Co-reporter:Subramaniam Kuppuswamy, Joshua D. Wofford, Chris Joseph, Zhu-Lin Xie, Azim K. Ali, Vincent M. Lynch, Paul A. Lindahl, and Michael J. Rose
Inorganic Chemistry May 15, 2017 Volume 56(Issue 10) pp:5998-5998
Publication Date(Web):April 25, 2017
DOI:10.1021/acs.inorgchem.7b00741
The syntheses, interconversions, and spectroscopic properties of a set of iron carbonyl clusters containing an interstitial carbide are reported. This includes the low temperature X-ray structures of the six-iron clusters (Y)2[Fe6(μ6-C)(μ2-CO)4(CO)12] (1a–c; where Y = NMe4, NEt4, PPh4); the five-iron cluster [Fe5(μ5-C)(CO)15] (3); and the novel formulation of the five-iron cluster (NMe4)2[Fe5(μ5-C)(μ2-CO)(CO)13] (4). Also included in this set is the novel charge-neutral cluster, [Fe6(μ6-C)(CO)18] (2), for which we were unable to obtain a crystallographic structure. As synthetic proof for the identity of 2, we performed a closed loop of interconversions within a family of crystallographically defined species (1, 3, and 4): [Fe6]2– → [Fe6]0 → [Fe5]0 → [Fe5]2– → [Fe6]2–. The structural, spectroscopic, and electronic properties of this “missing link” cluster 2 were investigated by IR, Raman, XPS, and Mössbauer spectroscopies—as well as by DFT calculations. A single νCO feature (1965 cm–1) in the IR spectrum of 2, as well as a prominent Raman feature (νsymm = 1550 cm–1), are consistent with the presence of terminal carbonyls and a {(μ6-C)Fe6} arrangement of iron centers around the central carbide. The XPS of 2 exhibits a higher energy Fe 2p3/2 feature (707.4 eV) as compared to that of 1 (705.5 eV), consistent with the two-electron oxidation induced by treatment of 1 with two equivalents of [Fc](PF6) under CO atmosphere (for the two added CO ligands). DFT calculations indicate two axial and four equatorial Fe sites in 1, all of which have the same or similar oxidation states, for example, two Fe(0) and four Fe(+0.5). These assignments are supported by Mössbauer spectra for 1, which exhibit two closely spaced quadrupole doublets with δ = 0.076 and 0.064 mm s–1. The high-field Mössbauer spectrum of 2 (4.2 K) exhibits three prominent quadrupole doublets with δ = −0.18, −0.11, and +0.41 mm s−1. This indicates three pairs of chemically equivalent Fe sites. The first two pairs arise from irons of a similar oxidation state, while the last pair arises from irons in a different oxidation state, indicating a mixed-valent cluster. Variable field Mössbauer spectra for 2 were simulated assuming these two groups and a diamagnetic ground state. Taken together, the Mössbauer results and DFT calculations for 2 indicate two axial Fe(II) sites and four equatorial sites of lower valence, probably Fe(0). In the DFT optimized pentagonal bipyramidal structure for 2, the Fe(II)–Ccarbide distances are compressed (∼1.84 Å), while the Fe(0)–Ccarbide distances are elongated (∼2.05 Å). Analysis of the formulations for 1 (closo-square bipyramid) and 2 (nido-pentagonal bipyramid) is considered in the context of the textbook electron-counting rules of 14n+2 and 14n+4 for closo and nido clusters, respectively. This redox-dependent intracluster disproportionation of Fe oxidation states is concluded to arise from changes in bonding to the central carbide. A similar phenomenon may be promoted by the central carbide of the FeMoco cluster of nitrogenase, which may in turn stimulate N2 reduction.
Co-reporter:J. Patrick Shupp;Amber R. Rose
Dalton Transactions 2017 vol. 46(Issue 28) pp:9163-9171
Publication Date(Web):2017/07/18
DOI:10.1039/C7DT01506B
We report the synthesis, interconversions and X-ray structures of a set of [mFe–nS]-type carbonyl clusters (where S = S2−, S22− or RS−; m = 2–3; n = 1–2). All of the clusters have been identified and characterized by single crystal X-ray diffraction, IR and 13C NMR. Reduction of the parent neutral dimer [μ2-(SPh)2Fe2(CO)6] (1) with KC8 affords an easily separable ∼1 : 1 mixture of the anionic, dimeric thiolate dimer K[Fe2(SPh)(CO)6(μ-CO)] (2) and the dianionic, sulfido trimer [K(benzo-15-crown-5)2]2[Fe3(μ3-S)(CO)9] (3). Oxidation of 2 with diphenyl-disulfide (Ph2S2) cleanly returns the starting material 1. The Ph–S bond in 1 can be cleaved to form sulfide trimer 3. Oxidation of sulfido trimer 3 with [Fc](PF6) in the presence of S8 cleanly affords the all-inorganic persulfide dimer [μ2-(S)2Fe2(CO)6] (4), a thermodynamically stable product. The inverse reactions to form 3 (dianion) from 4 (neutral) were not successful, and other products were obtained. For example, reduction of 4 with KC8 afforded the mixed valence Fe(I)/Fe(II) species [((FeI2S2)(CO)6)2FeII]2− (5), in which the two {Fe2S2(CO)6}2− units serve as bidendate ligands to a Fe(II) center. Another isolated product (THF insoluble portion) was recrystallized in MeCN to afford [K(benzo-15-crown-5)2]2[((Fe2S)(CO)6)2(μ-S)2] (6), in which a persulfide dianion bridges two {2Fe–S} moieties (dimer of dimers). Finally, to close the interconversion loop, we converted the persulfide dimer 4 into the thiolate dimer 1 by reduction with KC8 followed by reaction with the diphenyl iodonium salt [Ph2I](PF6), in modest yield. These reactions underscore the thermodynamic stability of the dimers 1 and 4, as well as the synthetic and crystallization versatility of using the crown/K+ counterion system for obtaining structural information on highly reduced iron–sulfur–carbonyl clusters.
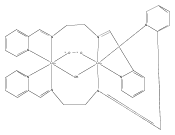
Co-reporter:Taylor A. Manes, Michael J. Rose
Coordination Chemistry Reviews 2017 Volume 353(Volume 353) pp:
Publication Date(Web):15 December 2017
DOI:10.1016/j.ccr.2017.09.022
•Survey of scaffold based ligands for geometric control and reactivity preferences.•Address the use of anthracene as a viable scaffold for fac chelate systems.•Comparison of DBF and anthracene scaffolds utilizing para and meta donor groups.•Survey of biological systems incorporating facial donor fields.•Survey of structural data to provide a framework for rational ligand design.•Review of the potential utility of anthracene for modeling the active site of Hmd.Herein we examine the use of scaffold-based ligands for organometallic catalysis and bio-inorganic modeling studies. The use of scaffolds in catalyst development and complex design stems from researchers’ desire to install specific donor geometries (e.g., cis vs trans, fac vs mer) to generate desired structures and corresponding reactivities in mononuclear metal complexes. Starting from the use of polyaryl ligands for asymmetric catalysis (primarily Ru), we review successive implementations of scaffold-based ligands of ever-increasing complexity. Particular attention is paid to rigidly planar anthracene-based (and related) ligands that support both precious metal (Pd, Rh, Re) and base metal (Mn, Fe) centers in structural and reactivity studies. Previous work in scaffold design by others (Lu, Gelman) is considered in concert with our own contributions to this field. Ultimately, the complexity of such scaffolds has evolved to include non-symmetric anthracene scaffolds relevant to bio-inorganic synthetic modeling. As an illustrative example, work regarding the enzyme mono-iron hydrogenase is documented, wherein the ligand scaffold provides a biomimetic CNS chelate (containing an organometallic acyl-C donor) for structural and functional synthetic models. A quantitative analysis of structural relationships among torsion angles, donor atom distances, and bite angles of the ligand systems and resulting metal complexes is presented. This provides a foundation for a rational, target-driven syntheses of metal complexes derived from rigid, tricyclic scaffold ligands.Download high-res image (92KB)Download full-size image
Co-reporter:Junhyeok Seo, Timothy E. Sotman, Eileen R. Sullivan, Bryan D. Ellis, Truc Phung, Michael J. Rose
Tetrahedron 2017 Volume 73, Issue 31(Issue 31) pp:
Publication Date(Web):3 August 2017
DOI:10.1016/j.tet.2017.05.082
We report regioselective functionalization of pyridones and pyrones via electrophilic bromination (Br2) or radical trifluoromethylation (NaSO2CF3/tBuOOH) at the 3-position. Counter-intuitively, the 3-position EW groups decreased the carbonyl stretching energy by 6–23 cm−1; however, 3,5-dibromination increased the CO frequency by 10–22 cm−1 compared to the 3-Br pyridones. X-ray crystallography revealed pyridone tautomers with contracted CO bond metrics. pKa values and 1H NMR shifts for the series 3-H→Br→CF3 revealed the expected trend of increasing acidity (pKa = 8.85 → 8.33→6.78, MeOH) and increasing chemical shifts (10.97 → 11.42→11.71 DMSO-d6). We conclude that the paradoxical decrease in CO stretching frequencies by the 3-positoin EW groups is explained by an ‘assistive’ electron-withdrawing effect, whereby the 3-position EW group assists the electronegative oxygen atom in recruiting more electron density, and – as a result – attaining more oxyanion character (decreased the CO bond strength).Download high-res image (134KB)Download full-size image
Co-reporter:Daniel W. Redman, Hark Jin Kim, Keith J. Stevenson and Michael J. Rose
Journal of Materials Chemistry A 2016 vol. 4(Issue 18) pp:7027-7035
Publication Date(Web):25 Mar 2016
DOI:10.1039/C5TA09684G
This work reports on the synergistic utility of ionic liquid-based, photo-assisted electrodeposition of MoSx onto organic-functionalized silicon photolelectrodes for dihydrogen (H2) evolution under 1-sun illumination. The surface linker 3,5-dimethoxyphenyl covalently attached to Si(111) enhances conductivity at the substrate|electrolyte interface (impedance spectroscopy) and provides improved physico-chemical support for MoSx deposition (SEM, Raman, PEC-deposition, XPS), to generate a functional composite device that results in 0.33% efficiency for solar → H2 conversion. We report a new method of ionic liquid-based MoSx deposition that avoids complications in analogous aqueous procedures and facilitates the deposition of a uniform catalytic film. This work provides a generalized framework for the optimization of non-platinum catalysts for solar → H2 conversion.
Co-reporter:Owen M. Williams, Justin W. Shi and Michael J. Rose
Chemical Communications 2016 vol. 52(Issue 58) pp:9145-9148
Publication Date(Web):12 May 2016
DOI:10.1039/C6CC00703A
We report a photocathode device consisting of GaP, a metal oxide (Al2O3 or ZnO), a phosphonate-C12-thiol monolayer, and gold nanoparticles (AuNPs). The AuNPs enhance electron transfer: in non-aqueous electrochemistry (EtV2+ in MeCN), p-GaP|Al2O3|O3PC12S|AuNP and …ZnO|…|AuNP rescued the photocurrent (24%, 59% of Jmax-etch). Aqueous experiments (CO2 saturated KCl) using the optimized ZnO-functionalized device exhibited H+ → H2 (FY = 66%) and CO2 → CO (FY = 6%).
Co-reporter:Hark Jin Kim, Junhyeok Seo, and Michael J. Rose
ACS Applied Materials & Interfaces 2016 Volume 8(Issue 2) pp:1061
Publication Date(Web):January 7, 2016
DOI:10.1021/acsami.5b09902
We report the fabrication of a {semiconductor}|{metal oxide}|{molecular catalyst} construct for the photogeneration of dihydrogen (H2) under illumination, including band-edge modulation of the semiconductor electrode depending on the identity of Si(111)–R and the metal oxide. Briefly, a synergistic band-edge modulation is observed upon (i) the introduction of a p-Si|n-AZO heterojunction and (ii) introduction of an organic dimethoxyphenyl (diMeOPh) group at the heterojunction interface; the AZO also serves as a transparent and conductive conduit, which was capped with an ultrathin layer (20 Å) of amorphous TiO2 for stability. A phosphonate-appended PNP ligand and its Ni complex were then adsorbed to the p/n heterojunction for photoelectrochemical H2 generation (figures of merit: Vonset ≈ + 0.03 V vs NHE, Jmax ≈ 8 mA cm–2 at 60 mM TsOH).Keywords: AZO; band-edge modulation; Ni-PNP; photoelectrochemical H2 evolution; Si(111) semiconductor; TiO2
Co-reporter:Gummadi Durgaprasad; Zhu-Lin Xie
Inorganic Chemistry 2016 Volume 55(Issue 2) pp:386-389
Publication Date(Web):September 25, 2015
DOI:10.1021/acs.inorgchem.5b01733
We report the identification and reactivity of an iron hydride species in a synthetic model complex of monoiron hydrogenase. The hydride complex is derived from a phosphine-free CNS chelate that includes a Fe–CNH(═O) bond (carbamoyl) as a mimic of the active site iron acyl. The reaction of [(O═CHNNpySMe)Fe(CO)2(Br)] (1) with NaHBEt3 generates the iron hydride intermediate [(O═CHNNpySMe)Fe(H)(CO)2] (2; δFe–H = −5.08 ppm). Above −40 °C, the hydride species extrudes CH3S– via intramolecular hydride transfer, which is stoichiometrically trapped in the structurally characterized dimer μ2-(CH3S)2-[(O═CHNNPh)Fe(CO)2]2 (3). Alternately, when activated by base (tBuOK), 1 undergoes desulfurization to form a cyclometalated species, [(O═CNHNCPh)Fe(CO)2] (5); derivatization of 5 with PPh3 affords the structurally characterized species [(O═CNHNC)Fe(CO)(PPh3)2] (6), indicating complex 6 as the common intermediate along each pathway of desulfurization.
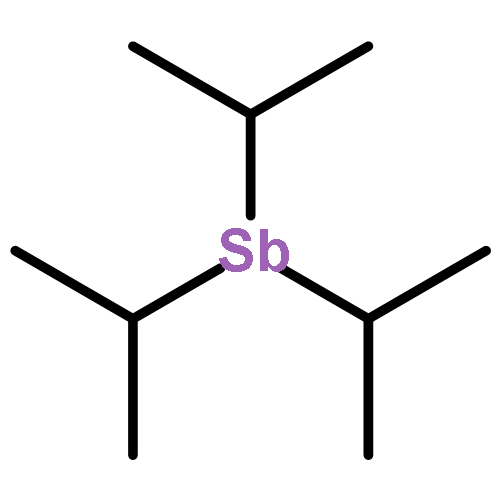
Co-reporter:William V. Taylor, Ulysses H. Soto, Vincent M. Lynch, and Michael J. Rose
Inorganic Chemistry 2016 Volume 55(Issue 7) pp:3206-3208
Publication Date(Web):March 24, 2016
DOI:10.1021/acs.inorgchem.5b02933
Herein we report the synthesis, temperature-dependent X-ray structures (100, 150, 200, 250, 273, and 300 K), and solid-state emissive properties of the antimony–copper(I)–iodo cluster [Cu4(I)4(SbiPr3)4] (1), supported by the trialkylantimony donor SbiPr3. Overall, 1 exhibits a distorted cuboidal structure, wherein a twisting of the “cube” generates an interconnected Cu4 tetrahedron, as well as long Cu–I and Cu–Sb bonds [e.g., 2.707(2) and 2.571(2) Å]; in the 100 K X-ray structure, the Cu–Cu bonds are quite short [2.761(3) Å]. Solid-state emission spectra of 1 were obtained over a range of temperatures (163–298 K), wherein 1 exhibits only a low-energy emission feature centered near 700 nm (λEm = 711 nm, fwhm = 150 nm, and λEx = 390 nm). Because 1 contains no aryl units, the emission spectrum (and absorption) can be unambiguously attributed to the Cu4I4 core [no L (pnictogen ligand) component]. The luminescence is sharply attenuated upon warming from 150 to 200 K. An overlaid plot of the emissive properties of 1 and its Cu–Cu bond distances [2.761(3)–2.836(4) Å] reveal that ∼2.80 Å represents a critical crossing point for low-energy thermoluminescence in Cu4-based clusters.
Co-reporter:Taylor A. Manes and Michael J. Rose
Inorganic Chemistry 2016 Volume 55(Issue 11) pp:5127-5138
Publication Date(Web):May 19, 2016
DOI:10.1021/acs.inorgchem.5b02737
Presented herein is a synthetic scheme to generate symmetric and asymmetric ligands based on a 1,8-disubstituted anthracene scaffold. The metal-binding scaffolds were prepared by aryl chloride activation of 1,8-dichloroanthracene using Suzuki-type couplings facilitated by [Pd(dba)2] as a Pd source; the choice of cocatalyst (XPhos or SPhos) yielded symmetrically or asymmetrically substituted scaffolds (respectively): namely, Anth-SMe2 (3), Anth-N2 (4), and Anth-NSMe (6). The ligands exhibit a nonplanar geometry in the solid state (X-ray), owing to steric hindrance between the anthracene scaffold and the coupled aryl units. To determine the flexibility and binding characteristics of the anthracene-based ligands, the symmetric scaffolds were complexed with [Mn(CO)5Br] to afford the mononuclear species [(Anth-SMe2)Mn(CO)3Br] (8) and [(Anth-N2)Mn(CO)3Br] (9), in which the donor moieties chelate the Mn center in a cis fashion. The asymmetric ligand Anth-NSMe (6) binds preferentially through the py moieties, affording the bis-ligated complex [(Anth-NSMe)2Mn(CO)3Br] (10), wherein the thioether-S donors remain unbound. Alternatively, deprotection of the thioether in 6 affords the free thiol ligand Anth-NSH (7), which more readily binds the Mn center. Complexation of 7 ultimately affords the mixed-valence MnI/MnII dimer of formula [(Anth-NS)3Mn2(CO)3] (11), which exhibits a fac-{Mn(CO)3} unit supported by a triad of bridging thiolates, which are in turn ligated to a supporting Mn(II) center (EPR: |D| = 0.053 cm–1, E/|D| = 0.3, Aiso = −150 MHz). All of the metal complexes have been characterized by single-crystal X-ray diffraction, IR spectroscopy and NMR/EPR measurements—all of which demonstrate that the meta-linked, anthracene-based ligand scaffold is a viable approach for the coordination of metal carbonyls.
Co-reporter:Yae In Cho, Meredith L. Ward and Michael J. Rose
Dalton Transactions 2016 vol. 45(Issue 34) pp:13466-13476
Publication Date(Web):05 Aug 2016
DOI:10.1039/C6DT02104B
We report the synthesis of two fluoride bridged cobalt(II) dimers – [CoII2(μ-F)(pnN4-PhCl)2(OH2)(MeCN)](BF4)3 (1) and [CoII2(μ-F)2(pnN4-PhCl)2](BF4)2 (2) – and related complexes derived from propyl-bridged N4 Schiff base plus pyridine ligands. Notably, the bridging fluoride ion(s) emanate from B–F abstraction processes on the BF4 anions in the starting salt, [Co(H2O)6](BF4)2. Two types of bridging motifs are generated – mono-bridged (μ-F) or di-bridged (μ-F)2 – synthetically differentiated by the absence or presence of pyridine, respectively, during metalation. The synergistic roles of pyridine and the ClPhN4 ligand in promoting B–F abstraction were clarified by the isolation and crystallization of the simple tetrakis-pyridine monomeric complex [Co(py)4(MeCN)2](BF4)2 (4) [no B–F abstraction]; subsequent addition of the ClPhN4 ligand to 4 resulted in formation of the dimeric, di-bridged complex 2. Omission of pyridine during metalation resulted in formation of the mono-bridged dimer 1. The bulky chlorophenyl substituents were obligate for B–F abstraction, as metalation of the unsubstituted N4 ligand resulted in the non-fluoride-bridged dimer, [CoII2(pnN4)3](BF4)4 (3). In magnetic studies, complexes 1 (μeff = 6.24μB, 298 K) and 2 (μeff = 7.70μB, 298 K) both exhibit antiferromagnetic (AFM) coupling, but to different extents. Temperature-dependent magnetic susceptibility measurements (SQUID, 2 → 300 K) reveal that the linearity of the mono-fluoride bridge in 1 [∠Co–F–Co = 159.47(11)°] results in very strong AFM coupling (J = −14.9 cm−1). In contrast, the more acute Co2F2 diamond core [∠Co–F–Co = 98.8(2)°, 99.1(2)°] results in a smaller extent of AFM coupling (J = −2.97 cm−1). Overall, the results indicate the ‘non-innocence’ of the BF4 counterion in cobalt(II) chemistry, and dimers 1 and 2 affirm the effect of the geometry of the bridging fluoride ion(s) in determining the extent of AFM coupling.
Co-reporter:Hark Jin Kim, Kara L. Kearney, Luc H. Le, Zachary J. Haber, Angus A. Rockett, and Michael J. Rose
The Journal of Physical Chemistry C 2016 Volume 120(Issue 45) pp:25697-25708
Publication Date(Web):November 3, 2016
DOI:10.1021/acs.jpcc.6b08096
Ultrathin film amorphous (a-TiO2) and anatase crystalline (c-TiO2) titanium dioxide were investigated as corrosion passivation layers on n-type Si(111). Varying thicknesses of TiO2 (5–60 Å) were deposited on n-Si(111)–CH3 substrates by atomic layer deposition (ALD) at 150 and 240 °C, thus yielding a-TiO2 and c-TiO2, respectively. The phase and morphology of the TiO2 films were determined using a combination of XRD, Raman spectroscopy, XPS, AFM and SEM. The electronic properties of a-TiO2 and c-TiO2 films were compared using 4-point sheet resistance and electrochemical impedance spectroscopy. Substrates functionalized with c-TiO2 exhibited higher conductivity, lower charge transfer resistance, and comparable anticorrosion behavior to a-TiO2. The photoelectrochemical response of n-Si(111)–CH3|TiO2 electrodes as a function of TiO2 thickness was characterized using cyclic voltammetry with ferrocene in acetonitrile. A thickness of 20 or 40 Å was required to block charge transport through a-TiO2 and c-TiO2, respectively. Lastly, the charge transport behaviors of both the amorphous and crystalline n-Si(111)–CH3|TiO2 constructs were enhanced via the deposition of platinum nanoparticles (ALD) on the TiO2 layer. Using a solid-state drift diffusion simulation package (wxAMPS), a theoretical basis for the charge-transport behavior was developed. The experimental thickness-dependence results were used as a basis of comparison to determine the charge transfer mechanism across the n-Si(111)–CH3|TiO2 electrodes. The simulations suggest that the charges conduct via field-assisted thermionic emission across the Si(111)–CH3|TiO2 interface, utilizing a defect band that is consistent with the “leaky dielectric” attribute of TiO2 films. In addition, the simulations suggest that the defects present in a-TiO2 behave as trap states, while the defects present in c-TiO2 behave as recombination centers; this is derived from the observed difference in photoelectrochemical behavior between the two films.
Co-reporter:Junhyeok Seo; Hark Jin Kim; Ryan T. Pekarek
Journal of the American Chemical Society 2015 Volume 137(Issue 9) pp:3173-3176
Publication Date(Web):February 26, 2015
DOI:10.1021/ja5126287
The efficient generation of dihydrogen on molecularly modified p-Si(111) has remained a challenge due to the low barrier heights observed on such surfaces. The band-edge and barrier height challenge is a primary obstruction to progress in the area of integration of molecular H2 electrocatalysts with silicon photoelectrodes. In this work, we demonstrate that an optimal combination of organic passivating agent and inorganic metal oxide leads to H2 evolution at photovoltages positive of RHE. Modulation of the passivating R group [CH3 → Ph → Naph → Anth → Ph(OMe)2] improves both the band-edge position and ΔV (Vonset – VJmax). Subsequent atomic layer deposition (ALD) of Al2O3 or TiO2 along with ALD-Pt deposition results in to our knowledge the first example of a positive H2 operating potential on molecularly modified Si(111). Mott–Schottky analyses reveal that the flat-band potential of the stable Ph(OMe)2 surface approaches that of the native (but unstable) hydride-terminated surface. The series resistance is diminished by the methoxy functional groups on the phenyl unit, due to its chemical and electronic connectivity with the TiO2 layer. Overall, judicious choice of the R group in conjunction with TiO2|Pt effects H2 generation on p-Si(111) photoelectrodes (Voc = 207 ± 5.2 mV; Jsc = −21.7 mA/cm2; ff = 0.22; ηH2 = 0.99%). These results provide a viable hybrid strategy toward the operation of catalysts on molecularly modified p-Si(111).
Co-reporter:Hark Jin Kim, Kara L. Kearney, Luc H. Le, Ryan T. Pekarek, and Michael J. Rose
ACS Applied Materials & Interfaces 2015 Volume 7(Issue 16) pp:8572
Publication Date(Web):April 16, 2015
DOI:10.1021/acsami.5b00376
We report the preparation, stability, and utility of Si(111)−CH3 photoelectrodes protected with thin films of aluminum oxide (Al2O3) prepared by atomic layer deposition (ALD). The photoelectrodes have been characterized by X-ray photoelectron spectroscopy (XPS), photoelectrochemistry (Fc in MeCN, Fc–OH in H2O), electrochemical impedance spectroscopy (EIS), and cyclic voltammetry (CV) simulation. XPS analysis of the growing Al2O3 layer affords both the thickness, and information regarding two-dimensional versus three-dimensional mode of growth. Impedance measurements on Si(111)|CH3|Al2O3 devices reveal that the nascent films (5–30 Å) exhibit significant capacitance, which is attenuated upon surpassing the bulk threshold (∼30 Å). The Al2O3 layer provides enhanced photoelectrochemical (PEC) stability evidenced by an increase in the anodic window of operation in MeCN (up to +0.5 V vs Ag) and enhanced stability in aqueous electrolyte (up to +0.2 V vs Ag). XPS analysis before and after PEC confirms the Al2O3 layer is persistent and prevents surface corrosion (SiOx). Sweep-rate dependent CVs in MeCN at varying thicknesses exhibit a trend of increasingly broad features, characteristic of slow electron transport kinetics. Simulations were modeled as slow electron transfer through a partially resistive and electroactive Al2O3 layer. Lastly, we find that the Al2O3 ultrathin film serves as a support for the ALD deposition of Pt nanoparticles (d ≈ 8 nm) that enhance electron transfer through the Al2O3 layer. Surface recombination velocity (SRV) measurements on the assembled Si(111)|CH3|Al2O3-15 device affords an S value of 4170 cm s–1 (τ = 4.2 μs) comparable to the bare Si(111)–CH3 surface (3950 cm s–1; τ = 4.4 μs). Overall, the results indicate that high electronic quality and low surface defect densities can be retained throughout a multistep assembly of an integrated and passivated semiconductor|thin-film|metal device.Keywords: Al2O3 passivation; atomic layer deposition; electron transport; methyl-passivated Si(111); platinum (Pt) ALD;
Co-reporter:Junhyeok Seo, Ryan T. Pekarek and Michael J. Rose
Chemical Communications 2015 vol. 51(Issue 68) pp:13264-13267
Publication Date(Web):22 May 2015
DOI:10.1039/C5CC02802G
We demonstrate the covalent attachment and catalytic function of a nickel-phosphine H2 evolution catalyst to a p-Si(111) photoelectrode. The covalently assembled semiconductor|molecular construct achieves a synergistic improvement (ΔVonset = +200 mV) as compared to a solution of [(PNP)2Ni]2+ in contact with a p-Si(111)–CH3 photoelectrode.
Co-reporter:Owen M. Williams, Alan H. Cowley and Michael J. Rose
Dalton Transactions 2015 vol. 44(Issue 29) pp:13017-13029
Publication Date(Web):13 Apr 2015
DOI:10.1039/C5DT00924C
Reported here are the syntheses and characterization of cobaloximes that feature a bis(imino)acenaphthene (BIAN) appended ligand. The X-ray crystal structures and spectroscopy (1H NMR or EPR) of the complexes within the series [Co(aqdBF2)2(MeCN)2] (1), [Co(aqdBF2)(MeCN)2]− (2) and [Co(aqdBF2)2(MeCN)2]2− (3, 3′) are reported and the 3-electron reduced complex [Co(aqdBF2)2(MeCN)2]3− (4) has been prepared in situ and characterized by 1H NMR spectroscopy. The X-ray crystal structures revealed the presence of a 6-coordinate CoII species (1), a 5-coordinate CoI species (2), and a 4-coordinate complex (3, 3′). In the case of complex 3, evidence from single crystal EPR spectroscopy (g‖ = 2.017, g⊥ = 1.987; <10 G linewidths) in conjunction with DFT calculations indicate that the EPR signal originates from a delocalized ligand-based unpaired spin. The frontier orbitals obtained from DFT calculations on 1, 2, 3, & 4 support the electronic assignments that were observed spectroscopically. The cathodic cyclic voltammogram (CV) of the solvato congener in DMF solution, namely [Co(aqdBF2)2(DMF)2], exhibits three reversible redox events near −1.0, −1.5 and −2.0 V vs. Fc/Fc+. Catalytic proton reduction was observed by CV near the third redox peak. Compared with other cobaloximes (Ecat = −1.0 V), the delay of catalytic onset arises from the existence of a series of resonance-stabilized intermediates.
Co-reporter:Keren A. Thomas Muthiah;Gummadi Durgaprasad;Zhu-Lin Xie;Owen M. Williams;Christopher Joseph;Vincent M. Lynch
European Journal of Inorganic Chemistry 2015 Volume 2015( Issue 10) pp:1675-1691
Publication Date(Web):
DOI:10.1002/ejic.201403013
Abstract
We report the syntheses and characterization of dicarbonyliron complexes derived from tridentate, ortho-substituted Schiff base pyridine/thioether ligands (RNNS). Metalation reactions of RNNS (R = CH3, OCH3) at low temperature (–78 °C) with [Fe(CO)4(Br)2] afforded the desired complexes [(RNNS)Fe(CO)2Br]Br (2-COBr, 3-COBr). Reactions under similar conditions with more sterically demanding ligands [R = quinoline (Q), ClPh] afforded complex salts of the form [(RNNS)Fe(CO)2Br][Fe(CO)3(Br)3] (4-COFe and 5-COFe, respectively). Alternatively, the metalation of the RNNS ligands (for all R ≠ H) with [Fe(CO)4(Br)2] in Et2O at room temperature reliably affords the complex species of type [(RNNS)Fe(CO)2Br][Fe(CO)3(Br)3] (2-COFe, 3-COFe, 5-COFe). The metalation reactions of RNNS at only moderately low temperatures (–20 to 0 °C) result in the loss of CO to form the corresponding trigonal-bipyramidal iron(II) dibromide species of type [(RNNS)FeBr2] (2-Br, 4-Br, 5-Br; μeff ≈ 5.3 μB, S = 2). The IR spectrum of each dicarbonyl cation exhibits two ν(CO) stretches at ≈ 2070 and 2030 cm–1. Low-temperature 1H NMR spectroscopy measurements of 2-CO to 5-CO in CD3CN (–35 to 5 °C) revealed sharp resonances in the diamagnetic region. Under dark conditions, each dicarbonyl species is relatively stable (<10 % loss of CO, 1–2 h). However, photolysis revealed varying extents of photostability (stability rank: R = OMe > Me ≈ Q > ClPh). An examination of the structural parameters reveals that higher photostabilities correlate with shorter Fe–C(O) bond lengths, which are induced by variation of the ortho substituent of the pyridine ring. DFT calculations along the putative photolysis pathway revealed that the bulky ligand substituent (in 5-CO) destabilizes the monocarbonyl intermediate, and this is a likely explanation for its more rapid rate of CO photodissociation. Relevance to a possible “apo-active site” of mono-iron hydrogenase (pre-acyl formation) is discussed.
Co-reporter:Taylor A. Manes, Michael J. Rose
Inorganic Chemistry Communications 2015 Volume 61() pp:221-224
Publication Date(Web):November 2015
DOI:10.1016/j.inoche.2015.10.012
We report the synthesis of a novel bis-pyridine chelate derived from an anthracene scaffold. Complexation of the N2 ligand with rhenium carbonyl affords the Re tricarbonyl bromide complex [(Anth-N2)Re(CO)3Br]. In THF solution, the title complex exhibits two quasi-reversible reductions located near − 2.1 and − 2.5 V vs Fc/Fc+, which are similar to those observed in the free Anth-N2 ligand, suggesting the non-innocent behavior of the complexed anthracene scaffold. Under CO2 atmosphere, the title complex exhibits an electrocatalytic response consistent with CO2 → CO reduction, and the presence of electrocatalytically generated CO was confirmed by bulk electrolysis. These results suggest that alternate locations of redox activity (other than at the Re metal center, pyr donors, or bpy framework) can lead to interesting electrochemical behavior.
Co-reporter:Feng Li, Victoria M. Basile, and Michael J. Rose
Langmuir 2015 Volume 31(Issue 28) pp:7712-7716
Publication Date(Web):July 8, 2015
DOI:10.1021/acs.langmuir.5b02121
We report the surface growth of oligophenylene molecular wires on Si(111) substrates and their electron-transfer (ET) properties. Iterative wire growth of biphenylene was achieved via Pd-catalyzed Negishi reactions for lengths of nphenyl = 1, 2, 4, 6, 8, and 12 (d ≈ 5–50 Å). The triflato-capped wires were functionalized with vinylferrocene for potentiometric studies. For the oligophenylenes of nphenyl = 1, 2, and 4 (wire length d ≈ 5–20 Å), there was a strong distance dependence (kapp = 22.6, 16.0, 8.40 s–1, respectively), correlated to β = 0.07 Å–1. In contrast, longer oligophenylenes for nphenyl = 4–12 (d ≈ 20–50 Å) displayed a negligible distance dependence with an ET rate of kapp ≈ 10.0 ± 1.6 s–1. These data suggest a distance-dependent tunneling mechanism at short lengths (d < 20 Å) and a distance-independent ET at longer lengths (d > 20 Å).
Co-reporter:Feng Li, Victoria M. Basile, Ryan T. Pekarek, and Michael J. Rose
ACS Applied Materials & Interfaces 2014 Volume 6(Issue 22) pp:20557
Publication Date(Web):October 29, 2014
DOI:10.1021/am506244m
Surfaces with high photoelectrochemical and electronic quality can be prepared by tethering small molecules to single-crystalline Si(111) surfaces using a two-step halogenation/alkylation method (by Lewis and co-workers).1−7 We report here that the surface coverage of custom-synthesized, phenyl-based molecular linkers can be controlled by varying the steric size of R-groups (R = CH3, C6H11, 2-ethylhexyl) at the periphery of the linker. Additionally, the linkers possess a para triflate group (−O2SCF3) that serves as a convenient analytical marker and as a point of covalent attachment for a redox active label. Quantitative X-ray photoelectron spectroscopy (XPS) measurements revealed that the surface coverage systematically varies according to the steric size of the linker: CH3 (6.7 ± 0.8%), CyHex (2.9 ± 1.2%), EtHex (2.1 ± 0.9%). The stability of the photoelectrochemical cyclic voltammetry (PEC-CV) behavior was dependent on an additional methylation step (with CH3MgCl) to passivate residual Si(111)–Cl bonds. Subsequently, the triflate functional group was utilized to perform Pd-catalyzed Heck coupling of vinylferrocene to the surface-attached linkers. Ferrocene surface coverages measured from cyclic voltammetry on the ferrocene-functionalized surfaces Si(111)–8a/CH3–Fc (R = CH3) and Si(111)–8c/CH3–Fc (R = 2-EtHex) are consistent with the corresponding Fe 2p XPS coverages and suggest a ∼1:1 conversion of surface triflate groups to vinyl-Fc sites. The surface defect densities of the linker/CH3 modified surfaces are dependent on the coverage and composition of the organic layer. Surface recombination velocity (SRV) measurements indicated that n-Si(111)–8a/CH3 and the ferrocene coupled n-Si(111)–8a/CH3–Fc exhibited relatively high surface carrier lifetimes (4.51 and 3.88 μs, respectively) and correspondingly low S values (3880 and 4510 cm s–1). Thus, the multistep, linker/Fc functionalized surfaces exhibit analogously low trap state densities as compared to the fully passivated n-Si(111)–CH3 surface.Keywords: covalent attachment; passivation; photoelectrochemistry; Si(111); steric spacing; surface functionalization
Co-reporter:Simone E. A. Lumsden, Gummadi Durgaprasad, Keren A. Thomas Muthiah and Michael J. Rose
Dalton Transactions 2014 vol. 43(Issue 28) pp:10725-10738
Publication Date(Web):13 May 2014
DOI:10.1039/C4DT00600C
We have investigated the coordination modes of NNS Schiff base, thioether ligands to manganese(I) carbonyls. The ligands contain ortho substituted pyridines (H, CH3, OCH3, fluorophenyl) and varying substituents (H, CH3) at the Schiff base linkage. In general, reaction of [Mn(CO)5Br] with a tridentate NNS ligand in CH2Cl2 affords species in which the thioether-S may be bound or unbound to the manganese center, depending on the steric and electronic substitution in the ligand framework; as a result, the complexes exhibit two or three carbonyl ligands, respectively. Aldehyde-derived ligand frames (R1NHNS) generally afford complexes of type [(RNNS)Mn(CO)3Br] (1CO, 2CO, 3CO; R = H, OCH3, CH3) that exhibit incomplete ligation of the chelate (S not bound) in X-ray structures. In contrast, use of the iminomethyl ligand (NMeNS) affords a complex of formula [(NMeNS)Mn(CO)2Br] (4CO), in which the mixed N/thioether-S stabilizes the {Mn(CO)2}+ fragment. In solid state IR spectra, complexes of type [(RNNS)Mn(CO)3Br] (1CO through 3CO) afford three ν(CO) in the range ∼2060–1865 cm−1; the dicarbonyl complex [(NMeNS)Mn(CO)2Br] (4CO) exhibits two carbonyl stretches in the range ∼1920–1845 cm−1. Prolonged storage of the tricarbonyl [(MeNNS)Mn(CO)3Br] (3CO) in presence of trace dioxygen affords the dibromide species [(MeNNS)Mn(Br)2] (3Br), in which the thioether S reliably binds to the Mn(II) center. Complexes 1CO–3CO exhibit simple, diamagnetic 1H NMR spectra in CD2Cl2. The S-ligated complex 4CO exhibits spectra consistent with a mixture of an S-bound (6-coordinate) and S-unbound (5-coordinate) species as represented by [(NMeNS)Mn(CO)2Br] ↔ [(NMeNS)Mn(CO)2Br]. Lastly, we obtained crystal structures of the S-bound and S-unbound conformers derived from the same ligand – the fluorophenyl derived FPhNNS, namely [(FPhNNS)Mn(CO)3Br] (5CO-a) and [(FPhNNS)Mn(CO)2Br] (5CO-b). This report represents several examples of a thioether-stabilized {Mn(CO)2}+ fragment, a deviation from the usual ‘piano stool’ Mn(I) tricarbonyl motif. We highlight that coordination of these NNS ligands to Mn(I) carbonyls occurs on a soft conformational landscape, and that ligand substituents can be rationally employed to favor the desired coordination mode.
Co-reporter:Yae In Cho ; David M. Joseph
Inorganic Chemistry 2013 Volume 52(Issue 23) pp:13298-13300
Publication Date(Web):November 15, 2013
DOI:10.1021/ic402391f
We report the synthesis and structural characterization of a dicobalt(III) complex with a μ-OH,μ-O2 core, namely μ-OH,μ-O2-[Co(enN4)]2(X)3 [1(ClO4)3 and 1(BF4)3]. The dinuclear core is cross-linked by two N4 Schiff base ligands that span each cobalt center. The formally CoIII–CoIII dimer is formed spontaneously upon exposure of the mononuclear Co(II) complex to air and exhibits a ν(O–O) value at 882 cm–1 that shifts to 833 cm–1 upon substitution with 18O2. The CV of 1(BF4)3 exhibits a reversible {CoIII–CoIII}↔{CoIII–CoIV} redox process, and we have investigated the oxidized {CoIII–CoIV} species by EPR spectroscopy (g = 2.02, 2.06; S = 1/2 signal) and DFT calculations.
Co-reporter:J. Patrick Shupp, Amber R. Rose and Michael J. Rose
Dalton Transactions 2017 - vol. 46(Issue 28) pp:NaN9171-9171
Publication Date(Web):2017/07/04
DOI:10.1039/C7DT01506B
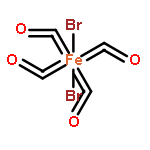
We report the synthesis, interconversions and X-ray structures of a set of [mFe–nS]-type carbonyl clusters (where S = S2−, S22− or RS−; m = 2–3; n = 1–2). All of the clusters have been identified and characterized by single crystal X-ray diffraction, IR and 13C NMR. Reduction of the parent neutral dimer [μ2-(SPh)2Fe2(CO)6] (1) with KC8 affords an easily separable ∼1:1 mixture of the anionic, dimeric thiolate dimer K[Fe2(SPh)(CO)6(μ-CO)] (2) and the dianionic, sulfido trimer [K(benzo-15-crown-5)2]2[Fe3(μ3-S)(CO)9] (3). Oxidation of 2 with diphenyl-disulfide (Ph2S2) cleanly returns the starting material 1. The Ph–S bond in 1 can be cleaved to form sulfide trimer 3. Oxidation of sulfido trimer 3 with [Fc](PF6) in the presence of S8 cleanly affords the all-inorganic persulfide dimer [μ2-(S)2Fe2(CO)6] (4), a thermodynamically stable product. The inverse reactions to form 3 (dianion) from 4 (neutral) were not successful, and other products were obtained. For example, reduction of 4 with KC8 afforded the mixed valence Fe(I)/Fe(II) species [((FeI2S2)(CO)6)2FeII]2− (5), in which the two {Fe2S2(CO)6}2− units serve as bidendate ligands to a Fe(II) center. Another isolated product (THF insoluble portion) was recrystallized in MeCN to afford [K(benzo-15-crown-5)2]2[((Fe2S)(CO)6)2(μ-S)2] (6), in which a persulfide dianion bridges two {2Fe–S} moieties (dimer of dimers). Finally, to close the interconversion loop, we converted the persulfide dimer 4 into the thiolate dimer 1 by reduction with KC8 followed by reaction with the diphenyl iodonium salt [Ph2I](PF6), in modest yield. These reactions underscore the thermodynamic stability of the dimers 1 and 4, as well as the synthetic and crystallization versatility of using the crown/K+ counterion system for obtaining structural information on highly reduced iron–sulfur–carbonyl clusters.
Co-reporter:Junhyeok Seo, Ryan T. Pekarek and Michael J. Rose
Chemical Communications 2015 - vol. 51(Issue 68) pp:NaN13267-13267
Publication Date(Web):2015/05/22
DOI:10.1039/C5CC02802G
We demonstrate the covalent attachment and catalytic function of a nickel-phosphine H2 evolution catalyst to a p-Si(111) photoelectrode. The covalently assembled semiconductor|molecular construct achieves a synergistic improvement (ΔVonset = +200 mV) as compared to a solution of [(PNP)2Ni]2+ in contact with a p-Si(111)–CH3 photoelectrode.
Co-reporter:Owen M. Williams, Alan H. Cowley and Michael J. Rose
Dalton Transactions 2015 - vol. 44(Issue 29) pp:NaN13029-13029
Publication Date(Web):2015/04/13
DOI:10.1039/C5DT00924C
Reported here are the syntheses and characterization of cobaloximes that feature a bis(imino)acenaphthene (BIAN) appended ligand. The X-ray crystal structures and spectroscopy (1H NMR or EPR) of the complexes within the series [Co(aqdBF2)2(MeCN)2] (1), [Co(aqdBF2)(MeCN)2]− (2) and [Co(aqdBF2)2(MeCN)2]2− (3, 3′) are reported and the 3-electron reduced complex [Co(aqdBF2)2(MeCN)2]3− (4) has been prepared in situ and characterized by 1H NMR spectroscopy. The X-ray crystal structures revealed the presence of a 6-coordinate CoII species (1), a 5-coordinate CoI species (2), and a 4-coordinate complex (3, 3′). In the case of complex 3, evidence from single crystal EPR spectroscopy (g‖ = 2.017, g⊥ = 1.987; <10 G linewidths) in conjunction with DFT calculations indicate that the EPR signal originates from a delocalized ligand-based unpaired spin. The frontier orbitals obtained from DFT calculations on 1, 2, 3, & 4 support the electronic assignments that were observed spectroscopically. The cathodic cyclic voltammogram (CV) of the solvato congener in DMF solution, namely [Co(aqdBF2)2(DMF)2], exhibits three reversible redox events near −1.0, −1.5 and −2.0 V vs. Fc/Fc+. Catalytic proton reduction was observed by CV near the third redox peak. Compared with other cobaloximes (Ecat = −1.0 V), the delay of catalytic onset arises from the existence of a series of resonance-stabilized intermediates.
Co-reporter:Daniel W. Redman, Hark Jin Kim, Keith J. Stevenson and Michael J. Rose
Journal of Materials Chemistry A 2016 - vol. 4(Issue 18) pp:NaN7035-7035
Publication Date(Web):2016/03/25
DOI:10.1039/C5TA09684G
This work reports on the synergistic utility of ionic liquid-based, photo-assisted electrodeposition of MoSx onto organic-functionalized silicon photolelectrodes for dihydrogen (H2) evolution under 1-sun illumination. The surface linker 3,5-dimethoxyphenyl covalently attached to Si(111) enhances conductivity at the substrate|electrolyte interface (impedance spectroscopy) and provides improved physico-chemical support for MoSx deposition (SEM, Raman, PEC-deposition, XPS), to generate a functional composite device that results in 0.33% efficiency for solar → H2 conversion. We report a new method of ionic liquid-based MoSx deposition that avoids complications in analogous aqueous procedures and facilitates the deposition of a uniform catalytic film. This work provides a generalized framework for the optimization of non-platinum catalysts for solar → H2 conversion.
Co-reporter:Simone E. A. Lumsden, Gummadi Durgaprasad, Keren A. Thomas Muthiah and Michael J. Rose
Dalton Transactions 2014 - vol. 43(Issue 28) pp:NaN10738-10738
Publication Date(Web):2014/05/13
DOI:10.1039/C4DT00600C
We have investigated the coordination modes of NNS Schiff base, thioether ligands to manganese(I) carbonyls. The ligands contain ortho substituted pyridines (H, CH3, OCH3, fluorophenyl) and varying substituents (H, CH3) at the Schiff base linkage. In general, reaction of [Mn(CO)5Br] with a tridentate NNS ligand in CH2Cl2 affords species in which the thioether-S may be bound or unbound to the manganese center, depending on the steric and electronic substitution in the ligand framework; as a result, the complexes exhibit two or three carbonyl ligands, respectively. Aldehyde-derived ligand frames (R1NHNS) generally afford complexes of type [(RNNS)Mn(CO)3Br] (1CO, 2CO, 3CO; R = H, OCH3, CH3) that exhibit incomplete ligation of the chelate (S not bound) in X-ray structures. In contrast, use of the iminomethyl ligand (NMeNS) affords a complex of formula [(NMeNS)Mn(CO)2Br] (4CO), in which the mixed N/thioether-S stabilizes the {Mn(CO)2}+ fragment. In solid state IR spectra, complexes of type [(RNNS)Mn(CO)3Br] (1CO through 3CO) afford three ν(CO) in the range ∼2060–1865 cm−1; the dicarbonyl complex [(NMeNS)Mn(CO)2Br] (4CO) exhibits two carbonyl stretches in the range ∼1920–1845 cm−1. Prolonged storage of the tricarbonyl [(MeNNS)Mn(CO)3Br] (3CO) in presence of trace dioxygen affords the dibromide species [(MeNNS)Mn(Br)2] (3Br), in which the thioether S reliably binds to the Mn(II) center. Complexes 1CO–3CO exhibit simple, diamagnetic 1H NMR spectra in CD2Cl2. The S-ligated complex 4CO exhibits spectra consistent with a mixture of an S-bound (6-coordinate) and S-unbound (5-coordinate) species as represented by [(NMeNS)Mn(CO)2Br] ↔ [(NMeNS)Mn(CO)2Br]. Lastly, we obtained crystal structures of the S-bound and S-unbound conformers derived from the same ligand – the fluorophenyl derived FPhNNS, namely [(FPhNNS)Mn(CO)3Br] (5CO-a) and [(FPhNNS)Mn(CO)2Br] (5CO-b). This report represents several examples of a thioether-stabilized {Mn(CO)2}+ fragment, a deviation from the usual ‘piano stool’ Mn(I) tricarbonyl motif. We highlight that coordination of these NNS ligands to Mn(I) carbonyls occurs on a soft conformational landscape, and that ligand substituents can be rationally employed to favor the desired coordination mode.
Co-reporter:Yae In Cho, Meredith L. Ward and Michael J. Rose
Dalton Transactions 2016 - vol. 45(Issue 34) pp:NaN13476-13476
Publication Date(Web):2016/08/05
DOI:10.1039/C6DT02104B
We report the synthesis of two fluoride bridged cobalt(II) dimers – [CoII2(μ-F)(pnN4-PhCl)2(OH2)(MeCN)](BF4)3 (1) and [CoII2(μ-F)2(pnN4-PhCl)2](BF4)2 (2) – and related complexes derived from propyl-bridged N4 Schiff base plus pyridine ligands. Notably, the bridging fluoride ion(s) emanate from B–F abstraction processes on the BF4 anions in the starting salt, [Co(H2O)6](BF4)2. Two types of bridging motifs are generated – mono-bridged (μ-F) or di-bridged (μ-F)2 – synthetically differentiated by the absence or presence of pyridine, respectively, during metalation. The synergistic roles of pyridine and the ClPhN4 ligand in promoting B–F abstraction were clarified by the isolation and crystallization of the simple tetrakis-pyridine monomeric complex [Co(py)4(MeCN)2](BF4)2 (4) [no B–F abstraction]; subsequent addition of the ClPhN4 ligand to 4 resulted in formation of the dimeric, di-bridged complex 2. Omission of pyridine during metalation resulted in formation of the mono-bridged dimer 1. The bulky chlorophenyl substituents were obligate for B–F abstraction, as metalation of the unsubstituted N4 ligand resulted in the non-fluoride-bridged dimer, [CoII2(pnN4)3](BF4)4 (3). In magnetic studies, complexes 1 (μeff = 6.24μB, 298 K) and 2 (μeff = 7.70μB, 298 K) both exhibit antiferromagnetic (AFM) coupling, but to different extents. Temperature-dependent magnetic susceptibility measurements (SQUID, 2 → 300 K) reveal that the linearity of the mono-fluoride bridge in 1 [∠Co–F–Co = 159.47(11)°] results in very strong AFM coupling (J = −14.9 cm−1). In contrast, the more acute Co2F2 diamond core [∠Co–F–Co = 98.8(2)°, 99.1(2)°] results in a smaller extent of AFM coupling (J = −2.97 cm−1). Overall, the results indicate the ‘non-innocence’ of the BF4 counterion in cobalt(II) chemistry, and dimers 1 and 2 affirm the effect of the geometry of the bridging fluoride ion(s) in determining the extent of AFM coupling.
Co-reporter:Owen M. Williams, Justin W. Shi and Michael J. Rose
Chemical Communications 2016 - vol. 52(Issue 58) pp:NaN9148-9148
Publication Date(Web):2016/05/12
DOI:10.1039/C6CC00703A
We report a photocathode device consisting of GaP, a metal oxide (Al2O3 or ZnO), a phosphonate-C12-thiol monolayer, and gold nanoparticles (AuNPs). The AuNPs enhance electron transfer: in non-aqueous electrochemistry (EtV2+ in MeCN), p-GaP|Al2O3|O3PC12S|AuNP and …ZnO|…|AuNP rescued the photocurrent (24%, 59% of Jmax-etch). Aqueous experiments (CO2 saturated KCl) using the optimized ZnO-functionalized device exhibited H+ → H2 (FY = 66%) and CO2 → CO (FY = 6%).

















![Benzenamine, 4-bromo-N,N-bis[(diphenylphosphino)methyl]-](http://img.cochemist.com/ccimg/592600/592531-36-7.png)
![Benzenamine, 4-bromo-N,N-bis[(diphenylphosphino)methyl]-](http://img.cochemist.com/ccimg/592600/592531-36-7_b.png)