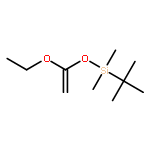
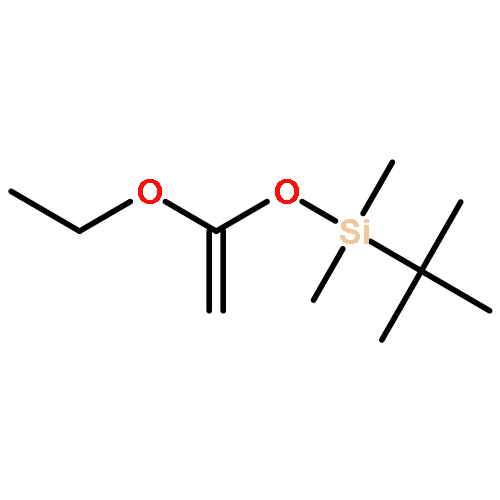
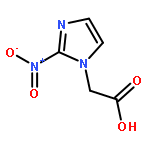
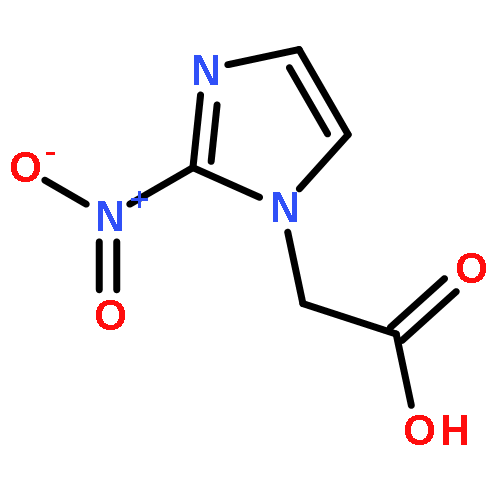
Co-reporter:Julia Kunath, Nicolas Delaroque, Michael Szardenings, Ines Neundorf, Rainer H Straub
Life Sciences 2017 Volume 168(Volume 168) pp:
Publication Date(Web):1 January 2017
DOI:10.1016/j.lfs.2016.11.009
AimsIn rheumatoid arthritis and collagen type II arthritis (CIA), sympathetic nerve fibers get lost in inflamed tissue. The process is probably induced by nerve repellent factors like semaphorin 3F (SEMA3F). Repulsion of sympathetic nerve fibers in inflamed tissue has proinflammatory effects due to the loss of anti-inflammatory neurotransmitters. We hypothesized that design molecules like antibodies and specific peptides that inhibit nerve fiber repulsion can ameliorate CIA.Materials and methodsTwo blocking antibodies were used and four blocking peptides were generated using the phage display technique with the targets of SEMA3F and plexin-A2. All blocking molecules were tested in vitro using a sympathetic neurite outgrowth assay. CIA was induced by collagen type II in mice.Key findingsIn the neurite outgrowth assay, the two antibodies against plexin-A2 and neuropilin-2 as well as the four blocking peptides – two SEMA3F analogous peptides (WLFQRDPGDR, QATVKWLFQRDPGDRR) and two plexin A2 analogous peptides (DSSDQFSFDYELEQN, DSSIQFFSFEKDKERI) - were able to block sympathetic nerve fiber repulsion in vitro (at 150–600 nmol/l). Administration of the two antibodies prophylactically on day 4 after immunization did not change clinical CIA. Similarly, using the top candidate antibody to plexin-A2 after CIA onset (mild score of 4 points, maximum = 52 points), did not ameliorate CIA. The tested blocking peptides were not recovered in peripheral blood after i.v. and i.p. administration.SignificanceWhile designer molecules blocked nerve fiber repulsion in vitro, therapeutic administration in vivo did not change CIA. Possible strategies to overcome negative effects demonstrated in vivo are discussed.
Co-reporter:M. Horn, F. Reichart, S. Natividad-Tietz, D. Diaz and I. Neundorf
Chemical Communications 2016 vol. 52(Issue 11) pp:2261-2264
Publication Date(Web):10 Dec 2015
DOI:10.1039/C5CC08938G
Cell-penetrating peptides (CPPs) present a versatile alternative to viral gene delivery vectors, addressing the still challenging task to suitably transport the desired gene to the target cell. In this work, the rational design of triazole-bridged CPPs and their detailed investigation concerning peptide/lipid interactions, using also NMR-based structure calculations, are reported.
Co-reporter:Jennifer Hochscherf, Dirk Lindenblatt, Michaela Steinkrüger, Eungyoung Yoo, Özlem Ulucan, Stefan Herzig, Olaf-Georg Issinger, Volkhard Helms, Claudia Götz, Ines Neundorf, Karsten Niefind, Markus Pietsch
Analytical Biochemistry 2015 Volume 468() pp:4-14
Publication Date(Web):1 January 2015
DOI:10.1016/j.ab.2014.09.003
Abstract
Increased activity of protein kinase CK2 is associated with various types of cancer, neurodegenerative diseases, and chronic inflammation. In the search for CK2 inhibitors, attention has expanded toward compounds disturbing the interaction between CK2α and CK2β in addition to established active site-directed approaches. The current article describes the development of a fluorescence anisotropy-based assay that mimics the principle of CK2 subunit interaction by using CK2α1–335 and the fluorescent probe CF-Ahx-Pc as a CK2β analog. In addition, we identified new inhibitors able to displace the fluorescent probe from the subunit interface on CK2α1–335. Both CF-Ahx-Pc and the inhibitors I-Pc and Cl-Pc were derived from the cyclic peptide Pc, a mimetic of the C-terminal CK2α-binding motif of CK2β. The design of the two inhibitors was based on docking studies using the known crystal structure of the Pc/CK2α1–335 complex. The dissociation constants obtained in the fluorescence anisotropy assay for binding of all compounds to human CK2α1–335 were validated by isothermal titration calorimetry. I-Pc was identified as the tightest binding ligand with a KD value of 240 nM and was shown to inhibit the CK2 holoenzyme-dependent phosphorylation of PDX-1, a substrate requiring the presence of CK2β, with an IC50 value of 92 μM.
Co-reporter:A. Reinhardt, M. Horn, J. Pieper gen. Schmauck, A. Bröhl, R. Giernoth, C. Oelkrug, A. Schubert, and I. Neundorf
Bioconjugate Chemistry 2014 Volume 25(Issue 12) pp:2166
Publication Date(Web):November 27, 2014
DOI:10.1021/bc500510c
Our study presents innovative research dealing with the synthesis and biological evaluation of conjugates out of antimicrobial peptides (AMPs) and imidazolium cations that are derived from ionic liquids. AMPs are considered as promising alternatives to common antibiotics due to their different activity mechanisms. Antibacterial effects have also been described for ionic liquids bearing imidazolium cations . Besides single coupling of carboxy-functionalized imidazolium cations to the peptide N-terminal we also developed conjugates bearing multiple copies of imidazolium cations. The combination of both compounds resulted in synergistic effects that were most pronounced when more imidazolium cations were attached to the peptides. In addition, antibacterial activity even in drug-resistant bacterial strains could be observed. Moreover, the novel compounds showed good selectivity only against bacterial cells, an observation that was further proven by lipid interaction studies using giant unilamellar vesicles.
Co-reporter:Jan Hoyer
BIOspektrum 2013 Volume 19( Issue 1) pp:33-35
Publication Date(Web):2013 February
DOI:10.1007/s12268-013-0268-2
Metallocenes have been in the focus of intense research, due to their promising activity as anti-cancer therapeutics. However, these compounds suffer from only poor bioavailability that is caused by limited water solubility and restricted uptake in living cells. We describe one concept to circumvent these drawbacks using cell-penetrating peptides as tools to modulate physical properties and biological activity of organometallic complexes.
Co-reporter:Jan Hoyer and Ines Neundorf
Accounts of Chemical Research 2012 Volume 45(Issue 7) pp:1048
Publication Date(Web):March 28, 2012
DOI:10.1021/ar2002304
Over the past two decades, gene therapy has garnered tremendous attention and is heralded by many as the ultimate cure to treat diseases such as cancer, viral infections, and inherited genetic disorders. However, the therapeutic applications of nucleic acids extend beyond the delivery of double-stranded DNA and subsequent expression of deficient gene products in diseased tissue. Other strategies include antisense oligonucleotides and most notably RNA interference (RNAi). Antisense strategies bear great potential for the treatment of diseases that are caused by misspliced mRNA, and RNAi is a universal and extraordinarily efficient tool to knock down the expression of virtually any gene by specific degradation of the desired target mRNA.However, because of the hurdles associated with effective delivery of nucleic acids across a cell membrane, the initial euphoria surrounding siRNA therapy soon subsided. The ability of oligonucleotides to cross the plasma membrane is hampered by their size and highly negative charge. Viral vectors have long been the gold standard to overcome this barrier, but they are associated with severe immunogenic effects and possible tumorigenesis. Cell-penetrating peptides (CPPs), cationic peptides that can translocate through the cell membrane independent of receptors and can transport cargo including proteins, small organic molecules, nanoparticles, and oligonucleotides, represent a promising class of nonviral delivery vectors.This Account focuses on peptide carrier systems for the cellular delivery of various types of therapeutic nucleic acids with a special emphasis on cell-penetrating peptides. We also emphasize the clinical relevance of this research through examples of promising in vivo studies. Although CPPs are often derived from naturally occurring protein transduction domains, they can also be artificially designed. Because CPPs typically include many positively charged amino acids, those electrostatic interactions facilitate the formation of complexes between the carriers and the oligonucleotides. One drawback of CPP-mediated delivery includes entrapment of the cargo in endosomes because uptake tends to be endocytic: coupling of fatty acids or endosome-disruptive peptides to the CPPs can overcome this problem. CPPs can also lack specificity for a single cell type, which can be addressed through the use of targeting moieties, such as peptide ligands that bind to specific receptors. Researchers have also applied these strategies to cationic carrier systems for nonviral oligonucleotide delivery, such as liposomes or polymers, but CPPs tend to be less cytotoxic than other delivery vehicles.
Co-reporter:Jan Hoyer, Ines Neundorf
Journal of Controlled Release 2012 Volume 161(Issue 3) pp:826-834
Publication Date(Web):10 August 2012
DOI:10.1016/j.jconrel.2012.05.017
In recent years, therapeutic applications of siRNAs have come into the focus of pharmaceutical research owing to their potential to specifically regulate gene expression. However, oligonucleotides have to overcome a series of extracellular and intracellular barriers which is why delivery systems helping to overcome these barriers are desperately needed. A promising approach to transport nucleic acids beyond cellular membranes is the use of cell-penetrating peptides (CPPs), which are able to autonomously cross the plasma membrane. Recently, we synthesized branched derivatives of truncated human calcitonin (hCT) and identified them as efficient vehicles for non-covalent gene delivery. Here we describe two novel branched hCT-derivatives that are optimized for efficient intracellular delivery of siRNA by conjugation with either a fatty acid or an endosomolytic peptide sequence. As target we chose the human NPY Y1 receptor (NPY1R), which belongs to the family of G protein-coupled receptors and thus constitutes a model for complex therapeutic targets related to various disorders. For instance, knockdown of Y1 receptor expression offers a potential therapy for osteoporosis. We present a read-out system that allows for the quantitation of the induced knockdown of receptor expression on the protein as well as on the mRNA level. As a result of this study, we could show that the herein presented cell-penetrating peptides effectively transport siRNA into HEK-293 cells without inducing cytotoxicity and that the knockdown rates are comparable to those obtained by lipofection.
Co-reporter:Jan Hoyer, Andrea Hunold, Hans-Günther Schmalz and Ines Neundorf
Dalton Transactions 2012 vol. 41(Issue 21) pp:6396-6398
Publication Date(Web):14 Mar 2012
DOI:10.1039/C2DT12211A
The conjugation of a ferrocenyl amino acid to the cell-penetrating peptide hCT(9–32) does not impair its ability to efficiently translocate into cells. Furthermore, the bioconjugate does not induce any cytotoxicity, thus presenting a potential electrochemical sensor suitable for the detection of living cells.
Co-reporter:Katrin Splith;Dr. Ralf Bergmann;Dr. Jens Pietzsch;Dr. Ines Neundorf
ChemMedChem 2012 Volume 7( Issue 1) pp:57-61
Publication Date(Web):
DOI:10.1002/cmdc.201100401
Co-reporter:Jan Hoyer, Andrea Hunold, Hans-Günther Schmalz and Ines Neundorf
Dalton Transactions 2012 - vol. 41(Issue 21) pp:NaN6398-6398
Publication Date(Web):2012/03/14
DOI:10.1039/C2DT12211A
The conjugation of a ferrocenyl amino acid to the cell-penetrating peptide hCT(9–32) does not impair its ability to efficiently translocate into cells. Furthermore, the bioconjugate does not induce any cytotoxicity, thus presenting a potential electrochemical sensor suitable for the detection of living cells.
Co-reporter:M. Horn, F. Reichart, S. Natividad-Tietz, D. Diaz and I. Neundorf
Chemical Communications 2016 - vol. 52(Issue 11) pp:NaN2264-2264
Publication Date(Web):2015/12/10
DOI:10.1039/C5CC08938G
Cell-penetrating peptides (CPPs) present a versatile alternative to viral gene delivery vectors, addressing the still challenging task to suitably transport the desired gene to the target cell. In this work, the rational design of triazole-bridged CPPs and their detailed investigation concerning peptide/lipid interactions, using also NMR-based structure calculations, are reported.





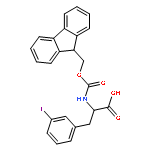
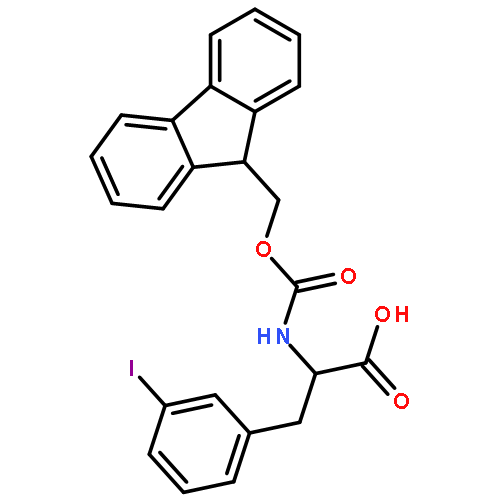
![4-Pentynoic acid, 2-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-](http://img.cochemist.com/ccimg/191300/191215-87-9.png)
![4-Pentynoic acid, 2-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-](http://img.cochemist.com/ccimg/191300/191215-87-9_b.png)
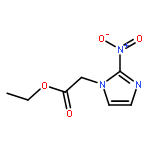
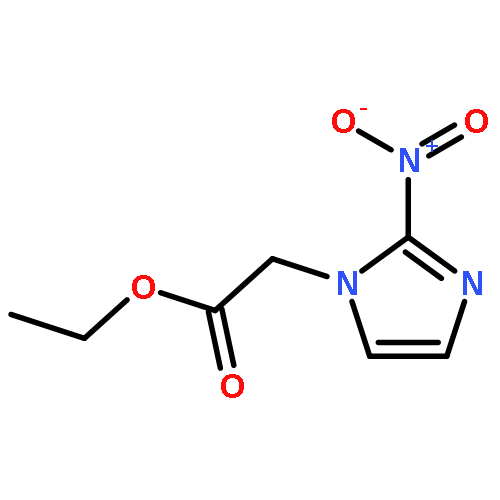
![L-Norleucine, 6-azido-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/116900/159610-89-6.png)
![L-Norleucine, 6-azido-N-[(9H-fluoren-9-ylmethoxy)carbonyl]-](/data/chemimg/116900/159610-89-6_b.png)