2013
DOI:10.1016/j.bmcl.2012.11.076
Phosphoinositide 3-kinases (PI3K) hold significant therapeutic potential as novel targets for the treatment of cancer. ZSTK474 (4a) is a potent, pan-PI3K inhibitor currently under clinical evaluation for the treatment of cancer. Structural studies have shown that derivatisation at the 5- or 6-position of the benzimidazole ring may influence potency and isoform selectivity. However, synthesis of these derivatives by the traditional route results in a mixture of the two regioisomers. We have developed a straightforward regioselective synthesis that gave convenient access to 5- and 6-methoxysubstituted benzimidazole derivatives of ZSTK474. While 5-methoxy substitution abolished activity at all isoforms, the 6-methoxy substitution is consistently 10-fold more potent. This synthesis will allow convenient access to further 6-position derivatives, thus allowing the full scope of the structure-activity relationships of ZSTK474 to be probed.
Co-reporter:Mengjie Liu;Simon J. Mountford;Lei Zhang
International Journal of Peptide Research and Therapeutics 2013 Volume 19( Issue 1) pp:33-41
Publication Date(Web):2013 March
DOI:10.1007/s10989-012-9330-z
Neuropeptide Y (NPY) Y1 receptors are overexpressed in human breast carcinomas. They also have important functional roles in breast tumour growth and metastasis. This study investigates the synthesis of 15 truncated NPY analogues as models for Y1 receptor specific radiopharmaceuticals, using competition radioreceptor binding assays from brain tissue homogenates from Y2Y4-double knockout mice. These peptides are based on the previously reported BVD15 scaffold. Different measures to improve Y1 affinity and plasma metabolic stability were investigated. Extending from the previously reported [Lys(DOTA)4]BVD15 analogue, it was found that lysine4 is capable of tolerating various modifications, including prosthetic groups and other bifunctional chelators, but also that [Lys4]BVD15 has improved Y1 affinity, relative to BVD15 itself. Substitution of lysine4 for side chain shortened analogues retains Y1 receptor affinity of the analogues. Furthermore, modifications at the N-terminal isoleucine resulted in dramatic reduction of Y1 affinity.
Co-reporter:Jo-Anne Pinson;Dr. Oleg Schmidt-Kittler;Dr. Jiuxiang Zhu;Dr. Ian G. Jennings; Kenneth W. Kinzler; Bert Vogelstein;Dr. David K. Chalmers; Philip E. Thompson
ChemMedChem 2011 Volume 6( Issue 3) pp:514-522
Publication Date(Web):
DOI:10.1002/cmdc.201000467
Abstract
A series of synthesized and commercially available compounds were assessed against PI3Kα for in vitro inhibitory activity and the results compared to binding calculated in silico. Using published crystal structures of PI3Kγ and PI3Kδ co-crystallized with inhibitors as a template, docking was able to identify the majority of potent inhibitors from a decoy set of 1000 compounds. On the other hand, PI3Kα in the apo-form, modeled by induced fit docking, or built as a homology model gave only poor results. A PI3Kα homology model derived from a ligand-bound PI3Kδ crystal structure was developed that has a good ability to identify active compounds. The docking results identified binding poses for active compounds that differ from those identified to date and can contribute to our understanding of structure–activity relationships for PI3K inhibitors.
Co-reporter:Simon J. Mountford, Mengjie Liu, Lei Zhang, Marleen Groenen, Herbert Herzog, Nicholas D. Holliday and Philip E. Thompson
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 20) pp:NaN3281-3281
Publication Date(Web):2014/04/02
DOI:10.1039/C4OB00176A
The potent Y1 receptor antagonist, 1229U91 has an unusual cyclic dimer structure that makes syntheses of analogue series quite challenging. We have examined three new routes to the synthesis of such peptides that has given access to novel structural variants including heterodimeric compounds, ring size variants and labelled conjugates. These compounds, including a fluorescently labelled analogue VIII show potent antagonism that can be utilised in studying Y1 receptor pharmacology.
Co-reporter:Michelle A. Camerino, David K. Chalmers and Philip E. Thompson
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 16) pp:NaN2573-2573
Publication Date(Web):2013/02/27
DOI:10.1039/C3OB40218E
An efficient synthesis of the enantiomers of fluorenylethylchloroformate (FLEC) has been achieved that allows the routine application of the reagent for the resolution of chiral amines including unusual amino acids. The utility of the fluorenylethoxycarbonyl (Feoc) group as a chiral Fmoc equivalent, for combined resolution and protection of amino acids, in solid phase peptide synthesis is also shown.

![4-(4-Chloro-6-(2-(difluoromethyl)-1H-benzo[d]imidazol-1-yl)-1,3,5-triazin-2-yl)morpholine](http://img.cochemist.com/ccimg/475200/475111-38-7.png)
![4-(4-Chloro-6-(2-(difluoromethyl)-1H-benzo[d]imidazol-1-yl)-1,3,5-triazin-2-yl)morpholine](http://img.cochemist.com/ccimg/475200/475111-38-7_b.png)
![5-(3-Chloro-benzyl)-3-isopropyl-2,6-dihydro-pyrazolo[4,3-d]pyrimidin-7-one](http://img.cochemist.com/ccimg/521300/521296-91-3.png)
![5-(3-Chloro-benzyl)-3-isopropyl-2,6-dihydro-pyrazolo[4,3-d]pyrimidin-7-one](http://img.cochemist.com/ccimg/521300/521296-91-3_b.png)





























![2-AMINO-N-[4-AMINO-1-OXO-1-[[6,9,18-TRIS(2-AMINOETHYL)-3-(1-HYDROXYETHYL)-12,15-BIS(2-METHYLPROPYL)-2,5,8,11,14,17,20-HEPTAOXO-1,4,7,10,13,16,19-HEPTAZACYCLOTRICOS-21-YL]AMINO]BUTAN-2-YL]-3-HYDROXYBUTANAMIDE](http://img.cochemist.com/ccimg/32600/32530-94-2.png)
![2-AMINO-N-[4-AMINO-1-OXO-1-[[6,9,18-TRIS(2-AMINOETHYL)-3-(1-HYDROXYETHYL)-12,15-BIS(2-METHYLPROPYL)-2,5,8,11,14,17,20-HEPTAOXO-1,4,7,10,13,16,19-HEPTAZACYCLOTRICOS-21-YL]AMINO]BUTAN-2-YL]-3-HYDROXYBUTANAMIDE](http://img.cochemist.com/ccimg/32600/32530-94-2_b.png)












![N-[2-({2-[DIMETHYL(PHENYL)SILYL]ETHYL}SULFANYL)ETHYL]-5-FLUORO-2,<WBR />4-DIOXO-3,4-DIHYDRO-1(2H)-PYRIMIDINECARBOXAMIDE](http://img.cochemist.com/ccimg/7300/7239-48-7.png)
![N-[2-({2-[DIMETHYL(PHENYL)SILYL]ETHYL}SULFANYL)ETHYL]-5-FLUORO-2,<WBR />4-DIOXO-3,4-DIHYDRO-1(2H)-PYRIMIDINECARBOXAMIDE](http://img.cochemist.com/ccimg/7300/7239-48-7_b.png)