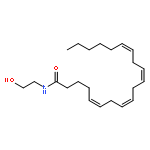
Co-reporter:Beatrice Castellani;Eleonora Diamanti;Daniela Pizzirani;Piero Tardia;Martina Maccesi;Natalia Realini;Paola Magotti;Gianpiero Garau;Thomas Bakkum;Silvia Rivara;Marco Mor
Chemical Communications 2017 vol. 53(Issue 95) pp:12814-12817
Publication Date(Web):2017/11/28
DOI:10.1039/C7CC07582K
N-Acylphosphatidylethanolamine phospholipase D (NAPE-PLD) is a membrane-associated zinc enzyme that catalyzes the hydrolysis of N-acylphosphatidylethanolamines (NAPEs) into fatty acid ethanolamides (FAEs). Here, we describe the identification of the first small-molecule NAPE-PLD inhibitor, the quinazoline sulfonamide derivative 2,4-dioxo-N-[4-(4-pyridyl)phenyl]-1H-quinazoline-6-sulfonamide, ARN19874.
Co-reporter:Rita Petracca;Elisa Romeo;Marc P. Baggelaar;Marta Artola;Silvia Pontis;Stefano Ponzano;Herman S. Overkleeft;Mario van der Stelt
Chemical Communications 2017 vol. 53(Issue 86) pp:11810-11813
Publication Date(Web):2017/10/26
DOI:10.1039/C7CC06838G
The cysteine hydrolase, N-acylethanolamine acid amidase (NAAA) is a promising target for analgesic and anti-inflammatory drugs. Here, we describe the development of two unprecedented NAAA-reactive activity-based probes as research tools for application in the discovery of new inhibitors and for the in-depth characterization of NAAA in its cellular environment.
Co-reporter:Valentina Vozella, Abdul Basit, Alessandra Misto, Daniele Piomelli
Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 2017 Volume 1862, Issue 12(Issue 12) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.bbalip.2017.08.008
•Hippocampal sphingolipids of young, middle-aged and old mice were quantified.•Age and sex-dependent changes were observed.•Changes in nervonic acid-containing sphingolipids were especially prominent.Sphingolipids have been implicated in age-related neurodegeneration. Previous studies have reported elevated ceramide levels in the brain of old rodents, but a systematic investigation of the impact of age on brain sphingolipid metabolism is still lacking. Here we quantified 17 key sphingolipid species in the hippocampus of young (3 months), middle-aged (12 months) and old (21 months) male and female mice. Lipids were extracted and quantified by liquid chromatography/mass spectrometry; transcription of enzymes involved in sphingolipid biosynthesis was evaluated by qPCR. Age-dependent changes of multiple sphingolipid species - including ceramide (d18:1/18:0), sphingomyelin (d34:1), hexosylceramide (d18:1/16:0), ceramide (d18:1/24:0) - were found in mice of both sexes. Moreover, sex-dependent changes were seen with hexosylceramide (d18:1/18:0), ceramide (d18:1/22:0), sphingomyelin (d36:1) and sphingomyelin (d42:1). Importantly, an age-dependent accumulation of sphingolipids containing nervonic acid (24:1) was observed in 21 month-old male (p = 0.04) and female mice (p < 0.0001). Consistent with this increase, transcription of the nervonic acid-synthesizing enzyme, stearoyl-CoA desaturase (Scd1 and Scd2), was upregulated in 21 month-old female mice (Scd1 p = 0.006; Scd2 p = 0.009); a similar trend was observed in males (Scd1 p = 0.07). In conclusion, the results suggest that aging is associated with profound sex-dependent and -independent changes in hippocampal sphingolipid profile. The results also highlight the need to examine the contribution of sphingolipids, and particularly of those containing nervonic acid, in normal and pathological brain aging.
Co-reporter:Don Wei, Stephen Allsop, Kay Tye, Daniele Piomelli
Trends in Neurosciences 2017 Volume 40, Issue 7(Issue 7) pp:
Publication Date(Web):1 July 2017
DOI:10.1016/j.tins.2017.04.005
Many mammalian species, including humans, exhibit social behavior and form complex social groups. Mechanistic studies in animal models have revealed important roles for the endocannabinoid signaling system, comprising G protein-coupled cannabinoid receptors and their endogenous lipid-derived agonists, in the control of neural processes that underpin social anxiety and social reward, two key aspects of social behavior. An emergent insight from these studies is that endocannabinoid signaling in specific circuits of the brain is context dependent and selectively recruited. These insights open new vistas on the neural basis of social behavior and social impairment.
Co-reporter:Miki Igarashi, Vidya Narayanaswami, Virginia Kimonis, Pietro M. Galassetti, Fariba Oveisi, Kwang-Mook Jung, Daniele Piomelli
Pharmacological Research 2017 Volume 117(Volume 117) pp:
Publication Date(Web):1 March 2017
DOI:10.1016/j.phrs.2016.12.024
Prader-Willi syndrome (PWS), the leading genetic cause of obesity, is characterized by a striking hyperphagic behavior that can lead to obesity, type-2 diabetes, cardiovascular disease and death. The molecular mechanism underlying impaired satiety in PWS is unknown. Oleoylethanolamide (OEA) is a lipid mediator involved in the control of feeding, body weight and energy metabolism. OEA produced by small-intestinal enterocytes during dietary fat digestion activates type-α peroxisome proliferator-activated receptors (PPAR-α) to trigger an afferent signal that causes satiety. Emerging evidence from genetic and human laboratory studies suggests that deficits in OEA-mediated signaling might be implicated in human obesity. In the present study, we investigated whether OEA contributes to feeding dysregulation in Magel2m+/p− (Magel2 KO) mice, an animal model of PWS. Fasted/refed male Magel2 KO mice eat more than do their wild-type littermates and become overweight with age. Meal pattern analyses show that hyperphagia in Magel2 KO is due to increased meal size and meal duration rather than to lengthening of the intermeal interval, which is suggestive of a defect in mechanisms underlying satiation. Food-dependent OEA accumulation in jejunum and fasting OEA levels in plasma are significantly greater in Magel2 KO mice than in wild-type controls. Together, these findings indicate that deletion of the Magel2 gene is accompanied by marked changes in OEA signaling. Importantly, intraperitoneal administration of OEA (10 mg/kg) significantly reduces food intake in fasted/refed Magel2 KO mice, pointing to a possible use of this natural compound to control hunger in PWS.Download high-res image (93KB)Download full-size image
Co-reporter:James Lim, Miki Igarashi, Kwang-Mook Jung, Stefania Butini, Giuseppe Campiani and Daniele Piomelli
Neuropsychopharmacology 2016 41(5) pp:1329-1339
Publication Date(Web):September 30, 2015
DOI:10.1038/npp.2015.284
Individuals who experience life-threatening psychological trauma are at risk of developing a series of chronic neuropsychiatric pathologies that include generalized anxiety, depression, and drug addiction. The endocannabinoid system has been implicated in the modulation of these responses by regulating the activity of the amygdala and the hypothalamic–pituitary–adrenal axis. However, the relevance of this signaling complex to the long-term consequences of traumatic events is unclear. Here we use an animal model of predatory stress-induced anxiety-like behavior to investigate the role of the endocannabinoid system in the development of persistent anxiety states. Our main finding is that rats exposed to the fox pheromone 2,5-dihydro-2,4,5-trimethylthiazoline (TMT), a life-threatening stimulus for rodents, display a marked and selective increase in the mobilization of the endocannabinoid, 2-arachidonoyl-sn-glycerol (2-AG), in the amygdala. This effect lasts for at least 14 days after the stress has occurred. In addition, systemic or local pharmacological inhibition of monoacylglycerol lipase (MGL)—a lipid hydrolase that degrades 2-AG in presynaptic nerve terminals—elevates 2-AG levels and suppresses the anxiety-like behavior elicited by exposure to TMT. The results suggest that predator threat triggers long-term changes in 2-AG-mediated endocannabinoid signaling in the amygdala, and that pharmacological interventions targeting MGL might provide a therapeutic strategy for the treatment of chronic brain disorders initiated by trauma.
Co-reporter:Marco Migliore, Damien Habrant, Oscar Sasso, Clara Albani, Sine Mandrup Bertozzi, Andrea Armirotti, Daniele Piomelli, Rita Scarpelli
European Journal of Medicinal Chemistry 2016 Volume 109() pp:216-237
Publication Date(Web):15 February 2016
DOI:10.1016/j.ejmech.2015.12.036
•Rational design - SAR exploration of the first class of potent multitarget inhibitors of FAAH and COX enzymes.•Focused SAR studies around 10r (ARN2508) identified novel leads 18b and 29a-c, e.•Stereochemical and pharmacological studies of 10r enantiomers are reported.•Animal studies indicate that (S)-(+)-10r strongly inhibits FAAH and COX activities in vivo.Non-steroidal anti-inflammatory drugs (NSAIDs) exert their pharmacological effects by inhibiting cyclooxygenase (COX)-1 and COX-2. Though widely prescribed for pain and inflammation, these agents have limited utility in chronic diseases due to serious mechanism-based adverse events such as gastrointestinal damage. Concomitant blockade of fatty acid amide hydrolase (FAAH) enhances the therapeutic effects of the NSAIDs while attenuating their propensity to cause gastrointestinal injury. This favorable interaction is attributed to the accumulation of protective FAAH substrates, such as the endocannabinoid anandamide, and suggests that agents simultaneously targeting COX and FAAH might provide an innovative strategy to combat pain and inflammation with reduced side effects. Here, we describe the rational design and structure-active relationship (SAR) properties of the first class of potent multitarget FAAH-COX inhibitors. A focused SAR exploration around the prototype 10r (ARN2508) led to the identification of achiral (18b) as well as racemic (29a-c and 29e) analogs. Absolute configurational assignment and pharmacological evaluation of single enantiomers of 10r are also presented. (S)-(+)-10r is the first highly potent and selective chiral inhibitor of FAAH-COX with marked in vivo activity, and represents a promising lead to discover novel analgesics and anti-inflammatory drugs.
Co-reporter:Laura Scalvini, Daniele Piomelli, Marco Mor
Chemistry and Physics of Lipids 2016 Volume 197() pp:13-24
Publication Date(Web):May 2016
DOI:10.1016/j.chemphyslip.2015.07.011
•Monoglyceride lipase (MGL) is the main responsible for degradation of 2-arachidonoylglycerol, the major endocannabinoid.•Available crystal structures of MGL are here described, with focus on the role of the flexible lid domain.•Different inhibitors of MGL have been reported. They can address the catalytic site or regulatory cysteines on the anzyme surface.Monoglyceride lipase (MGL), the main enzyme responsible for the hydrolytic deactivation of the endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG), is an intracellular serine hydrolase that plays critical roles in many physiological and pathological processes, such as pain, inflammation, neuroprotection and cancer. The crystal structures of MGL that are currently available provide valuable information about how this enzyme might function and interact with site-directed small-molecule inhibitors. On the other hand, its conformational equilibria and the contribution of regulatory cysteine residues present within the substrate-binding pocket or on protein surface remain open issues. Several classes of MGL inhibitors have been developed, from early reversible ones, such as URB602 and pristimerin, to carbamoylating agents that react with the catalytic serine, such as JZL184 and more recent O-hexafluoroisopropyl carbamates. Other inhibitors that modulate MGL activity by interacting with conserved regulatory cysteines act through mechanisms that deserve to be more thoroughly investigated.
Co-reporter:Oscar Sasso;Silvia Pontis;Andrea Armirotti;Giorgia Cardinali;Daniela Kovacs;Maria Summa;Marco Migliore;Guillermo Moreno-Sanz;Mauro Picardo
PNAS 2016 Volume 113 (Issue 30 ) pp:E4397-E4406
Publication Date(Web):2016-07-26
DOI:10.1073/pnas.1605578113
The intracellular serine amidase, fatty acid amide hydrolase (FAAH), degrades a heterogeneous family of lipid-derived bioactive
molecules that include amides of long-chain fatty acids with taurine [N-acyl-taurines (NATs)]. The physiological functions of the NATs are unknown. Here we show that genetic or pharmacological
disruption of FAAH activity accelerates skin wound healing in mice and stimulates motogenesis of human keratinocytes and differentiation
of human fibroblasts in primary cultures. Using untargeted and targeted lipidomics strategies, we identify two long-chain
saturated NATs—N-tetracosanoyl-taurine [NAT(24:0)] and N-eicosanoyl-taurine [NAT(20:0)]—as primary substrates for FAAH in mouse skin, and show that the levels of these substances
sharply decrease at the margins of a freshly inflicted wound to increase again as healing begins. Additionally, we demonstrate
that local administration of synthetic NATs accelerates wound closure in mice and stimulates repair-associated responses in
primary cultures of human keratinocytes and fibroblasts, through a mechanism that involves tyrosine phosphorylation of the
epidermal growth factor receptor and an increase in intracellular calcium levels, under the permissive control of transient
receptor potential vanilloid-1 receptors. The results point to FAAH-regulated NAT signaling as an unprecedented lipid-based
mechanism of wound-healing control in mammalian skin, which might be targeted for chronic wound therapy.
Co-reporter:Don Wei;DaYeon Lee;Dandan Li;Jennifer Daglian;Kwang-Mook Jung
Psychopharmacology 2016 Volume 233( Issue 10) pp:1911-1919
Publication Date(Web):2016 May
DOI:10.1007/s00213-016-4222-0
The endocannabinoid system is an important modulator of brain reward signaling. Investigations have focused on cannabinoid (CB1) receptors, because dissection of specific contributions of individual endocannabinoids has been limited by the available toolset. While we recently described an important role for the endocannabinoid anandamide in the regulation of social reward, it remains to be determined whether the other major endocannabinoid, 2-arachidonoyl-sn-glycerol (2-AG), serves a similar or different function.To study the role of 2-AG in natural reward, we used a transgenic mouse model (MGL-Tg mice) in which forebrain 2-AG levels are selectively reduced. We complemented behavioral analysis with measurements of brain 2-AG levels.We tested male MGL-Tg mice in conditioned place preference (CPP) tasks for high-fat food, social contact, and cocaine. We measured 2-AG content in the brain regions of interest by liquid chromatography/mass spectrometry.Male MGL-Tg mice are impaired in developing CPP for high-fat food and social interaction, but do develop CPP for cocaine. Furthermore, compared to isolated mice, levels of 2-AG in socially stimulated wild-type mice are higher in the nucleus accumbens and ventral hippocampus (183 and 140 % of controls, respectively), but unchanged in the medial prefrontal cortex.The results suggest that reducing 2-AG-mediated endocannabinoid signaling impairs social and high-fat food reward in male mice, and that social stimulation mobilizes 2-AG in key brain regions implicated in the control of motivated behavior. The time course of this response differentiates 2-AG from anandamide, whose role in mediating social reward was previously documented.
Co-reporter:Emmanuel Y. Dotsey, Kwang-Mook Jung, Abdul Basit, Don Wei, Jennifer Daglian, Federica Vacondio, Andrea Armirotti, Marco Mor, Daniele Piomelli
Chemistry & Biology 2015 Volume 22(Issue 5) pp:619-628
Publication Date(Web):21 May 2015
DOI:10.1016/j.chembiol.2015.04.013
•The second messenger hydrogen peroxide inhibits monoacylglycerol lipase (MGL)•Hydrogen peroxide sulfenylates cysteines C201 and C208 in MGL•MGL sulfenylation elevates 2-AG-mediated endocannabinoid signaling in neurons•MGL sulfenylation may serve a presynaptic control point for endocannabinoid signalingThe second messenger hydrogen peroxide transduces changes in the cellular redox state by reversibly oxidizing protein cysteine residues to sulfenic acid. This signaling event regulates many cellular processes but has never been shown to occur in the brain. Here, we report that hydrogen peroxide heightens endocannabinoid signaling in brain neurons through sulfenylation of cysteines C201 and C208 in monoacylglycerol lipase (MGL), a serine hydrolase that deactivates the endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG) in nerve terminals. The results suggest that MGL sulfenylation may provide a presynaptic control point for 2-AG-mediated endocannabinoid signaling.Figure optionsDownload full-size imageDownload high-quality image (196 K)Download as PowerPoint slide
Co-reporter:Anders Bach; Daniela Pizzirani; Natalia Realini; Valentina Vozella; Debora Russo; Ilaria Penna; Laurin Melzig; Rita Scarpelli
Journal of Medicinal Chemistry 2015 Volume 58(Issue 23) pp:9258-9272
Publication Date(Web):November 11, 2015
DOI:10.1021/acs.jmedchem.5b01188
Ceramides are lipid-derived intracellular messengers involved in the control of senescence, inflammation, and apoptosis. The cysteine amidase, acid ceramidase (AC), hydrolyzes these substances into sphingosine and fatty acid and, by doing so, regulates their signaling activity. AC inhibitors may be useful in the treatment of pathological conditions, such as cancer, in which ceramide levels are abnormally reduced. Here, we present a systematic SAR investigation of the benzoxazolone carboxamides, a recently described class of AC inhibitors that display high potency and systemic activity in mice. We examined a diverse series of substitutions on both benzoxazolone ring and carboxamide side chain. Several modifications enhanced potency and stability, and one key compound with a balanced activity–stability profile (14) was found to inhibit AC activity in mouse lungs and cerebral cortex after systemic administration. The results expand our arsenal of AC inhibitors, thereby facilitating the use of these compounds as pharmacological tools and their potential development as drug leads.
Co-reporter:Elisa Romeo, Stefano Ponzano, Andrea Armirotti, Maria Summa, Fabio Bertozzi, Gianpiero Garau, Tiziano Bandiera, and Daniele Piomelli
ACS Chemical Biology 2015 Volume 10(Issue 9) pp:2057
Publication Date(Web):June 23, 2015
DOI:10.1021/acschembio.5b00197
N-Acylethanolamine acid amidase (NAAA) is a lysosomal cysteine hydrolase involved in the degradation of saturated and monounsaturated fatty acid ethanolamides (FAEs), a family of endogenous lipid signaling molecules that includes oleoylethanolamide (OEA) and palmitoylethanolamide (PEA). Among the reported NAAA inhibitors, α-amino-β-lactone (3-aminooxetan-2-one) derivatives have been shown to prevent FAE hydrolysis in innate-immune and neural cells and to reduce reactions to inflammatory stimuli. Recently, we disclosed two potent and selective NAAA inhibitors, the compounds ARN077 (5-phenylpentyl-N-[(2S,3R)-2-methyl-4-oxo-oxetan-3-yl]carbamate) and ARN726 (4-cyclohexylbutyl-N-[(S)-2-oxoazetidin-3-yl]carbamate). The former is active in vivo by topical administration in rodent models of hyperalgesia and allodynia, while the latter exerts systemic anti-inflammatory effects in mouse models of lung inflammation. In the present study, we designed and validated a derivative of ARN726 as the first activity-based protein profiling (ABPP) probe for the in vivo detection of NAAA. The newly synthesized molecule 1 is an effective in vitro and in vivo click-chemistry activity based probe (ABP), which is able to capture the catalytically active form of NAAA in Human Embryonic Kidney 293 (HEK293) cells overexpressing human NAAA as well as in rat lung tissue. Competitive ABPP with 1 confirmed that ARN726 and ARN077 inhibit NAAA in vitro and in vivo. Compound 1 is a useful new tool to identify activated NAAA both in vitro and in vivo and to investigate the physiological and pathological roles of this enzyme.
Co-reporter:Alison Ribeiro, Silvia Pontis, Luisa Mengatto, Andrea Armirotti, Valerio Chiurchiù, Valeria Capurro, Annalisa Fiasella, Andrea Nuzzi, Elisa Romeo, Guillermo Moreno-Sanz, Mauro Maccarrone, Angelo Reggiani, Giorgio Tarzia, Marco Mor, Fabio Bertozzi, Tiziano Bandiera, and Daniele Piomelli
ACS Chemical Biology 2015 Volume 10(Issue 8) pp:1838
Publication Date(Web):April 15, 2015
DOI:10.1021/acschembio.5b00114
Fatty acid ethanolamides such as palmitoylethanolamide (PEA) and oleoylethanolamide (OEA) are lipid-derived mediators that potently inhibit pain and inflammation by ligating type-α peroxisome proliferator-activated receptors (PPAR-α). These bioactive substances are preferentially degraded by the cysteine hydrolase, N-acylethanolamine acid amidase (NAAA), which is highly expressed in macrophages. Here, we describe a new class of β-lactam derivatives that are potent, selective, and systemically active inhibitors of intracellular NAAA activity. The prototype of this class deactivates NAAA by covalently binding the enzyme’s catalytic cysteine and exerts profound anti-inflammatory effects in both mouse models and human macrophages. This agent may be used to probe the functions of NAAA in health and disease and as a starting point to discover better anti-inflammatory drugs.
Co-reporter:Dr. Daniela Pizzirani;Dr. Anders Bach;Dr. Natalia Realini;Dr. Andrea Armirotti;Dr. Luisa Mengatto;Dr. Inga Bauer;Dr. Stefania Girotto;Dr. Chiara Pagliuca;Dr. Marco DeVivo;Dr. Maria Summa;Dr. Alison Ribeiro; Daniele Piomelli
Angewandte Chemie 2015 Volume 127( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/ange.201410931
Co-reporter:Dr. Daniela Pizzirani;Dr. Anders Bach;Dr. Natalia Realini;Dr. Andrea Armirotti;Dr. Luisa Mengatto;Dr. Inga Bauer;Dr. Stefania Girotto;Dr. Chiara Pagliuca;Dr. Marco DeVivo;Dr. Maria Summa;Dr. Alison Ribeiro; Daniele Piomelli
Angewandte Chemie 2015 Volume 127( Issue 2) pp:495-499
Publication Date(Web):
DOI:10.1002/ange.201409042
Abstract
The ceramides are a family of bioactive lipid-derived messengers involved in the control of cellular senescence, inflammation, and apoptosis. Ceramide hydrolysis by acid ceramidase (AC) stops the biological activity of these substances and influences survival and function of normal and neoplastic cells. Because of its central role in the ceramide metabolism, AC may offer a novel molecular target in disorders with dysfunctional ceramide-mediated signaling. Here, a class of benzoxazolone carboxamides is identified as the first potent and systemically active inhibitors of AC. Prototype members of this class inhibit AC with low nanomolar potency by covalent binding to the catalytic cysteine. Their metabolic stability and high in vivo efficacy suggest that these compounds may be used as probes to investigate the roles of ceramide in health and disease, and that this scaffold may represent a promising starting point for the development of novel therapeutic agents.
Co-reporter:Dr. Daniela Pizzirani;Dr. Anders Bach;Dr. Natalia Realini;Dr. Andrea Armirotti;Dr. Luisa Mengatto;Dr. Inga Bauer;Dr. Stefania Girotto;Dr. Chiara Pagliuca;Dr. Marco DeVivo;Dr. Maria Summa;Dr. Alison Ribeiro; Daniele Piomelli
Angewandte Chemie International Edition 2015 Volume 54( Issue 2) pp:
Publication Date(Web):
DOI:10.1002/anie.201410931
Co-reporter:Dr. Daniela Pizzirani;Dr. Anders Bach;Dr. Natalia Realini;Dr. Andrea Armirotti;Dr. Luisa Mengatto;Dr. Inga Bauer;Dr. Stefania Girotto;Dr. Chiara Pagliuca;Dr. Marco DeVivo;Dr. Maria Summa;Dr. Alison Ribeiro; Daniele Piomelli
Angewandte Chemie International Edition 2015 Volume 54( Issue 2) pp:485-489
Publication Date(Web):
DOI:10.1002/anie.201409042
Abstract
The ceramides are a family of bioactive lipid-derived messengers involved in the control of cellular senescence, inflammation, and apoptosis. Ceramide hydrolysis by acid ceramidase (AC) stops the biological activity of these substances and influences survival and function of normal and neoplastic cells. Because of its central role in the ceramide metabolism, AC may offer a novel molecular target in disorders with dysfunctional ceramide-mediated signaling. Here, a class of benzoxazolone carboxamides is identified as the first potent and systemically active inhibitors of AC. Prototype members of this class inhibit AC with low nanomolar potency by covalent binding to the catalytic cysteine. Their metabolic stability and high in vivo efficacy suggest that these compounds may be used as probes to investigate the roles of ceramide in health and disease, and that this scaffold may represent a promising starting point for the development of novel therapeutic agents.
Co-reporter:DaYeon Lee;Carley A. Karsten;Daniel H. Geschwind;Christine M. Gall;Don Wei;Conor D. Cox;Olga Peñagarikano
PNAS 2015 Volume 112 (Issue 45 ) pp:14084-14089
Publication Date(Web):2015-11-10
DOI:10.1073/pnas.1509795112
Marijuana exerts profound effects on human social behavior, but the neural substrates underlying such effects are unknown.
Here we report that social contact increases, whereas isolation decreases, the mobilization of the endogenous marijuana-like
neurotransmitter, anandamide, in the mouse nucleus accumbens (NAc), a brain structure that regulates motivated behavior. Pharmacological
and genetic experiments show that anandamide mobilization and consequent activation of CB1 cannabinoid receptors are necessary and sufficient to express the rewarding properties of social interactions, assessed using
a socially conditioned place preference test. We further show that oxytocin, a neuropeptide that reinforces parental and social
bonding, drives anandamide mobilization in the NAc. Pharmacological blockade of oxytocin receptors stops this response, whereas
chemogenetic, site-selective activation of oxytocin neurons in the paraventricular nucleus of the hypothalamus stimulates
it. Genetic or pharmacological interruption of anandamide degradation offsets the effects of oxytocin receptor blockade on
both social place preference and cFos expression in the NAc. The results indicate that anandamide-mediated signaling at CB1 receptors, driven by oxytocin, controls social reward. Deficits in this signaling mechanism may contribute to social impairment
in autism spectrum disorders and might offer an avenue to treat these conditions.
Co-reporter:Andrea Armirotti, Abdul Basit, Natalia Realini, Carlo Caltagirone, Paola Bossù, Gianfranco Spalletta, Daniele Piomelli
Analytical Biochemistry 2014 Volume 455() pp:48-54
Publication Date(Web):15 June 2014
DOI:10.1016/j.ab.2014.03.019
Abstract
We describe a simple protocol for the preparation and orthogonal hydrophobic/hydrophilic LC–MS/MS analysis of mouse and human plasma samples, which enables the untargeted (“shotgun”) or targeted profiling of hydrophilic, amphipathic, and hydrophobic constituents of plasma metabolome. The protocol is rapid, efficient, and reliable, and offers several advantages compared to current procedures. When applied to a training set of human plasma samples, the protocol allowed for the rapid acquisition of full LogP metabolic profiles in plasma samples obtained from cognitively healthy human subjects and age-matched subjects with mild cognitive impairment or Alzheimer’s disease (n = 15 each). Targeted analyses confirmed these findings, which are consistent with data previously published by other groups.
Co-reporter:Stefano Ponzano ; Fabio Bertozzi ; Luisa Mengatto ; Mauro Dionisi ; Andrea Armirotti ; Elisa Romeo ; Anna Berteotti ; Claudio Fiorelli ; Glauco Tarozzo ; Angelo Reggiani ; Andrea Duranti ; Giorgio Tarzia ; Marco Mor ; Andrea Cavalli ; Daniele Piomelli ;Tiziano Bandiera
Journal of Medicinal Chemistry 2013 Volume 56(Issue 17) pp:6917-6934
Publication Date(Web):August 30, 2013
DOI:10.1021/jm400739u
N-Acylethanolamine acid amidase (NAAA) is a lysosomal cysteine hydrolase involved in the degradation of saturated and monounsaturated fatty acid ethanolamides (FAEs), a family of endogenous lipid agonists of peroxisome proliferator-activated receptor-α, which include oleoylethanolamide (OEA) and palmitoylethanolamide (PEA). The β-lactone derivatives (S)-N-(2-oxo-3-oxetanyl)-3-phenylpropionamide (2) and (S)-N-(2-oxo-3-oxetanyl)-biphenyl-4-carboxamide (3) inhibit NAAA, prevent FAE hydrolysis in activated inflammatory cells, and reduce tissue reactions to pro-inflammatory stimuli. Recently, our group disclosed ARN077 (4), a potent NAAA inhibitor that is active in vivo by topical administration in rodent models of hyperalgesia and allodynia. In the present study, we investigated the structure–activity relationship (SAR) of threonine-derived β-lactone analogues of compound 4. The main results of this work were an enhancement of the inhibitory potency of β-lactone carbamate derivatives for NAAA and the identification of (4-phenylphenyl)-methyl-N-[(2S,3R)-2-methyl-4-oxo-oxetan-3-yl]carbamate (14q) as the first single-digit nanomolar inhibitor of intracellular NAAA activity (IC50 = 7 nM on both rat NAAA and human NAAA).
Co-reporter:Daniela Pizzirani ; Chiara Pagliuca ; Natalia Realini ; Davide Branduardi ; Giovanni Bottegoni ; Marco Mor ; Fabio Bertozzi ; Rita Scarpelli ; Daniele Piomelli ;Tiziano Bandiera
Journal of Medicinal Chemistry 2013 Volume 56(Issue 9) pp:3518-3530
Publication Date(Web):April 24, 2013
DOI:10.1021/jm301879g
Acid ceramidase (AC) is an intracellular cysteine amidase that catalyzes the hydrolysis of the lipid messenger ceramide. By regulating ceramide levels in cells, AC may contribute to the regulation of cancer cell proliferation and senescence and to the response to cancer therapy. We recently identified the antitumoral agent carmofur (4a) as the first nanomolar inhibitor of intracellular AC activity (rat AC, IC50 = 0.029 μM). In the present work, we expanded our initial structure–activity relationship (SAR) studies around 4a by synthesizing and testing a series of 2,4-dioxopyrimidine-1-carboxamides. Our investigations provided a first elucidation of the structural features of uracil derivatives that are critical for AC inhibition and led us to identify the first single-digit nanomolar inhibitors of this enzyme. The present results confirm that substituted 2,4-dioxopyrimidine-1-carboxamides are a novel class of potent inhibitors of AC. Selected compounds of this class may represent useful probes to further characterize the functional roles of AC.
Co-reporter:F M Leweke, D Piomelli, F Pahlisch, D Muhl, C W Gerth, C Hoyer, J Klosterkötter, M Hellmich and D Koethe
Translational Psychiatry 2012 Volume 2(Mar) pp:e94
Publication Date(Web):2012-03-01
DOI:10.1038/tp.2012.15
Cannabidiol is a component of marijuana that does not activate cannabinoid receptors, but moderately inhibits the degradation of the endocannabinoid anandamide. We previously reported that an elevation of anandamide levels in cerebrospinal fluid inversely correlated to psychotic symptoms. Furthermore, enhanced anandamide signaling let to a lower transition rate from initial prodromal states into frank psychosis as well as postponed transition. In our translational approach, we performed a double-blind, randomized clinical trial of cannabidiol vs amisulpride, a potent antipsychotic, in acute schizophrenia to evaluate the clinical relevance of our initial findings. Either treatment was safe and led to significant clinical improvement, but cannabidiol displayed a markedly superior side-effect profile. Moreover, cannabidiol treatment was accompanied by a significant increase in serum anandamide levels, which was significantly associated with clinical improvement. The results suggest that inhibition of anandamide deactivation may contribute to the antipsychotic effects of cannabidiol potentially representing a completely new mechanism in the treatment of schizophrenia.
Co-reporter:G Moreno-Sanz;O Sasso;A Guijarro;O Oluyemi;R Bertorelli;A Reggiani;D Piomelli
British Journal of Pharmacology 2012 Volume 167( Issue 8) pp:1620-1628
Publication Date(Web):
DOI:10.1111/j.1476-5381.2012.02098.x
BACKGROUND AND PURPOSE
URB937 is a peripherally restricted inhibitor of the anandamide-deactivating enzyme fatty-acid amide hydrolase (FAAH). Despite its limited access to the CNS, URB937 produces marked antinociceptive effects in rodents. URB937 is actively extruded from the CNS by the ATP-binding cassette (ABC) membrane transporter, Abcg2. Tissue Abcg2 levels are markedly different between males and females, and this transporter is known to limit the access of xenobiotics to the fetoplacental unit in gestating female rodents. In the present study, we investigated the tissue distribution and antinociceptive properties of URB937 in female mice and rats.
EXPERIMENTAL APPROACH
We studied the systemic disposition of URB937 in female mice and the antinociceptive effects of this compound in models of visceral (acetic acid-induced writhing) and inflammatory nociception (carrageenan-induced hyperalgesia) in female mice and rats. Furthermore, we evaluated the interaction of URB937 with the blood-placenta barrier in gestating mice and rats.
KEY RESULTS
Abcg2 restricted the access of URB937 to the CNS of female mice and rats. Nevertheless, URB937 produced a high degree of antinociception in female mice and rats in models of visceral and inflammatory pain. Moreover, the compound displayed a restricted access to placental and fetal tissues in pregnant mice and rats.
CONCLUSIONS AND IMPLICATIONS
Peripheral FAAH blockade with URB937 reduces nociception in female mice and rats, as previously shown for males of the same species. In female mice and rats, Abcg2 limits the access of URB937, not only to the CNS, but also to the fetoplacental unit.
Co-reporter:Giuseppe Astarita, Daniele Piomelli
Journal of Chromatography B 2011 Volume 879(Issue 20) pp:1844
Publication Date(Web):15 June 2011
DOI:10.1016/j.jchromb.2011.05.001
Co-reporter:Nicholas V. DiPatrizio;Giuseppe Astarita;Gary Schwartz;Xiaosong Li;
Proceedings of the National Academy of Sciences 2011 108(31) pp:12904-12908
Publication Date(Web):July 5, 2011
DOI:10.1073/pnas.1104675108
Oral sensory signals drive dietary fat intake, but the neural mechanisms underlying this process are largely unknown. The
endocannabinoid system has gained recent attention for its central and peripheral roles in regulating food intake, energy
balance, and reward. Here, we used a sham-feeding paradigm, which isolates orosensory from postingestive influences of foods,
to examine whether endocannabinoid signaling participates in the positive feedback control of fat intake. Sham feeding a lipid-based
meal stimulated endocannabinoid mobilization in the rat proximal small intestine by altering enzymatic activities that control
endocannabinoid metabolism. This effect was abolished by surgical transection of the vagus nerve and was not observed in other
peripheral organs or in brain regions that control feeding. Sham feeding of a nutritionally complete liquid meal produced
a similar response to that of fat, whereas protein or carbohydrate alone had no such effect. Local infusion of the CB1-cannabinoid receptor antagonist, rimonabant, into the duodenum markedly reduced fat sham feeding. Similarly to rimonabant,
systemic administration of the peripherally restricted CB1-receptor antagonist, URB 447, attenuated sham feeding of lipid. Collectively, the results suggest that the endocannabinoid
system in the gut exerts a powerful regulatory control over fat intake and might be a target for antiobesity drugs.
Co-reporter:Carlos Solorzano ; Francesca Antonietti ; Andrea Duranti ; Andrea Tontini ; Silvia Rivara ; Alessio Lodola ; Federica Vacondio ; Giorgio Tarzia ; Daniele Piomelli ;Marco Mor
Journal of Medicinal Chemistry 2010 Volume 53(Issue 15) pp:5770-5781
Publication Date(Web):July 6, 2010
DOI:10.1021/jm100582w
The fatty acid ethanolamides (FAEs) are a family of bioactive lipid mediators that include the endogenous agonist of peroxisome proliferator-activated receptor-α, palmitoylethanolamide (PEA). FAEs are hydrolyzed intracellularly by either fatty acid amide hydrolase or N-acylethanolamine-hydrolyzing acid amidase (NAAA). Selective inhibition of NAAA by (S)-N-(2-oxo-3-oxetanyl)-3-phenylpropionamide [(S)-OOPP, 7a] prevents PEA degradation in mouse leukocytes and attenuates responses to proinflammatory stimuli. Starting from the structure of 7a, a series of β-lactones was prepared and tested on recombinant rat NAAA to explore structure−activity relationships (SARs) for this class of inhibitors and improve their in vitro potency. Following the hypothesis that these compounds inhibit NAAA by acylation of the catalytic cysteine, we identified several requirements for recognition at the active site and obtained new potent inhibitors. In particular, (S)-N-(2-oxo-3-oxetanyl)biphenyl-4-carboxamide (7h) was more potent than 7a at inhibiting recombinant rat NAAA activity (7a, IC50 = 420 nM; 7h, IC50 = 115 nM) in vitro and at reducing carrageenan-induced leukocyte infiltration in vivo.
Co-reporter:Alvin R. King, Emmanuel Y. Dotsey, Alessio Lodola, Kwang Mook Jung, Azar Ghomian, Yan Qiu, Jin Fu, Marco Mor, Daniele Piomelli
Chemistry & Biology 2009 Volume 16(Issue 10) pp:1045-1052
Publication Date(Web):30 October 2009
DOI:10.1016/j.chembiol.2009.09.012
Monoacylglycerol lipase (MGL) is a serine hydrolase involved in the biological deactivation of the endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG). Previous efforts to design MGL inhibitors have focused on chemical scaffolds that irreversibly block the activity of this enzyme. Here, we describe two naturally occurring terpenoids, pristimerin and euphol, which inhibit MGL activity with high potency (median effective concentration, IC50 = 93 nM and 315 nM, respectively) through a reversible mechanism. Mutational and modeling studies suggest that the two agents occupy a common hydrophobic pocket located within the putative lid domain of MGL, and each reversibly interacts with one of two adjacent cysteine residues (Cys201 and Cys208) flanking such pocket. This previously unrecognized regulatory region might offer a molecular target for potent and reversible inhibitors of MGL.
Co-reporter:AR King;A Lodola;C Carmi;J Fu;M Mor;D Piomelli
British Journal of Pharmacology 2009 Volume 157( Issue 6) pp:974-983
Publication Date(Web):
DOI:10.1111/j.1476-5381.2009.00276.x
Background and purpose: Monoacylglycerol lipase (MGL) is a presynaptic serine hydrolase that inactivates the endocannabinoid neurotransmitter, 2-arachidonoyl-sn-glycerol. Recent studies suggest that cysteine residues proximal to the enzyme active site are important for MGL function. In the present study, we characterize the role of cysteines in MGL function and identify a series of cysteine-reactive agents that inhibit MGL activity with nanomolar potencies by interacting with cysteine residue 208.
Experimental approach: A series of cysteine traps were screened for the ability to inhibit MGL in vitro. Rapid dilution assays were performed to determine reversibility of inhibition. Molecular modelling and site-directed mutagenesis were utilized to identify cysteine residues targeted by the inhibitors.
Key results: The screening revealed that 2-octyl-4-isothiazolin-3-one (octhilinone) inhibited purified rat recombinant MGL (IC50= 88 ± 12 nM) through a partially reversible mechanism. Initial structure–activity relationship studies showed that substitution of the n-octyl group of octhilinone with a more lipophilic oleoyl group increased inhibitor potency (IC50= 43 ± 8 nM), while substitution with a methyl group produced the opposite effect (IC50= 239 ± 68 nM). The inhibitory potency of octhilinone was selectively decreased by mutating cysteine 208 in MGL to glycine (IC50; wild-type, 151 ± 17 nM; C208G, 722 ± 74 nM), but not by mutation of other cysteine residues (C32, C55, C201, C208 and C242).
Conclusions and implications: The results indicated that cysteine 208 plays an important role in MGL function and identified a novel class of isothiazolinone-based MGL inhibitors with nanomolar potency in vitro.
Co-reporter:Jesse LoVerme, Andrea Duranti, Andrea Tontini, Gilberto Spadoni, Marco Mor, Silvia Rivara, Nephi Stella, Cong Xu, Giorgio Tarzia, Daniele Piomelli
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 3) pp:639-643
Publication Date(Web):1 February 2009
DOI:10.1016/j.bmcl.2008.12.059
Cannabinoid CB1 receptor antagonists reduce body weight in rodents and humans, but their clinical utility as anti-obesity agents is limited by centrally mediated side effects. Here, we describe the first mixed CB1 antagonist/CB2 agonist, URB447 ([4-amino-1-(4-chlorobenzyl)-2-methyl-5-phenyl-1H-pyrrol-3-yl](phenyl)methanone), which lowers food intake and body-weight gain in mice without entering the brain or antagonizing central CB1-dependent responses. URB447 may provide a useful pharmacological tool for investigating the cannabinoid system, and might serve as a starting point for developing clinically viable CB1 antagonists devoid of central side effects.Synthesis and in vivo pharmacology of the first mixed CB1 antagonist/CB2 agonist, URB447, which reduces food intake in rats with a peripheral mechanism, are reported.
Co-reporter:Giuseppe Astarita, Daniele Piomelli
Journal of Chromatography B 2009 Volume 877(Issue 26) pp:2755-2767
Publication Date(Web):15 September 2009
DOI:10.1016/j.jchromb.2009.01.008
The endocannabinoids are signaling lipids present in many living organisms. They activate G protein-coupled cannabinoid receptors to modulate a broad range of biological processes that include emotion, cognition, inflammation and reproduction. The endocannabinoids are embedded in an interconnected network of cellular lipid pathways, the regulation of which is likely to control the strength and duration of endocannabinoid signals. Therefore, physiopathological or pharmacological perturbations of these pathways may indirectly affect endocannabinoid activity and, vice versa, endocannabinoid activity may influence lipid pathways involved in other metabolic and signaling events. Recent progress in liquid chromatography and mass spectrometry has fueled the development of targeted lipidomic approaches, which allow researchers to examine complex lipid interactions in cells and gain a broader view of the endocannabinoid system. Here, we review these new developments from the perspective of our laboratory's experience in the field.
Co-reporter:JasonR. Clapper;Federica Vacondio Dr.;AlvinR. King;Andrea Duranti Dr.;Andrea Tontini Dr.;Claudia Silva ;Silvano Sanchini Dr.;Giorgio Tarzia ;Marco Mor
ChemMedChem 2009 Volume 4( Issue 9) pp:1505-1513
Publication Date(Web):
DOI:10.1002/cmdc.200900210
Abstract
The fatty acid ethanolamides are a class of signaling lipids that include agonists at cannabinoid and α type peroxisome proliferator-activated receptors (PPARα). In the brain, these compounds are primarily hydrolyzed by the intracellular serine enzyme fatty acid amide hydrolase (FAAH). O-aryl carbamate FAAH inhibitors such as URB597 are being evaluated clinically for the treatment of pain and anxiety, but interactions with carboxylesterases in liver might limit their usefulness. Here we explore two strategies aimed at overcoming this limitation. Lipophilic N-terminal substitutions, which enhance FAAH recognition, yield potent inhibitors but render such compounds susceptible to attack by broad-spectrum hydrolases and inactive in vivo. By contrast, polar electron-donating O-aryl substituents, which decrease carbamate reactivity, yield compounds, such as URB694, that are highly potent FAAH inhibitors in vivo and less reactive with off-target carboxylesterases. The results suggest that an approach balancing inhibitor reactivity with target recognition produces FAAH inhibitors that display significantly improved drug-likeness.
Co-reporter:Regina A. Mangieri;Kwang-Ik A. Hong;Rajita Sinha
Psychopharmacology 2009 Volume 205( Issue 1) pp:63-72
Publication Date(Web):2009 July
DOI:10.1007/s00213-009-1518-3
Alcoholics report persistent alcohol craving that is heightened by cognitive cues, stressful situations, and abstinence. The role of endogenous cannabinoids in human alcohol craving—though long suspected—remains elusive.We employed laboratory exposure to stress, alcohol cue, and neutral relaxed situations through guided imagery procedures to evoke alcohol desire and craving in healthy social drinkers (n = 11) and in treatment-engaged, recently abstinent alcoholic subjects (n = 12) and assessed alcohol craving, heart rate, and changes in circulating endocannabinoid levels. Subjective anxiety was also measured as a manipulation check for the procedures.In healthy social drinkers, alcohol cue imagery increased circulating levels of the endocannabinoid anandamide, whereas neutral and stress-related imagery had no such effect. Notably, baseline and response anandamide levels in these subjects were negatively and positively correlated with self-reported alcohol craving scores, respectively. Cue-induced increases in heart rate were also correlated with anandamide responses. By contrast, no imagery-induced anandamide mobilization was observed in alcoholics, whose baseline anandamide levels were markedly reduced compared to healthy drinkers and were uncorrelated to either alcohol craving or heart rate.The results suggest that plasma anandamide levels provide a marker of the desire for alcohol in social drinkers, which is suppressed in recently abstinent alcoholics.
Co-reporter:Carlos Solorzano;Chenggang Zhu;Natalia Battista;Giuseppe Astarita;Alessio Lodola;Silvia Rivara;Marco Mor;Roberto Russo;Mauro Maccarrone;Francesca Antonietti;Andrea Duranti;Andrea Tontini;Salvatore Cuzzocrea;Giorgio Tarzia
PNAS 2009 106 (49 ) pp:20966-20971
Publication Date(Web):2009-12-08
DOI:10.1073/pnas.0907417106
Identifying points of control in inflammation is essential to discovering safe and effective antiinflammatory medicines. Palmitoylethanolamide
(PEA) is a naturally occurring lipid amide that, when administered as a drug, inhibits inflammatory responses by engaging
peroxisome proliferator-activated receptor-α (PPAR-α). PEA is preferentially hydrolyzed by the cysteine amidase N-acylethanolamine-hydrolyzing acid amidase (NAAA), which is highly expressed in macrophages. Here we report the discovery
of a potent and selective NAAA inhibitor, N-[(3S)-2-oxo-3-oxetanyl]-3-phenylpropanamide [(S)-OOPP], and show that this inhibitor increases PEA levels in activated leukocytes and blunts responses induced by inflammatory
stimuli both in vitro and in vivo. These effects are stereoselective, mimicked by exogenous PEA, and abolished by PPAR-α deletion.
(S)-OOPP also attenuates inflammation and tissue damage and improves recovery of motor function in mice subjected to spinal
cord trauma. The results suggest that PEA activation of PPAR-α in leukocytes serves as an early stop signal that contrasts
the progress of inflammation. The PEA-hydrolyzing amidase NAAA may provide a previously undescribed target for antiinflammatory
medicines.
Co-reporter:Patrizia Campolongo;Benno Roozendaal;Viviana Trezza;Vincenzo Cuomo;Giuseppe Astarita;Jin Fu;James L. McGaugh
PNAS 2009 Volume 106 (Issue 19 ) pp:8027-8031
Publication Date(Web):2009-05-12
DOI:10.1073/pnas.0903038106
The ability to remember contexts associated with aversive and rewarding experiences provides a clear adaptive advantage to
animals foraging in the wild. The present experiments investigated whether hormonal signals released during feeding might
enhance memory of recently experienced contextual information. Oleoylethanolamide (OEA) is an endogenous lipid mediator that
is released when dietary fat enters the small intestine. OEA mediates fat-induced satiety by engaging type-α peroxisome proliferator-activated
receptors (PPAR-α) in the gut and recruiting local afferents of the vagus nerve. Here we show that post-training administration
of OEA in rats improves retention in the inhibitory avoidance and Morris water maze tasks. These effects are blocked by infusions
of lidocaine into the nucleus tractus solitarii (NTS) and by propranolol infused into the basolateral complex of the amygdala
(BLA). These findings suggest that the memory-enhancing signal generated by OEA activates the brain via afferent autonomic
fibers and stimulates noradrenergic transmission in the BLA. The actions of OEA are mimicked by PPAR-α agonists and abolished
in mutant mice lacking PPAR-α. The results indicate that OEA, acting as a PPAR-α agonist, facilitates memory consolidation
through noradrenergic activation of the BLA, a mechanism that is also critically involved in memory enhancement induced by
emotional arousal.
Co-reporter:Daniele Piomelli,
Giuseppe Astarita
&
Rao Rapaka
Nature Reviews Neuroscience 2007 8(10) pp:743
Publication Date(Web):2007-10-01
DOI:10.1038/nrn2233
Nerve cells mould the lipid fabric of their membranes to ease vesicle fusion, regulate ion fluxes and create specialized microenvironments that contribute to cellular communication. The chemical diversity of membrane lipids controls protein traffic, facilitates recognition between cells and leads to the production of hundreds of molecules that carry information both within and across cells. With so many roles, it is no wonder that lipids make up half of the human brain in dry weight. The objective of neural lipidomics is to understand how these molecules work together; this difficult task will greatly benefit from technical advances that might enable the testing of emerging hypotheses.
Co-reporter:Alvin R. King, Andrea Duranti, Andrea Tontini, Silvia Rivara, Anja Rosengarth, Jason R. Clapper, Giuseppe Astarita, Jennifer A. Geaga, Hartmut Luecke, Marco Mor, Giorgio Tarzia, Daniele Piomelli
Chemistry & Biology 2007 Volume 14(Issue 12) pp:1357-1365
Publication Date(Web):26 December 2007
DOI:10.1016/j.chembiol.2007.10.017
The N-aryl carbamate URB602 (biphenyl-3-ylcarbamic acid cyclohexyl ester) is an inhibitor of monoacylglycerol lipase (MGL), a serine hydrolase involved in the biological deactivation of the endocannabinoid 2-arachidonoyl-sn-glycerol (2-AG). Here, we investigated the mechanism by which URB602 inhibits purified recombinant rat MGL by using a combination of biochemical and structure-activity relationship (SAR) approaches. We found that URB602 weakly inhibits recombinant MGL (IC50 = 223 ± 63 μM) through a rapid and noncompetitive mechanism. Dialysis experiments and SAR analyses suggest that URB602 acts through a partially reversible mechanism rather than by irreversible carbamoylation of MGL. Finally, URB602 (100 μM) elevates 2-AG levels in hippocampal slice cultures without affecting levels of other endocannabinoid-related substances. Thus, URB602 may provide a useful tool by which to investigate the physiological roles of 2-AG and explore the potential interest of MGL as a therapeutic target.
Co-reporter:Marco Bortolato, Patrizia Campolongo, Regina Anne Mangieri, Maria Luisa Scattoni, Roberto Frau, Viviana Trezza, Giovanna La Rana, Roberto Russo, Antonio Calignano, Gian Luigi Gessa, Vincenzo Cuomo and Daniele Piomelli
Neuropsychopharmacology 2006 31(12) pp:2652-2659
Publication Date(Web):March 15, 2006
DOI:10.1038/sj.npp.1301061
The endocannabinoids anandamide and 2-arachidonoyglycerol (2-AG) may contribute to the regulation of mood and emotion. In this study, we investigated the impact of the endocannabinoid transport inhibitor AM404 on three rat models of anxiety: elevated plus maze, defensive withdrawal and separation-induced ultrasonic vocalizations. AM404 (1–5 mg kg-1, intraperitoneal (i.p.)) exerted dose-dependent anxiolytic-like effects in the three models. These behavioral effects were associated with increased levels of anandamide, but not 2-AG, in the prefrontal cortex and were prevented by the CB1 cannabinoid antagonist rimonabant (SR141716A), suggesting that they were dependent on anandamide-mediated activation of CB1 cannabinoid receptors. We also evaluated whether AM404 might influence motivation (in the conditioned place preference (CPP) test), sensory reactivity (acoustic startle reflex) and sensorimotor gating (prepulse inhibition (PPI) of the startle reflex). In the CPP test, AM404 (1.25–10 mg kg-1, i.p.) elicited rewarding effects in rats housed under enriched conditions, but not in rats kept in standard cages. Moreover, AM404 did not alter reactivity to sensory stimuli or cause overt perceptual distortion, as suggested by its lack of effect on startle or PPI of startle. These results support a role of anandamide in the regulation of emotion and point to the anandamide transport system as a potential target for anxiolytic drugs.
Co-reporter:Andrea G. Hohmann;Richard L. Suplita;Nathan M. Bolton;Mark H. Neely;Darren Fegley;Regina Mangieri;Jocelyn F. Krey;J. Michael Walker;Philip V. Holmes;Jonathon D. Crystal;Andrea Duranti;Andrea Tontini;Marco Mor;Giorgio Tarzia
Nature 2005 435(7045) pp:1108-1112
Publication Date(Web):2005-06-23
DOI:10.1038/nature03658
Acute stress suppresses pain by activating brain pathways that engage opioid or non-opioid mechanisms. Here we show that an opioid-independent form of this phenomenon, termed stress-induced analgesia1, is mediated by the release of endogenous marijuana-like (cannabinoid) compounds in the brain. Blockade of cannabinoid CB1 receptors in the periaqueductal grey matter of the midbrain prevents non-opioid stress-induced analgesia. In this region, stress elicits the rapid formation of two endogenous cannabinoids, the lipids 2-arachidonoylglycerol2 (2-AG) and anandamide3. A newly developed inhibitor of the 2-AG-deactivating enzyme, monoacylglycerol lipase4, 5, selectively increases 2-AG concentrations and, when injected into the periaqueductal grey matter, enhances stress-induced analgesia in a CB1-dependent manner. Inhibitors of the anandamide-deactivating enzyme fatty-acid amide hydrolase6, which selectively elevate anandamide concentrations, exert similar effects. Our results indicate that the coordinated release of 2-AG and anandamide in the periaqueductal grey matter might mediate opioid-independent stress-induced analgesia. These studies also identify monoacylglycerol lipase as a previously unrecognized therapeutic target.
Co-reporter:F. R. Bambico;G. Gobbi;R. Mangieri;M. Bortolato;P. Campolongo;M. Solinas;T. Cassano;M. G. Morgese;G. Debonnel;A. Duranti;A. Tontini;G. Tarzia;M. Mor;V. Trezza;V. Cuomo;S. R. Goldberg;D. Piomelli
PNAS 2005 Volume 102 (Issue 51 ) pp:18620-18625
Publication Date(Web):2005-12-20
DOI:10.1073/pnas.0509591102
Although anecdotal reports suggest that cannabis may be used to alleviate symptoms of depression, the psychotropic effects
and abuse liability of this drug prevent its therapeutic application. The active constituent of cannabis, Δ9-tetrahydrocannabinol, acts by binding to brain CB1 cannabinoid receptors, but an alternative approach might be to develop agents that amplify the actions of endogenous cannabinoids
by blocking their deactivation. Here, we show that URB597, a selective inhibitor of the enzyme fatty-acid amide hydrolase,
which catalyzes the intracellular hydrolysis of the endocannabinoid anandamide, exerts potent antidepressant-like effects
in the mouse tail-suspension test and the rat forced-swim test. Moreover, URB597 increases firing activity of serotonergic
neurons in the dorsal raphe nucleus and noradrenergic neurons in the nucleus locus ceruleus. These actions are prevented by
the CB1 antagonist rimonabant, are accompanied by increased brain anandamide levels, and are maintained upon repeated URB597 administration.
Unlike direct CB1 agonists, URB597 does not exert rewarding effects in the conditioned place preference test or produce generalization to the
discriminative effects of Δ9-tetrahydrocannabinol in rats. The findings support a role for anandamide in mood regulation and point to fatty-acid amide
hydrolase as a previously uncharacterized target for antidepressant drugs.
Co-reporter:
Nature Medicine 2004 10(1) pp:19 - 20
Publication Date(Web):
DOI:10.1038/nm0104-19
Co-reporter:Andrea Giuffrida, F Markus Leweke, Christoph W Gerth, Daniela Schreiber, Dagmar Koethe, Johannes Faulhaber, Joachim Klosterkötter and Daniele Piomelli
Neuropsychopharmacology 2004 29(11) pp:2108-2114
Publication Date(Web):September 8, 2004
DOI:10.1038/sj.npp.1300558
The endocannabinoids are a family of bioactive lipids that activate CB1 cannabinoid receptors in the brain and exert intense emotional and cognitive effects. Here, we have examined the role of endocannabinoid signaling in psychotic states by measuring levels of the endocannabinoid anandamide in cerebrospinal fluid (CSF) of acute paranoid-type schizophrenic patients. We found that CSF anandamide levels are eight-fold higher in antipsychotic-naïve first-episode paranoid schizophrenics (n=47) than healthy controls (n=84), dementia patients (n=13) or affective disorder patients (n=22). Such an alteration is absent in schizophrenics treated with 'typical' antipsychotics (n=37), which antagonize dopamine D2-like receptors, but not in those treated with 'atypical' antipsychotics (n=34), which preferentially antagonize 5HT2A receptors. Furthermore, we found that, in nonmedicated acute schizophrenics, CSF anandamide is negatively correlated with psychotic symptoms (rS=-0.452, P=0.001). The results suggest that anandamide elevation in acute paranoid schizophrenia may reflect a compensatory adaptation to the disease state.
Co-reporter:D. Fegley;R. Mercier;C. Li;S. Kathuria;A. Goutopoulos;D. Piomelli;A. Makriyannis
PNAS 2004 Volume 101 (Issue 23 ) pp:8756-8761
Publication Date(Web):2004-06-08
DOI:10.1073/pnas.0400997101
The endogenous cannabinoid anandamide is removed from the synaptic space by a high-affinity transport system present in neurons
and astrocytes, which is inhibited by N-(4-hydroxyphenyl)-arachidonamide (AM404). After internalization, anandamide is hydrolyzed by fatty-acid amide hydrolase (FAAH),
an intracellular membrane-bound enzyme that also cleaves AM404. Based on kinetic evidence, it has recently been suggested
that anandamide internalization may be mediated by passive diffusion driven by FAAH activity. To test this possibility, in
the present study, we have investigated anandamide internalization in wild-type and FAAH-deficient (FAAH–/–) mice. Cortical neurons from either mouse strain internalized [3H]anandamide through a similar mechanism, i.e., via a rapid temperature-sensitive and saturable process, which was blocked
by AM404. Moreover, systemic administration of AM404 to either wild-type or FAAH–/– mice enhanced the hypothermic effects of exogenous anandamide, a response that was prevented by the CB1 cannabinoid antagonist rimonabant (SR141716A). The results indicate that anandamide internalization in mouse brain neurons
is independent of FAAH activity. In further support of this conclusion, the compound N-(5Z, 8Z, 11Z, 14Z eicosatetraenyl)-4-hydroxybenzamide (AM1172) blocked [3H]anandamide internalization in rodent cortical neurons and human astrocytoma cells without acting as a FAAH substrate or
inhibitor. AM1172 may serve as a prototype for novel anandamide transport inhibitors with increased metabolic stability.
Co-reporter:
Nature Medicine 2003 9(1) pp:76 - 81
Publication Date(Web):02 December 2002
DOI:10.1038/nm803
Co-reporter:Silvana Gaetani, Fariba Oveisi and Daniele Piomelli
Neuropsychopharmacology 2003 28(7) pp:1311-1316
Publication Date(Web):April 2, 2003
DOI:10.1038/sj.npp.1300166
Oleoylethanolamide (OEA) is a structural analog of the endogenous cannabinoid anandamide, which does not activate cannabinoid receptors. The biosynthesis of OEA in rat small intestine is increased by feeding and reduced by fasting. Moreover, OEA decreases food intake in food-deprived rats via a mechanism that requires intact sensory fibers (Rodríguez de Fonseca, 2001). These results suggest that OEA may contribute to the peripheral regulation of feeding. In the present study, we have investigated the effects of systemic OEA administration (1–20 mg/kg, intraperitoneal) on meal pattern in free-feeding and food-deprived rats. In free-feeding animals, OEA delayed feeding onset in a dose-dependent manner, but had no effect on meal size or postmeal interval. In food-deprived animals, OEA both delayed feeding onset and reduced meal size. The selective effects of OEA in free-feeding rats are strikingly different from those of the serotonergic anorexiant d-fenfluramine (which delayed feeding and reduced meal size) and the intestinal peptide cholecystokinin (which reduced meal size). These results suggest that OEA may participate in the regulation of satiety and may provide a chemical scaffold for the design of novel appetite-suppressing medications.
Co-reporter:Jin Fu,
Silvana Gaetani,
Fariba Oveisi,
Jesse Lo Verme,
Antonia Serrano,
Fernando Rodríguez de Fonseca,
Anja Rosengarth,
Hartmut Luecke,
Barbara Di Giacomo,
Giorgio Tarzia
and
Daniele Piomelli
Nature 2003 425(6953) pp:90
Publication Date(Web):
DOI:10.1038/nature01921
Co-reporter:Thien P. Dinh, Támas F. Freund, Daniele Piomelli
Chemistry and Physics of Lipids 2002 Volume 121(1–2) pp:149-158
Publication Date(Web):31 December 2002
DOI:10.1016/S0009-3084(02)00150-0
2-Arachidonoylglycerol (2-AG) is a naturally occurring monoglyceride that activates cannabinoid receptors and meets several key requisites of an endogenous cannabinoid substance. It is present in the brain (where its levels are 170-folds higher than those of anandamide), is produced by neurons in an activity- and calcium-dependent manner, and is rapidly eliminated. The mechanism of 2-AG inactivation is not completely understood, but is thought to involve carrier-mediated transport into cells followed by enzymatic hydrolysis. We examined the possible role of the serine hydrolase, monoglyceride lipase (MGL), in brain 2-AG inactivation. We identified by homology screening a cDNA sequence encoding for a 303-amino acid protein, which conferred MGL activity upon transfection to COS-7 cells. Northern blot and in situ hybridization analyses revealed that MGL mRNA is unevenly present in the rat brain, with highest levels in regions where CB1 cannabinoid receptors are also expressed (hippocampus, cortex, anterior thalamus and cerebellum). Immunohistochemical studies in the hippocampus showed that MGL distribution has striking laminar specificity, suggesting a presynaptic localization of the enzyme. Adenovirus-mediated transfer of MGL cDNA into rat cortical neurons increased the degradation of endogenously produced 2-AG in these cells, whereas no such effect was observed on anandamide degradation. These results indicate that hydrolysis via MGL may be a primary route of 2-AG inactivation in intact neuronal cells.
Co-reporter:
Nature Medicine 2001 7(10) pp:1099 - 1100
Publication Date(Web):
DOI:10.1038/nm1001-1099
Co-reporter:F. Rodríguez de Fonseca,
M. Navarro,
R. Gómez,
L. Escuredo,
F. Nava,
J. Fu,
E. Murillo-Rodríguez,
A. Giuffrida,
J. LoVerme,
S. Gaetani,
S. Kathuria,
C. Gall
and
D. Piomelli
Nature 2001 414(6860) pp:209
Publication Date(Web):
DOI:10.1038/35102582
Oleylethanolamide (OEA) is a natural analogue of the endogenous cannabinoid anandamide. Like anandamide, OEA is produced in cells in a stimulus-dependent manner and is rapidly eliminated by enzymatic hydrolysis, suggesting a function in cellular signalling1. However, OEA does not activate cannabinoid receptors and its biological functions are still unknown2. Here we show that, in rats, food deprivation markedly reduces OEA biosynthesis in the small intestine. Administration of OEA causes a potent and persistent decrease in food intake and gain in body mass. This anorexic effect is behaviourally selective and is associated with the discrete activation of brain regions (the paraventricular hypothalamic nucleus and the nucleus of the solitary tract) involved in the control of satiety. OEA does not affect food intake when injected into the brain ventricles, and its anorexic actions are prevented when peripheral sensory fibres are removed by treatment with capsaicin. These results indicate that OEA is a lipid mediator involved in the peripheral regulation of feeding.
Co-reporter:A. Calignano,
I. Kátona,
F. Désarnaud,
A. Giuffrida,
G. La Rana,
K. Mackie,
T. F. Freund
and
D. Piomelli
Nature 2000 408(6808) pp:96
Publication Date(Web):
DOI:10.1038/35040576
Smoking marijuana or administration of its main active constituent,
9-tetrahydrocannabinol (9-THC), may exert potent
dilating effects on human airways1, 2, 3, 4. But the physiological
significance of this observation and its potential therapeutic value are obscured
by the fact that some asthmatic patients respond to these compounds with
a paradoxical bronchospasm3, 5. The mechanisms underlying these
contrasting responses remain unresolved. Here we show that the endogenous
cannabinoid anandamide exerts dual effects on bronchial responsiveness in
rodents: it strongly inhibits bronchospasm and cough evoked by the chemical
irritant, capsaicin, but causes bronchospasm when the constricting tone exerted
by the vagus nerve is removed. Both effects are mediated through peripheral
CB1 cannabinoid receptors found on axon terminals of airway nerves. Biochemical
analyses indicate that anandamide is synthesized in lung tissue on calcium-ion
stimulation, suggesting that locally generated anandamide participates in
the intrinsic control of airway responsiveness. In support of this conclusion,
the CB1 antagonist SR141716A enhances capsaicin-evoked bronchospasm and cough.
Our results may account for the contrasting bronchial actions of cannabis-like
drugs in humans, and provide a framework for the development of more selective
cannabinoid-based agents for the treatment of respiratory pathologies.
Co-reporter:Daniele Piomelli
Nature Medicine 2000 6(3) pp:255-256
Publication Date(Web):2000-03-01
DOI:10.1038/73088
Cannabinoid agonists arrest tumor progression in a rodent model
of malignant glioma. Will these molecules provide a starting point for new
strategies for anti-cancer therapy (313−319)?
Co-reporter:
Nature Neuroscience 1999 2(4) pp:358-363
Publication Date(Web):
DOI:10.1038/7268
We measured endogenous cannabinoid release in dorsal striatum of freely
moving rats by microdialysis and gas chromatography/mass spectrometry. Neural
activity stimulated the release of anandamide, but not of other endogenous
cannabinoids such as 2-arachidonylglycerol. Moreover, anandamide release was
increased eightfold over baseline after local administration of the D
2-like (D2, D3, D4) dopamine receptor
agonist quinpirole, a response that was prevented by the D2-like
receptor antagonist raclopride. Administration of the D1-like (D
1, D5) receptor agonist SKF38393 had no such effect. These
results suggest that functional interactions between endocannabinoid and dopaminergic
systems may contribute to striatal signaling. In agreement with this hypothesis,
pretreatment with the cannabinoid antagonist SR141716A enhanced the stimulation
of motor behavior elicited by systemic administration of quinpirole. The endocannabinoid
system therefore may act as an inhibitory feedback mechanism countering dopamine-induced
facilitation of motor activity.
Co-reporter:Guillermo Moreno-Sanz, Borja Barrera, Andrea Armirotti, Sine M. Bertozzi, Rita Scarpelli, Tiziano Bandiera, Julio G. Prieto, Andrea Duranti, Giorgio Tarzia, Gracia Merino, Daniele Piomelli
Pharmacological Research (September 2014) Volume 87() pp:87-93
Publication Date(Web):1 September 2014
DOI:10.1016/j.phrs.2014.06.004
The blood–brain barrier (BBB) is the main entry route for chemicals into the mammalian central nervous system (CNS). Two transmembrane transporters of the ATP-binding cassette (ABC) family – breast cancer resistance protein (ABCG2 in humans, Abcg2 in rodents) and P-glycoprotein (ABCB1 in humans, Abcb1 in rodents) – play a key role in mediating this process. Pharmacological and genetic evidence suggests that Abcg2 prevents CNS access to a group of highly potent and selective O-arylcarbamate fatty-acid amidohydrolase (FAAH) inhibitors, which include the compound URB937 (cyclohexylcarbamic acid 3′-carbamoyl-6-hydroxybiphenyl-3-yl ester). To define structure-activity relationships of the interaction of these molecules with Abcg2, in the present study we tested various peripherally restricted and non-restricted O-arylcarbamate FAAH inhibitors for their ability to serve as transport substrates in monolayer cultures of Madin-Darby Canine Kidney-II (MDCKII) cells over-expressing Abcg2. Surprisingly, we found that the majority of compounds tested – even those able to enter the CNS in vivo – were substrates for Abcg2 in vitro. Additional experiments in MDCKII cells overexpressing ABCB1 revealed that only those compounds that were dual substrates for ABCB1 and Abcg2 in vitro were also peripherally restricted in vivo. The extent of such restriction seems to depend upon other physicochemical features of the compounds, in particular the polar surface area. Consistent with these in vitro results, we found that URB937 readily enters the brain in dual knockout mice lacking both Abcg2 and Abcb1, whereas it is either partially or completely excluded from the brain of mice lacking either transporter alone. The results suggest that Abcg2 and Abcb1 act together to restrict the access of URB937 to the CNS.Download high-res image (121KB)Download full-size image
Co-reporter:Guillermo Moreno-Sanz, Borja Barrera, Ana Guijarro, Ilaria d’Elia, Jon Andoni Otero, Ana I. Alvarez, Tiziano Bandiera, Gracia Merino, Daniele Piomelli
Pharmacological Research (October 2011) Volume 64(Issue 4) pp:359-363
Publication Date(Web):1 October 2011
DOI:10.1016/j.phrs.2011.07.001
The O-arylcarbamate URB937 is a potent inhibitor of fatty-acid amide hydrolase (FAAH), an intracellular serine hydrolase responsible for the deactivation of the endocannabinoid anandamide. URB937 is unique among FAAH inhibitors in that is actively extruded from the central nervous system (CNS), and therefore increases anandamide levels exclusively in peripheral tissues. Despite its limited distribution, URB937 exhibits marked analgesic properties in rodent models of pain. Pharmacological evidence suggests that the extrusion of URB937 from the CNS may be mediated by the ABC membrane transporter ABCG2 (also called Breast cancer resistance protein, BCRP). In the present study, we show that URB937 is a substrate for both mouse and human orthologues of ABCG2. The relative transport ratios for URB937 in Madin-Darby canine kidney (MDCKII) cells monolayers over-expressing either mouse Abcg2 or human ABCG2 were significantly higher compared to parental monolayers (13.6 and 13.1 vs. 1.5, respectively). Accumulation of the compound in the luminal/apical side was prevented by co-administration of the selective ABCG2 inhibitor, Ko-143. In vivo studies in mice showed that URB937 (25 mg kg−1) readily entered the brain and spinal cord of Abcg2-deficient mice following intraperitoneal administration, whereas the same dose of drug remained restricted to peripheral tissues in wild-type mice. By identifying ABCG2 as a transport mechanism responsible for the extrusion of URB937 from the CNS, the present results should facilitate the rational design of novel peripherally restricted FAAH inhibitors.Download full-size image
Co-reporter:Oscar Sasso, Rosalia Bertorelli, Tiziano Bandiera, Rita Scarpelli, Giampiero Colombano, Andrea Armirotti, Guillermo Moreno-Sanz, Angelo Reggiani, Daniele Piomelli
Pharmacological Research (May 2012) Volume 65(Issue 5) pp:553-563
Publication Date(Web):1 May 2012
DOI:10.1016/j.phrs.2012.02.012
Fatty-acid amide hydrolase (FAAH) catalyzes the intracellular hydrolysis of the endocannabinoid anandamide and other bioactive lipid amides. In the present study, we conducted a comparative characterization of the effects of the newly identified brain-impermeant FAAH inhibitor, URB937 ([3-(3-carbamoylphenyl)-4-hydroxy-phenyl] N-cyclohexylcarbamate), in various rodent models of acute and persistent pain. When administered by the oral route in mice, URB937 was highly active (median effective dose, ED50, to inhibit liver FAAH activity: 0.3 mg kg−1) and had a bioavailability of 5.3%. The antinociceptive effects of oral URB937 were investigated in mouse models of acute inflammation (carrageenan), peripheral nerve injury (chronic sciatic nerve ligation) and arthritis (complete Freund's adjuvant). In all models, URB937 was as effective or more effective than standard analgesic and anti-inflammatory drugs (indomethacin, gabapentin, dexamethasone) and reversed pain-related responses (mechanical hyperalgesia, thermal hyperalgesia, and mechanical allodynia) in a dose-dependent manner. ED50 values ranged from 0.2 to 10 mg kg−1, depending on model and readout. Importantly, URB937 was significantly more effective than two global FAAH inhibitors, URB597 and PF-04457845, in the complete Freund's adjuvant model. The effects of a combination of URB937 with the non-steroidal anti-inflammatory agent, indomethacin, were examined in the carrageenan and chronic sciatic nerve ligation models. Isobolographic analyses showed that the two compounds interacted synergistically to attenuate pain-related behaviors. Furthermore, URB937 reduced the number and severity of gastric lesions produced by indomethacin, while exerting no ulcerogenic effect when administered alone. The results indicate that the peripheral FAAH inhibitor URB937 is more effective than globally active FAAH inhibitors at inhibiting inflammatory pain. Our findings further suggest that FAAH and cyclooxygenase inhibitors interact functionally in peripheral tissues, to either enhance or hinder each other's actions.Download high-res image (116KB)Download full-size image
Co-reporter:Oscar Sasso, Guillermo Moreno-Sanz, Cataldo Martucci, Natalia Realini, ... Daniele Piomelli
PAIN® (March 2013) Volume 154(Issue 3) pp:350-360
Publication Date(Web):1 March 2013
DOI:10.1016/j.pain.2012.10.018
Fatty acid ethanolamides (FAEs), which include palmitoylethanolamide (PEA) and oleoylethanolamide (OEA), are endogenous agonists of peroxisome proliferator-activated receptor-α (PPAR-α) and important regulators of the inflammatory response. They are degraded in macrophages by the lysosomal cysteine amidase, N-acylethanolamine acid amidase (NAAA). Previous studies have shown that pharmacological inhibition of NAAA activity suppresses macrophage activation in vitro and causes marked anti-inflammatory effects in vivo, which is suggestive of a role for NAAA in the control of inflammation. It is still unknown, however, whether NAAA-mediated FAE deactivation might regulate pain signaling. The present study examined the effects of ARN077, a potent and selective NAAA inhibitor recently disclosed by our group, in rodent models of hyperalgesia and allodynia caused by inflammation or nerve damage. Topical administration of ARN077 attenuated, in a dose-dependent manner, heat hyperalgesia and mechanical allodynia elicited in mice by carrageenan injection or sciatic nerve ligation. The antinociceptive effects of ARN077 were prevented by the selective PPAR-α antagonist GW6471 and did not occur in PPAR-α–deficient mice. Furthermore, topical ARN077 reversed the allodynia caused by ultraviolet B radiation in rats, and this effect was blocked by pretreatment with GW6471. Sciatic nerve ligation or application of the proinflammatory phorbol ester 12-O-tetradecanoylphorbol 13-acetate decreased FAE levels in sciatic nerve and skin tissue, respectively. ARN077 reversed these biochemical effects. The results identify ARN077 as a potent inhibitor of intracellular NAAA activity, which is active in vivo by topical administration. The findings further suggest that NAAA regulates peripheral pain initiation by interrupting endogenous FAE signaling at PPAR-α.
Co-reporter:Daniele Piomelli
Molecular Metabolism (July 2014) Volume 3(Issue 4) pp:345-346
Publication Date(Web):1 July 2014
DOI:10.1016/j.molmet.2014.04.005
Co-reporter:Daniele Piomelli
Trends in Endocrinology & Metabolism (July 2013) Volume 24(Issue 7) pp:332-341
Publication Date(Web):1 July 2013
DOI:10.1016/j.tem.2013.03.001
The absorptive epithelium of the proximal small intestine converts oleic acid released during fat digestion into oleoylethanolamide (OEA), an endogenous high-affinity agonist of peroxisome proliferator-activated receptor-α (PPAR-α). OEA interacts with this receptor to cause a state of satiety characterized by prolonged inter-meal intervals and reduced feeding frequency. The two main branches of the autonomic nervous system, sympathetic and parasympathetic, contribute to this effect: the former by enabling OEA mobilization in the gut and the latter by relaying the OEA signal to brain structures, such as the hypothalamus, that are involved in feeding regulation. OEA signaling may be a key component of the physiological system devoted to the monitoring of dietary fat intake, and its dysfunction might contribute to overweight and obesity.
Co-reporter:Nicholas V. DiPatrizio, Daniele Piomelli
Trends in Neurosciences (July 2012) Volume 35(Issue 7) pp:403-411
Publication Date(Web):1 July 2012
DOI:10.1016/j.tins.2012.04.006
The ‘thrifty gene hypothesis’ posits that evolution preferentially selects physiological mechanisms that optimize energy storage to increase survival under alternating conditions of abundance and scarcity of food. Recent experiments suggest that endocannabinoids – a class of lipid-derived mediators that activate cannabinoid receptors in many cells of the body – are key agents of energy conservation. The new evidence indicates that these compounds increase energy intake and decrease energy expenditure by controlling the activity of peripheral and central neural pathways involved in the sensing and hedonic processing of sweet and fatty foods, as well as in the storage of their energy content for future use.
Co-reporter:Daniele Piomelli
Neuropharmacology (January 2014) Volume 76(Part B) pp:228-234
Publication Date(Web):1 January 2014
DOI:10.1016/j.neuropharm.2013.07.026
•This article outlines several unanswered questions about the endocannabinoid system.•The enzymes involved in anandamide biosynthesis are unknown.•This knowledge gap makes it difficult to localize neurons that use anandamide as a transmitter.•The enzymes involved in 2-AG biosynthesis and degradation have been identified.•2-AG production is localized to a signaling complex, whose exact composition remain unknown.The objective of this review is to point out some important facts that we don't know about endogenous cannabinoids — lipid-derived signaling molecules that activate CB1 cannabinoid receptors and play key roles in motivation, emotion and energy balance. The first endocannabinoid substance to be discovered, anandamide, was isolated from brain tissue in 1992. Research has shown that this molecule is a bona fide brain neurotransmitter involved in the regulation of stress responses and pain, but the molecular mechanisms that govern its formation and the neural pathways in which it is employed are still unknown. There is a general consensus that enzyme-mediated cleavage, catalyzed by fatty acid amide hydrolase (FAAH), terminates the biological actions of anandamide, but there are many reasons to believe that other as-yet-unidentified proteins are also involved in this process. We have made significant headway in understanding the second arrived in the endocannabinoid family, 2-arachidonoyl-sn-glycerol (2-AG), which was discovered three years after anandamide. Researchers have established some of the key molecular players involved in 2-AG formation and deactivation, localized them to specific synaptic components, and showed that their assembly into a multi-molecular protein complex (termed the ‘2-AG signalosome’) allows 2-AG to act as a retrograde messenger at excitatory synapses of the brain. Basic questions that remain to be answered pertain to the exact molecular composition of the 2-AG signalosome, its regulation by neural activity and its potential role in the actions of drugs of abuse such as Δ9-THC and cocaine.This article is part of a Special Issue entitled ‘NIDA 40th Anniversary Issue’.
Co-reporter:Daniele Piomelli
PAIN® (January 2012) Volume 153(Issue 1) pp:3-4
Publication Date(Web):1 January 2012
DOI:10.1016/j.pain.2011.09.026
Co-reporter:Ana Guijarro, Jin Fu, Giuseppe Astarita, Daniele Piomelli
Pharmacological Research (January 2010) Volume 61(Issue 1) pp:27-33
Publication Date(Web):1 January 2010
DOI:10.1016/j.phrs.2009.09.003
Oleoylethanolamide (OEA) is an endogenous lipid mediator that decreases food intake and enhances lipid catabolism. Dietary fat stimulates OEA mobilization in the proximal small intestine, through a mechanism that requires the participation of the membrane glycoprotein CD36 (fatty acid translocase, FAT). CD36 is highly expressed in small-intestinal enterocytes and is involved in fatty acid uptake and intracellular signaling. Here, we analyze the impact of genetic CD36 deletion on OEA production in various mouse tissues under free-feeding conditions and at different times of the light/dark cycle. CD36 ablation decreases OEA levels in jejunum and plasma during the dark phase, when mice consume most of their daily food. CD36 deletion is also associated with reduced OEA levels in kidney, but not in other tissues including duodenum, stomach, adrenals, white and brown fat, heart, liver, pancreas, skeletal muscle and brain. The results underscore the important role of CD36 in jejunal OEA production linked to feeding.
Co-reporter:Tiziano Bandiera, Stefano Ponzano, Daniele Piomelli
Pharmacological Research (August 2014) Volume 86() pp:11-17
Publication Date(Web):1 August 2014
DOI:10.1016/j.phrs.2014.04.011
N-Acylethanolamine acid amidase (NAAA) is a cysteine amidase that hydrolyzes saturated or monounsaturated fatty acid ethanolamides, such as palmitoylethanolamide (PEA) and oleoylethanolamide (OEA). PEA has been shown to exert analgesic and anti-inflammatory effects by engaging peroxisome proliferator-activated receptor-α. Like other fatty acid ethanolamides, PEA is not stored in cells, but produced on demand from cell membrane precursors, and its actions are terminated by intracellular hydrolysis by either fatty acid amide hydrolase or NAAA. Endogenous levels of PEA and OEA have been shown to decrease during inflammation. Modulation of the tissue levels of PEA by inhibition of enzymes responsible for the breakdown of this lipid mediator may represent therefore a new therapeutic strategy for the treatment of pain and inflammation. While a large number of inhibitors of fatty acid amide hydrolase have been discovered, few compounds have been reported to inhibit NAAA activity. Here, we describe the most representative NAAA inhibitors and briefly highlight their pharmacological profile. A recent study has shown that a NAAA inhibitor attenuated heat hyperalgesia and mechanical allodynia caused by local inflammation or nerve damage in animal models of pain and inflammation. This finding encourages further exploration of the pharmacology of NAAA inhibitors.Download high-res image (102KB)Download full-size image
Co-reporter:Kwang-Mook Jung, Giuseppe Astarita, Sevil Yasar, Vitaly Vasilevko, David H. Cribbs, Elizabeth Head, Carl W. Cotman, Daniele Piomelli
Neurobiology of Aging (August 2012) Volume 33(Issue 8) pp:1522-1532
Publication Date(Web):1 August 2012
DOI:10.1016/j.neurobiolaging.2011.03.012
The endocannabinoids and their attending cannabinoid (CB)1 receptors have been implicated in the control of cognition, but their possible roles in dementias are still unclear. In the present study, we used liquid chromatography/mass spectrometry to conduct an endocannabinoid-targeted lipidomic analysis of postmortem brain samples from 38 Alzheimer's disease (AD) patients and 17 control subjects, matched for age and postmortem interval. The analysis revealed that midfrontal and temporal cortex tissue from AD patients contains, relative to control subjects, significantly lower levels of the endocannabinoid anandamide and its precursor 1-stearoyl, 2-docosahexaenoyl-sn-glycero-phosphoethanolamine-N-arachidonoyl (NArPE). No such difference was observed with the endocannabinoid 2-arachidonoyl-sn-glycerol or 15 additional lipid species. In AD patients, but not in control subjects, statistically detectable positive correlations were found between (1) anandamide content in midfrontal cortex and scores of the Kendrick's Digit Copy test (p = 0.004, r = 0.81; n = 10), which measures speed of information processing; and (2) anandamide content in temporal cortex and scores of the Boston Naming test (p = 0.027, r = 0.52; n = 18), which assesses language facility. Furthermore, anandamide and NArPE levels in midfrontal cortex of the study subjects inversely correlated with levels of the neurotoxic amyloid peptide, amyloid β-protein (Aβ)42, while showing no association with Aβ40 levels, amyloid plaque load or tau protein phosphorylation. Finally, high endogenous levels of Aβ42 in Swedish mutant form of amyloid precursor protein (APPSWE)/Neuro-2a cells directly reduced anandamide and NArPE concentrations in cells lysates. The results suggest that an Aβ42-dependent impairment in brain anandamide mobilization contributes to cognitive dysfunction in AD.
Co-reporter:Paola Magotti, Inga Bauer, Miki Igarashi, Masih Babagoli, ... Gianpiero Garau
Structure (3 March 2015) Volume 23(Issue 3) pp:598-604
Publication Date(Web):3 March 2015
DOI:10.1016/j.str.2014.12.018
•NAPE-PLD in complex with phosphatidylethanolamine and deoxycholate•Bile acids bind the enzyme, enhance dimer assembly, and enable catalysis•NAPE-PLD might orchestrate crosstalk between bile acids and lipid amide signalsThe fatty acid ethanolamides (FAEs) are lipid mediators present in all organisms and involved in highly conserved biological functions, such as innate immunity, energy balance, and stress control. They are produced from membrane N-acylphosphatidylethanolamines (NAPEs) and include agonists for G protein-coupled receptors (e.g., cannabinoid receptors) and nuclear receptors (e.g., PPAR-α). Here, we report the crystal structure of human NAPE-hydrolyzing phospholipase D (NAPE-PLD) at 2.65 Å resolution, a membrane enzyme that catalyzes FAE formation in mammals. NAPE-PLD forms homodimers partly separated by an internal ∼9-Å-wide channel and uniquely adapted to associate with phospholipids. A hydrophobic cavity provides an entryway for NAPE into the active site, where a binuclear Zn2+ center orchestrates its hydrolysis. Bile acids bind with high affinity to selective pockets in this cavity, enhancing dimer assembly and enabling catalysis. These elements offer multiple targets for the design of small-molecule NAPE-PLD modulators with potential applications in inflammation and metabolic disorders.Download high-res image (203KB)Download full-size image
Co-reporter:Daniele Piomelli
Biological Psychiatry (15 March 2014) Volume 75(Issue 6) pp:432-434
Publication Date(Web):15 March 2014
DOI:10.1016/j.biopsych.2014.01.010

![Butanamide,2-amino-N-[(1S,2S,4R)-7,7-dimethyl-1-[[[4-(2-methylphenyl)-1-piperazinyl]sulfonyl]methyl]bicyclo[2.2.1]hept-2-yl]-4-(methylsulfonyl)-,(2S)-](http://img.cochemist.com/ccimg/149000/148927-60-0.png)
![Butanamide,2-amino-N-[(1S,2S,4R)-7,7-dimethyl-1-[[[4-(2-methylphenyl)-1-piperazinyl]sulfonyl]methyl]bicyclo[2.2.1]hept-2-yl]-4-(methylsulfonyl)-,(2S)-](http://img.cochemist.com/ccimg/149000/148927-60-0_b.png)








![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7.png)
![5H-Dibenzo[b,e][1,4]diazepine,8-chloro-11-(4-methyl-4-oxido-1-piperazinyl)-](http://img.cochemist.com/ccimg/34300/34233-69-7_b.png)
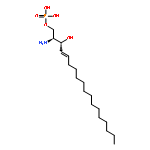
















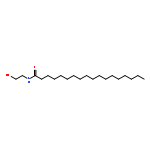

![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9.png)
![(3AR,4R,5R,6AS)-4-FORMYL-2-OXOHEXAHYDRO-2H-CYCLOPENTA[B]FURAN-5-Y<WBR />L 4-BIPHENYLCARBOXYLATE](http://img.cochemist.com/ccimg/100/72-89-9_b.png)








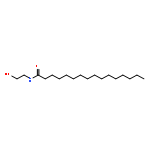



![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2.png)
![Methyl (3s,4r)-3-benzoyloxy-8-methyl-8-azabicyclo[3.2.1]octane-4-carboxylate](http://img.cochemist.com/ccimg/100/50-36-2_b.png)