Co-reporter:Hans-Joachim Wittmann
Naunyn-Schmiedeberg's Archives of Pharmacology 2017 Volume 390( Issue 6) pp:595-612
Publication Date(Web):20 February 2017
DOI:10.1007/s00210-017-1362-7
Within the last years, for several ligands, binding to G protein-coupled receptors or other target proteins, a binding of the ligand in two different orientations is described. One appropriate experimental technique to detect two different binding orientations is the crystallization of the ligand-protein-complex, but crystallization and subsequent X-ray analysis do not belong to the routine methods. By traditional competitive radioligand equilibrium binding assays, it is not possible to detect or to distinguish between two different binding orientations, but there is a possibility to identify two different binding orientations by performing kinetic competitive radioligand-binding assays. To study the limitations of this new technique, the related differential equations were defined and solved numerically for 8 different sets of rate constants, also considering an experimental error up to ~10%. In principal, the kinetic competitive radioligand binding assay is a suitable technique to detect two different ligand binding orientations. However, the present study shows that this is only possible under distinct conditions: (1) the rate constants of dissociation for both binding orientations of the cold ligand should at least be >> 10-fold different to each other and (2) the experimental error should be as small as possible. Although there are some limitations for the experimental usability of this method, it is worthwhile to perform kinetic competitive binding assays, especially if there are hints for two binding orientations of a ligand, e.g. based on molecular modelling studies.
Co-reporter:Andrea Strasser, Hans-Joachim Wittmann, Roland Seifert
Trends in Pharmacological Sciences 2017 Volume 38, Issue 8(Issue 8) pp:
Publication Date(Web):1 August 2017
DOI:10.1016/j.tips.2017.05.005
Previously, drugs were developed focusing on target affinity and selectivity. However, it is becoming evident that the drug–target residence time, related to the off-rate, is an important parameter for successful drug development. The residence time influences both the on-rate and overall effectiveness of drugs. Furthermore, ligand binding is now appreciated to be a multistep process because metastable and/or intermediate binding sites in the extracellular region have been identified. In this review, we summarize experimental ligand-binding data for G-protein-coupled receptors (GPCRs), and their binding pathways, analyzed by molecular dynamics (MD). The kinetics of drug binding to GPCRs are complex and depend on several factors, including charge distribution on the receptor surface, ligand–receptor interactions in the binding channel and the binding site, or solvation.
Co-reporter:Sebastian G. Hammer, Susanne Gobleder, Franziska Naporra, Hans-Joachim Wittmann, Sigurd Elz, Markus R. Heinrich, Andrea Strasser
Bioorganic & Medicinal Chemistry Letters 2016 Volume 26(Issue 2) pp:292-300
Publication Date(Web):15 January 2016
DOI:10.1016/j.bmcl.2015.12.035
Distinct diaminopyrimidines, for example, 4-(4-methylpiperazin-1-yl)-5,6-dihydrobenzo[h]quinazolin-2-amine are histamine H4 receptor (H4R) antagonists and show high affinity to the H4R, but only a moderate affinity to the histamine H1 receptor (H1R). Within previous studies it was shown that an aromatic side chain with a distinct distance to the basic amine and aromatic core is necessary for affinity to the human H1R (hH1R). Thus, a rigid aminopyrimidine with a tricyclic core was used as a lead structure. There, (1) the flexible aromatic side chain was introduced, (2) the substitution pattern of the pyrimidine core was exchanged and (3) rigidity was decreased by opening the tricyclic core. Within the present study, two compounds with similar affinity in the one digit μM range to the human H1R and H4R were identified. While the affinity at the hH1R increased about 4- to 8-fold compared to the parent diaminopyrimidine, the affinity to the hH4R decreased about 5- to 8-fold. In addition to the parent diaminopyrimidine, two selected compounds were docked into the H1R and H4R and molecular dynamic studies were performed to predict the binding mode and explain the experimental results on a molecular level. The two new compounds may be good lead structures for the development of dual H1/H4 receptor ligands with affinities in the same range.
Co-reporter:Hans-Joachim Wittmann, Andrea Strasser
Bioorganic & Medicinal Chemistry Letters 2015 Volume 25(Issue 6) pp:1259-1268
Publication Date(Web):15 March 2015
DOI:10.1016/j.bmcl.2015.01.052
Histamine binds with high affinity to the human histamine H4 receptor (hH4R). We are the first to examine the complete binding pathway of histamine from the extracellular side to the orthosteric binding site of the hH4R by means of unconstrained molecular dynamic simulation. Furthermore, the simulations show that the positively charged amine moiety of the histamine interacts electrostatically with the highly conserved Asp3.32, while the imidazole moiety forms a hydrogen bond interaction with Glu5.46 and Gln7.42.
Co-reporter:Andrea Strasser;Hans-Joachim Wittmann
Naunyn-Schmiedeberg's Archives of Pharmacology 2015 Volume 388( Issue 3) pp:363-380
Publication Date(Web):2015 March
DOI:10.1007/s00210-014-1073-2
The recent resolution of G-protein-coupled receptor (GPCR) structures in complex with Na+ bound to an allosteric modulatory site has renewed interest of the regulation of GPCRs by ions. Here, we summarise key data on ion modulation of GPCRs, obtained in pharmacological, crystallographic, mutagenesis and molecular modelling studies. We show that ion modulation is a highly complex process, involving not only cations but also, rather neglected until now, anions. Pharmacotherapeutic and toxicological aspects are discussed. We provide a mathematical framework for the analysis of ion effects. Finally, we discuss open questions in the field and future research directions. Most importantly, the in vivo relevance of the modulation of GPCR function by monovalent ions must be clarified.
Co-reporter:Eva Wagner;Hans-Joachim Wittmann
Naunyn-Schmiedeberg's Archives of Pharmacology 2014 Volume 387( Issue 3) pp:235-250
Publication Date(Web):2014 March
DOI:10.1007/s00210-013-0926-4
Astemizole, a H1R antagonist shows high affinity to the histamine H1 receptor but only a moderate affinity to the histamine H4 receptor. This study aims to modify the astemizole to keep high affinity to the histamine H1 receptor and to increase affinity to the histamine H4 receptor. Therefore, 13 astemizole-derived compounds and astemizole-JNJ7777120-derived hybrid compounds were synthesized and pharmacologically characterized at the histamine H1 and H4 receptors. The new compounds show affinity to the histamine H1 receptor in the pKi range from 5.3 to 8.8, whereas the affinity of these compounds to the histamine H4 receptor was surprisingly rather low (pKi from 4.4 to 5.6). Three representative compounds were docked into the histamine H1 receptor and molecular dynamic studies were performed to explain the binding mode and the experimental results on a molecular level. Furthermore, taking into account the binding mode of compounds with high affinity to the histamine H4 receptor, a H1/H4-pharmacophore hypothesis was developed.
Co-reporter:Eva Wagner, Hans-Joachim Wittmann, Sigurd Elz, Andrea Strasser
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 21) pp:6274-6280
Publication Date(Web):1 November 2011
DOI:10.1016/j.bmcl.2011.09.001
In literature, a synergism between histamine H1 and H4 receptor is discussed. Furthermore, it was shown, that the combined application of mepyramine, a H1 antagonist and JNJ7777120, a H4 receptor ligand leads to a synergistic effect in the acute murine asthma model. Thus, the aim of this study was to develop new hybrid ligands, containing one H1 and one H4 pharmacophor, connected by an appropriate spacer, in order to address both, H1R and H4R. Within this study, we synthesized nine hybrid compounds, which were pharmacologically characterized at hH1R and hH4R. The new compounds revealed (high) affinity to hH1R, but showed only low affinity to hH4R. Additionally, we performed molecular dynamic studies for some selected compounds at hH1R, in order to obtain information about the binding mode of these compounds on molecular level.
Co-reporter:Hans-Joachim Wittmann;Sigurd Elz
Naunyn-Schmiedeberg's Archives of Pharmacology 2011 Volume 384( Issue 3) pp:
Publication Date(Web):2011 September
DOI:10.1007/s00210-011-0671-5
Histamine H1-receptor agonists and antagonists exhibit affinity to the human histamine H4-receptor (hH4R). However, the pharmacological profiles between hH1R and hH4R exhibit similarities and differences. Since suprahistaprodifen and trifluoromethylphenylhistamine show significant affinity to hH4R, the aim of this study was to analyse a large number of new phenylhistamines, histaprodifens and phenoprodifens at hH4R to extend the pharmacological profile of these compound classes at hH4R. The hH4R-RGS19 fusion protein was co-expressed with Gαi2 and Gβ1γ2 in Sf9 insect cells, and [3H]histamine competition binding as well as GTPase assays were performed. Based on adequate crystal structures, homology models of hH4R were generated. Molecular modelling studies, including molecular dynamics and prediction of Gibbs energy of ligand binding, were performed in order to explain the pharmacological data at hH4R on molecular level. The exchange of the phenyl moiety of phenylhistamines into the diphenylpropyl moiety of histaprodifens acts, in contrast to hH1R, as partial agonism–inverse agonism switch at hH4R. Based on our studies, some phenylhistamine derivatives with significantly higher affinity at hH4R than at hH1R were identified. The molecular dynamic simulations revealed two different conformations for the highly conserved Trp6.48, suggested to be involved in receptor activation. Furthermore, the predicted Gibbs energy of ligand binding for six selected phenylhistamines was in very good agreement with the experimentally determined affinities. We identified phenylhistamine derivatives with higher affinity at hH4R than at hH1R. Besides, we have identified partial agonism–inverse agonism switch between phenylhistamines and histaprodifens at hH4R. These results are very important to understand selectivity between hH1R and hH4R and to design new potent H1R and/or H4R receptor ligands.
Co-reporter:Andrea Straßer;Hans-Joachim Wittmann
Journal of Computer-Aided Molecular Design 2010 Volume 24( Issue 9) pp:759-769
Publication Date(Web):2010 September
DOI:10.1007/s10822-010-9372-2
The binding of (partial) agonists in the binding pocket of biogenic amine receptors induces a conformational change from the inactive to the active state of the receptors. There is only little knowledge about the binding pathways of ligands into binding pocket on molecular level. So far, it was not possible with molecular dynamic simulations to observe the ligand binding and receptor activation. Furthermore, there is nearly nothing known, in which state of ligand binding, the receptor gets activated. The aim of this study was to get more detailed insight into the process of ligand binding and receptor activation. With the recently developed LigPath algorithm, we scanned the potential energy surface of the binding process of dimeric histaprodifen, a partial agonist at the histamine H1-receptor, into the guinea pig histamine H1-receptor, taking also into account the receptor activation. The calculations exhibited large conformational changes of Trp6.48 and Phe6.55 during ligand binding and receptor activation. Additionally, conformational changes were also observed for Phe6.52, Tyr6.51 and Phe6.44. Conformational changes of Trp6.48 and Phe6.52 are discussed in literature as rotamer toggle switch in context with receptor activation. Additionally, the calculations indicate that the binding of dimeric histaprodifen, accompanied by receptor activation is energetically preferred. In general, this study gives new, theoretical insights onto ligand binding and receptor activation on molecular level.
Co-reporter:Andrea Straßer;Hans-Joachim Wittmann
Journal of Molecular Modeling 2010 Volume 16( Issue 8) pp:1307-1318
Publication Date(Web):2010 August
DOI:10.1007/s00894-010-0646-3
The aim of this study was to perform an in silico analysis of the interaction of the human β2 adrenergic receptor with Gαs. In a first step, a systematic surface-interaction-scan between the inactive or active human β2 adrenergic receptor and Gαs was performed in order to gain knowledge about energetically preferred areas on the potential energy surface. Subsequently, two energetically favored regions for the active human β2 adrenergic receptor–Gαs complex were identified. Two representative complex structures were put into a POPC (1-palmitoyl-2-oleoyl-phosphatidylcholine) bilayer and solvated in order to perform molecular dynamic simulations. The simulations revealed that both conformations, which have comparable potential energy, are stable. A mean number of about 14 hydrogen bonds was observed between the active receptor and Gαs for both conformations. Based on these results, two energetically favored β2–Gαscomplexes can be proposed.
Co-reporter:Andrea Straßer;Hans-Joachim Wittmann
Journal of Computer-Aided Molecular Design 2007 Volume 21( Issue 9) pp:499-509
Publication Date(Web):2007 September
DOI:10.1007/s10822-007-9131-1
The Histamine H1-receptor (H1R), belonging to the amine receptor-class of family A of the G-protein coupled receptors (GPCRs) gets activated by agonists. The consequence is a conformational change of the receptor, which may involve the binding-pocket. So, for a good prediction of the binding-mode of an agonist, it is necessary to have knowledge about these conformational changes. Meanwhile some experimental data about the structural changes of GPCRs during activation exist. Based on homology modeling of the guinea-pig H1R (gpH1R), using the crystal structure of bovine rhodopsin as template, we performed several MD simulations with distance restraints in order to get an inactive and an active structure of the gpH1R. The calculations led to a Phe6.44/Trp6.48/Phe6.52-switch and linearization of the proline kinked transmembrane helix VI during receptor activation. Our calculations showed that the Trp6.48/Phe6.52-switch induces a conformational change in Phe6.44, which slides between transmembrane helices III and VI. Additionally we observed a hydrogen bond interaction of Ser3.39 with Asn7.45 in the inactive gpH1R, but because of a counterclockwise rotation of transmembrane helix III Ser3.39 establishes a water-mediated hydrogen bond to Asp2.50 in the active gpH1R. Additionally we simulated a possible mechanism for receptor activation with a modified LigPath-algorithm.
Co-reporter:Andrea Strasser, Hans-Joachim Wittmann, Armin Buschauer, Erich H. Schneider, Roland Seifert
Trends in Pharmacological Sciences (January 2013) Volume 34(Issue 1) pp:13-32
Publication Date(Web):1 January 2013
DOI:10.1016/j.tips.2012.10.004
Histamine is a biogenic amine that exerts its biological effects as a neurotransmitter and local mediator via four histamine receptor (HR) subtypes (HxRs) – H1R, H2R, H3R, and H4R – belonging to the superfamily of G-protein-coupled receptors (GPCRs). All four HxRs exhibit pronounced differences in agonist and/or antagonist pharmacology among various species orthologs. The species differences constitute a problem for animal experiments and drug development. This problem applies to GPCRs with diverse ligands. Here, we summarize our current knowledge on HxR orthologs as a case study for species-dependent activity of GPCR ligands. We show that species-specific pharmacology also provides unique opportunities to study important aspects of GPCR pharmacology in general, including ligand-binding sites, the roles of extracellular domains in ligand binding and receptor activation, agonist-independent (constitutive) receptor activity, thermodynamics of ligand/receptor interaction, receptor-activation mechanisms, and ligand-specific receptor conformations.

![2-(4-BROMO-2,3,6,7-TETRAHYDROFURO[2,3-F][1]BENZOFURAN-8-YL)-N-[(2-METHOXYPHENYL)METHYL]ETHANAMINE](http://img.cochemist.com/data/images/1335331-42-4.png)
![2-(4-BROMO-2,3,6,7-TETRAHYDROFURO[2,3-F][1]BENZOFURAN-8-YL)-N-[(2-METHOXYPHENYL)METHYL]ETHANAMINE](http://img.cochemist.com/data/images/1335331-42-4_b.png)
![N-cyano-N'-[4-(1H-imidazol-5-yl)butyl]-Guanidine](http://img.cochemist.com/ccimg/1192600/1192560-01-2.png)
![N-cyano-N'-[4-(1H-imidazol-5-yl)butyl]-Guanidine](http://img.cochemist.com/ccimg/1192600/1192560-01-2_b.png)

![2-[2,5-DIMETHOXY-4-(TRIFLUOROMETHYL)PHENYL]-N-[(2-METHOXYPHENYL)METHYL]ETHANAMINE](/data/chemimg/329000/1027161-33-6.png)
![2-[2,5-DIMETHOXY-4-(TRIFLUOROMETHYL)PHENYL]-N-[(2-METHOXYPHENYL)METHYL]ETHANAMINE](/data/chemimg/329000/1027161-33-6_b.png)

![Phenol, 2-[[[2-(4-iodo-2,5-dimethoxyphenyl)ethyl]amino]methyl]-](http://img.cochemist.com/ccimg/919800/919797-20-9.png)
![Phenol, 2-[[[2-(4-iodo-2,5-dimethoxyphenyl)ethyl]amino]methyl]-](http://img.cochemist.com/ccimg/919800/919797-20-9_b.png)


![Benzeneethanamine, 2,5-dimethoxy-N-[(2-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/919800/919797-16-3.png)
![Benzeneethanamine, 2,5-dimethoxy-N-[(2-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/919800/919797-16-3_b.png)




![PIPERIDINE, 1-[2-[2-[3-(TRIFLUOROMETHYL)PHENYL]-1H-IMIDAZOL-4-YL]ETHYL]-](http://img.cochemist.com/ccimg/588800/588707-23-7.png)
![PIPERIDINE, 1-[2-[2-[3-(TRIFLUOROMETHYL)PHENYL]-1H-IMIDAZOL-4-YL]ETHYL]-](http://img.cochemist.com/ccimg/588800/588707-23-7_b.png)
![1H-Imidazole-4-ethanamine, 2-[[(4-methylphenyl)thio]methyl]-](http://img.cochemist.com/ccimg/588800/588707-22-6.png)
![1H-Imidazole-4-ethanamine, 2-[[(4-methylphenyl)thio]methyl]-](http://img.cochemist.com/ccimg/588800/588707-22-6_b.png)


![PIPERAZINE, 1-[(5-AMINO-1H-INDOL-2-YL)CARBONYL]-4-METHYL-](http://img.cochemist.com/ccimg/459200/459169-41-6.png)
![PIPERAZINE, 1-[(5-AMINO-1H-INDOL-2-YL)CARBONYL]-4-METHYL-](http://img.cochemist.com/ccimg/459200/459169-41-6_b.png)




![1H-Imidazole,5-[3-(phenylmethoxy)propyl]-](http://img.cochemist.com/ccimg/177800/177708-09-7.png)
![1H-Imidazole,5-[3-(phenylmethoxy)propyl]-](http://img.cochemist.com/ccimg/177800/177708-09-7_b.png)





![Carbamimidothioic acid,N-[(4-chlorophenyl)methyl]-, 3-(1H-imidazol-5-yl)propyl ester](http://img.cochemist.com/ccimg/145300/145231-45-4.png)
![Carbamimidothioic acid,N-[(4-chlorophenyl)methyl]-, 3-(1H-imidazol-5-yl)propyl ester](http://img.cochemist.com/ccimg/145300/145231-45-4_b.png)

![Carbamimidothioic acid,N-[2-[4-(iodo-125I)phenyl]ethyl]-, 3-(1H-imidazol-5-yl)propyl ester](http://img.cochemist.com/ccimg/143500/143407-29-8.png)
![Carbamimidothioic acid,N-[2-[4-(iodo-125I)phenyl]ethyl]-, 3-(1H-imidazol-5-yl)propyl ester](http://img.cochemist.com/ccimg/143500/143407-29-8_b.png)





![1-Piperidinecarboxylic acid, 4-[[1-(2-phenylethyl)-1H-benzimidazol-2-yl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/112000/111921-78-9.png)
![1-Piperidinecarboxylic acid, 4-[[1-(2-phenylethyl)-1H-benzimidazol-2-yl]amino]-, ethyl ester](http://img.cochemist.com/ccimg/112000/111921-78-9_b.png)
![1,2-Ethanediamine,N1-[(4-methoxyphenyl)methyl]-N2-methyl-N1-2-pyridinyl-](http://img.cochemist.com/ccimg/104500/104499-47-0.png)
![1,2-Ethanediamine,N1-[(4-methoxyphenyl)methyl]-N2-methyl-N1-2-pyridinyl-](http://img.cochemist.com/ccimg/104500/104499-47-0_b.png)


![Benzoic acid, 2-[(ethoxycarbonyl)amino]-5-(phenylmethoxy)-](http://img.cochemist.com/ccimg/104400/104356-42-5.png)
![Benzoic acid, 2-[(ethoxycarbonyl)amino]-5-(phenylmethoxy)-](http://img.cochemist.com/ccimg/104400/104356-42-5_b.png)


![Carbamimidothioic acid,2-[(aminoiminomethyl)amino]ethyl ester](http://img.cochemist.com/ccimg/98100/98021-17-1.png)
![Carbamimidothioic acid,2-[(aminoiminomethyl)amino]ethyl ester](http://img.cochemist.com/ccimg/98100/98021-17-1_b.png)


![PYRROLO[2,1-B]QUINAZOLIN-9(1H)-ONE, 2,3-DIHYDRO-7-HYDROXY-](http://img.cochemist.com/ccimg/85700/85654-22-4.png)
![PYRROLO[2,1-B]QUINAZOLIN-9(1H)-ONE, 2,3-DIHYDRO-7-HYDROXY-](http://img.cochemist.com/ccimg/85700/85654-22-4_b.png)




![1-Piperidinecarboxylic acid, 4-[[1-[(2-fluorophenyl)methyl]-1H-benzimidazol-2-yl]amino]-, ethyl ester](/data/chemimg/1954500/73734-13-1.png)
![1-Piperidinecarboxylic acid, 4-[[1-[(2-fluorophenyl)methyl]-1H-benzimidazol-2-yl]amino]-, ethyl ester](/data/chemimg/1954500/73734-13-1_b.png)
![1-Piperidinecarboxylic acid, 4-[(6-chloro-1H-benzimidazol-2-yl)amino]-, ethyl ester](/data/chemimg/1957200/73734-08-4.png)
![1-Piperidinecarboxylic acid, 4-[(6-chloro-1H-benzimidazol-2-yl)amino]-, ethyl ester](/data/chemimg/1957200/73734-08-4_b.png)
![1-Piperidinecarboxylic acid, 4-[[[(2-amino-5-chlorophenyl)amino]thioxomethyl]amino]-, ethyl ester](/data/chemimg/1955800/73733-80-9.png)
![1-Piperidinecarboxylic acid, 4-[[[(2-amino-5-chlorophenyl)amino]thioxomethyl]amino]-, ethyl ester](/data/chemimg/1955800/73733-80-9_b.png)


![Guanidine,N-[2-[[[2-[(aminoiminomethyl)amino]-4-thiazolyl]methyl]thio]ethyl]-N'-cyano-N''-methyl-](http://img.cochemist.com/ccimg/69100/69014-14-8.png)
![Guanidine,N-[2-[[[2-[(aminoiminomethyl)amino]-4-thiazolyl]methyl]thio]ethyl]-N'-cyano-N''-methyl-](http://img.cochemist.com/ccimg/69100/69014-14-8_b.png)





![2-Pyridinamine,N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/52900/52818-63-0.png)
![2-Pyridinamine,N-[(4-methoxyphenyl)methyl]-](http://img.cochemist.com/ccimg/52900/52818-63-0_b.png)
![Guanidine, N-cyano-N'-[4-(1H-imidazol-4-yl)butyl]-N''-methyl-](http://img.cochemist.com/ccimg/52400/52378-57-1.png)
![Guanidine, N-cyano-N'-[4-(1H-imidazol-4-yl)butyl]-N''-methyl-](http://img.cochemist.com/ccimg/52400/52378-57-1_b.png)





![2-{[(5-METHYL-1H-IMIDAZOL-4-YL)METHYL]-THIO}ETHANAMINE](http://img.cochemist.com/ccimg/38600/38585-67-0.png)
![2-{[(5-METHYL-1H-IMIDAZOL-4-YL)METHYL]-THIO}ETHANAMINE](http://img.cochemist.com/ccimg/38600/38585-67-0_b.png)

![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7_b.png)








![Ethanone,1-(4-chlorophenyl)-2-[[3-(10,11-dihydro-5H-dibenz[b,f]azepin-5-yl)propyl]methylamino]-](http://img.cochemist.com/ccimg/23100/23047-25-8.png)
![Ethanone,1-(4-chlorophenyl)-2-[[3-(10,11-dihydro-5H-dibenz[b,f]azepin-5-yl)propyl]methylamino]-](http://img.cochemist.com/ccimg/23100/23047-25-8_b.png)







![10H-Phenothiazine,10-[2-(1-methyl-2-piperidinyl)ethyl]-2-(methylsulfonyl)-](http://img.cochemist.com/ccimg/14800/14759-06-9.png)
![10H-Phenothiazine,10-[2-(1-methyl-2-piperidinyl)ethyl]-2-(methylsulfonyl)-](http://img.cochemist.com/ccimg/14800/14759-06-9_b.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(1-piperazinyl)-](http://img.cochemist.com/ccimg/6200/6104-71-8.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(1-piperazinyl)-](http://img.cochemist.com/ccimg/6200/6104-71-8_b.png)

![10H-Phenothiazine,10-[2-(1-methyl-2-piperidinyl)ethyl]-2-(methylsulfinyl)-](http://img.cochemist.com/ccimg/5600/5588-33-0.png)
![10H-Phenothiazine,10-[2-(1-methyl-2-piperidinyl)ethyl]-2-(methylsulfinyl)-](http://img.cochemist.com/ccimg/5600/5588-33-0_b.png)
![11H-Dibenzo[b,e][1,4]diazepin-11-one,10-[2-(dimethylamino)ethyl]-5,10-dihydro-5-methyl-](http://img.cochemist.com/ccimg/4500/4498-32-2.png)
![11H-Dibenzo[b,e][1,4]diazepin-11-one,10-[2-(dimethylamino)ethyl]-5,10-dihydro-5-methyl-](http://img.cochemist.com/ccimg/4500/4498-32-2_b.png)



![Dibenz[b,f][1,4]oxazepine,2-chloro-11-(4-methyl-1-piperazinyl)-](http://img.cochemist.com/ccimg/2000/1977-10-2.png)
![Dibenz[b,f][1,4]oxazepine,2-chloro-11-(4-methyl-1-piperazinyl)-](http://img.cochemist.com/ccimg/2000/1977-10-2_b.png)








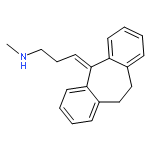









![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5.png)
![3-(10,11-Dihydro-5H-dibenzo[b,f]azepin-5-yl)-N-methylpropan-1-amine](http://img.cochemist.com/ccimg/100/50-47-5_b.png)




![ethyl 4-[[1-[(4-fluorophenyl)methyl]-1H-benzimidazol-2-yl]amino]piperidine-1-carboxylate](http://img.cochemist.com/ccimg/84600/84501-68-8.png)
![ethyl 4-[[1-[(4-fluorophenyl)methyl]-1H-benzimidazol-2-yl]amino]piperidine-1-carboxylate](http://img.cochemist.com/ccimg/84600/84501-68-8_b.png)

![1-[(4-FLUOROPHENYL)METHYL]-N-PIPERIDIN-4-YLBENZIMIDAZOL-2-AMINE](http://img.cochemist.com/ccimg/76000/75970-99-9.png)
![1-[(4-FLUOROPHENYL)METHYL]-N-PIPERIDIN-4-YLBENZIMIDAZOL-2-AMINE](http://img.cochemist.com/ccimg/76000/75970-99-9_b.png)






![1-Piperidinecarboxylic acid, 4-[[[(2-aminophenyl)amino]thioxomethyl]amino]-, ethyl ester](/data/chemimg/1957200/73733-81-0.png)
![1-Piperidinecarboxylic acid, 4-[[[(2-aminophenyl)amino]thioxomethyl]amino]-, ethyl ester](/data/chemimg/1957200/73733-81-0_b.png)

![1-{[2-(TRIMETHYLSILYL)ETHOXY]METHYL}-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/73200/73187-30-1.png)
![1-{[2-(TRIMETHYLSILYL)ETHOXY]METHYL}-1H-PYRROLO[2,3-B]PYRIDINE](http://img.cochemist.com/ccimg/73200/73187-30-1_b.png)
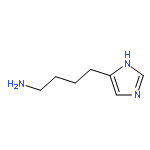
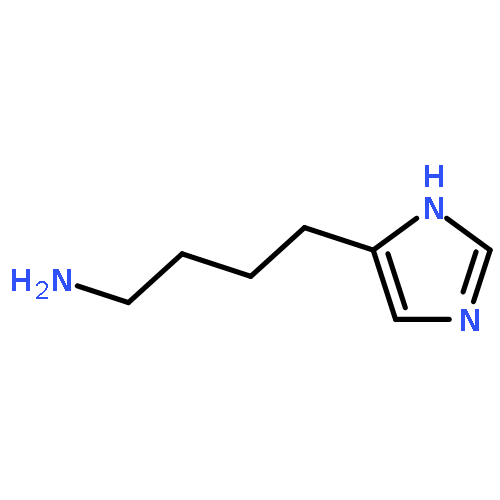


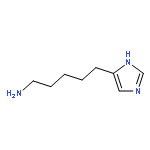

![10H-Benzo[4,5]cyclohepta[1,2-b]thiophen-10-one, 4,9-dihydro-4-(1-methyl-4-piperidinylidene)-](http://img.cochemist.com/ccimg/34600/34580-13-7.png)
![10H-Benzo[4,5]cyclohepta[1,2-b]thiophen-10-one, 4,9-dihydro-4-(1-methyl-4-piperidinylidene)-](http://img.cochemist.com/ccimg/34600/34580-13-7_b.png)
![Dibenzo[c,f]pyrazino[1,2-a]azepine,1,2,3,4,10,14b-hexahydro-2-methyl-](http://img.cochemist.com/ccimg/24300/24219-97-4.png)
![Dibenzo[c,f]pyrazino[1,2-a]azepine,1,2,3,4,10,14b-hexahydro-2-methyl-](http://img.cochemist.com/ccimg/24300/24219-97-4_b.png)


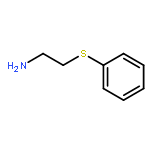

![3-(Dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine](http://img.cochemist.com/ccimg/1700/1668-19-5.png)
![3-(Dibenzo[b,e]oxepin-11(6H)-ylidene)-N,N-dimethylpropan-1-amine](http://img.cochemist.com/ccimg/1700/1668-19-5_b.png)

![4-(5H-Dibenzo[a,d][7]annulen-5-ylidene)-1-methylpiperidine](http://img.cochemist.com/ccimg/200/129-03-3.png)
![4-(5H-Dibenzo[a,d][7]annulen-5-ylidene)-1-methylpiperidine](http://img.cochemist.com/ccimg/200/129-03-3_b.png)






![1H-Imidazole-4-ethanamine, 2-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/162100/162049-82-3.png)
![1H-Imidazole-4-ethanamine, 2-[3-(trifluoromethyl)phenyl]-](http://img.cochemist.com/ccimg/162100/162049-82-3_b.png)

![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9.png)
![1-(4-Fluorobenzyl)-N-(1-(4-methoxyphenethyl)piperidin-4-yl)-1H-benzo[d]imidazol-2-amine](http://img.cochemist.com/ccimg/68900/68844-77-9_b.png)




![1,2-Ethanediamine,N1-[(4-methoxyphenyl)methyl]-N2,N2-dimethyl-N1-2-pyridinyl-](http://img.cochemist.com/ccimg/100/91-84-9.png)
![1,2-Ethanediamine,N1-[(4-methoxyphenyl)methyl]-N2,N2-dimethyl-N1-2-pyridinyl-](http://img.cochemist.com/ccimg/100/91-84-9_b.png)