Co-reporter:Christopher D. McNitt, Hazel Cheng, Susanne Ullrich, Vladimir V. Popik, and Matthew Bjerknes
Journal of the American Chemical Society October 11, 2017 Volume 139(Issue 40) pp:14029-14029
Publication Date(Web):September 19, 2017
DOI:10.1021/jacs.7b08472
Irradiation of cyclopropenone-masked dibenzocyclooctynes with near-infrared pulses from a femtosecond laser triggers photodecarbonylation via nonresonant two- or three-photon excitation. Multiphoton-generated cyclooctynes undergo a SPAAC reaction with organic azides, yielding the expected triazoles. Multiphoton-triggered SPAAC (MP-SPAAC) enables high resolution 3-D photoclick derivatization of hydrogels and tissues.
Co-reporter:Matthew Bjerknes, Hazel Cheng, Christopher D. McNitt, and Vladimir V. Popik
Bioconjugate Chemistry May 17, 2017 Volume 28(Issue 5) pp:1560-1560
Publication Date(Web):April 24, 2017
DOI:10.1021/acs.bioconjchem.7b00201
Little is known about the reactivity of strain-promoted alkyne–azide cycloaddition (SPAAC) reagents with inorganic azides. We explore the reactions of a variety of popular SPAAC reagents with sodium azide and hydrozoic acid. We find that the reactions proceed in water at rates comparable to those with organic azides, yielding in all cases a triazole adduct. The azide ion’s utility as a cyclooctyne quenching reagent is demonstrated by using it to spatially pattern uniformly doped hydrogels. The facile quenching of cyclooctynes demonstrated here should be useful in other bioorthogonal ligation techniques in which cyclooctynes are employed, including SPANC, Diels–Alder, and thiol–yne.
Co-reporter:Mariia V. Sutton;Matthew McKinley;Revathy Kulasekharan
Chemical Communications 2017 vol. 53(Issue 41) pp:5598-5601
Publication Date(Web):2017/05/18
DOI:10.1039/C7CC02056B
A new photocleavable analog of BAPTA chelating ligand has a high affinity towards Ca2+ ions (K = 2.5 × 106 M−1). The use of photolabile 3-(hydroxymethyl)-2-naphthol core in the design of photo-BAPTA allows for the efficient (Φ = 0. 63) and very fast (τ < 12 μs) release of Ca2+ ions upon 300 or 350 nm irradiation.
Co-reporter:Dewey A. Sutton, Seok-Ho Yu, Richard Steet and Vladimir V. Popik
Chemical Communications 2016 vol. 52(Issue 3) pp:553-556
Publication Date(Web):02 Nov 2015
DOI:10.1039/C5CC08106H
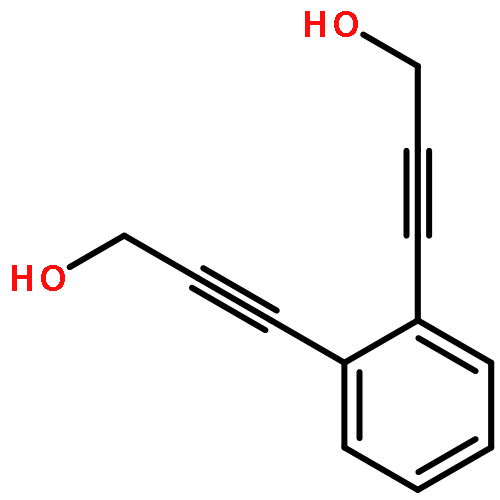
The first fully conjugated bis-cyclopropenone (photo-DIBOD), a derivative of dibenzo[a,e][8]annulene, has been synthesized. 350–420 nm irradiation of this robust compound results in the efficient formation of dibenzo [a,e] cyclooctadiyne, an unstable, but useful SPAAC cross-linking reagent. Since photo-DIBO doesn't react with organic azides, this method allows for the spatiotemporal control of the ligation of two azide-tagged substrates.
Co-reporter:Dewey A. Sutton and Vladimir V. Popik
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:8850-8857
Publication Date(Web):September 16, 2016
DOI:10.1021/acs.joc.6b01545
An order of magnitude difference in photoreactivity between bis- (photo-DIBOD, 1) and mono-cyclopropenone-caged dibenzocyclooctadiynes (MC-DIBOD, 5) allows for selective monodecarbonylation of 1. Alternatively, 5 is prepared by selective mono-cyclopropanation of dibenzo[a,e]cyclooctadiyne (DIBOD). MC-DIBOD (5) permits efficient sequential SPAAC cross-linking of azide-derivatized substrates. Cycloaddition to 5 converts an azide moiety into a photocaged form of triazole-fused dibenzo[a,e]cyclooctyne (3). While the azide reactivity of MC-DIBOD (5) and DIBOD is similar to that of other dibenzocyclooctynes, fusion of triazole to the dibenzocyclooctyne system in 3 results in a 3 orders of magnitude enhancement in SPAAC rates. In methanol, 3 reacts with butyl azide at an astonishing rate of 34 M–1 s–1, thus representing the most reactive cyclooctyne analogue reported so far. MC-DIBOD (5) was utilized in the preparation of mixed bis-triazoles and derivatization of the protein BSA with fluorescent dye and polyethylene glycol.
Co-reporter:Selvanathan Arumugam, Jun Guo, Ngalle Eric Mbua, Frédéric Friscourt, Nannan Lin, Emmanuel Nekongo, Geert-Jan Boons and Vladimir V. Popik
Chemical Science 2014 vol. 5(Issue 4) pp:1591-1598
Publication Date(Web):02 Jan 2014
DOI:10.1039/C3SC51691A
The selective derivatization of solvent-exposed cysteine residues in peptides and proteins is achieved by brief irradiation of an aqueous solution containing 3-(hydroxymethyl)-2-naphthol derivatives (NQMPs) with a 350 nm fluorescent lamp. NQMP can be conjugated with various moieties, such as PEG, dyes, carbohydrates, or possess a fragment for further selective derivatization, e.g., biotin, azide, alkyne, etc. Attractive features of this labeling approach include an exceptionally fast rate of the reaction and a requirement for a low equivalence of the reagent. The NQMP-thioether linkage is stable under ambient conditions, and survives protein digestion and MS analysis. Irradiation of an NQMP-labeled protein in a dilute solution (<40 μM) or in the presence of a vinyl ether results in a traceless release of the substrate. The reversible biotinylation of bovine serum albumin, as well as the capture and release of this protein using NeutrAvidin Agarose resin beads has been demonstrated.
Co-reporter:Selvanathan Arumugam and Vladimir V. Popik
The Journal of Organic Chemistry 2014 Volume 79(Issue 6) pp:2702-2708
Publication Date(Web):February 18, 2014
DOI:10.1021/jo500143v
Heterobifunctional linker allows for selective catalyst-free ligation of two different azide-tagged substrates via strained-promoted azide–alkyne cycloaddition (SPAAC). The linker contains an azadibenzocyclooctyne (ADIBO) moiety on one end and a cyclopropenone-masked dibenzocyclooctyne (photo-DIBO) group on the other. The first azide-derivatized substrate reacts only at the ADIBO end of the linker as the photo-DIBO moiety is azide-inert. After the completion of the first SPAAC step, photo-DIBO is activated by brief exposure to 350 nm light from a fluorescent UV lamp. The unmasked DIBO group then reacts with the second azide-tagged substrate. Both click reactions are fast (k = 0.4 and 0.07 M–1 s–1, respectively) and produce quantitative yield of ligation in organic solvents or aqueous solutions. The utility of the new cross-linker has been demonstrated by conjugation of azide functionalized bovine serum albumin (azido-BSA) with azido-fluorescein and by the immobilization of the latter protein on azide-derivatized silica beads. The BSA–bead linker was designed to incorporate hydrolytically labile fragment, which permits release of protein under the action of dilute acid. UV activation of the second click reaction permits spatiotemporal control of the ligation process.
Co-reporter:Emmanuel E. Nekongo and Vladimir V. Popik
The Journal of Organic Chemistry 2014 Volume 79(Issue 16) pp:7665-7671
Publication Date(Web):July 18, 2014
DOI:10.1021/jo501116g
The (3-hydroxy-2-naphthalenyl)methyl (NQMP) group represents an efficient photocage for fluorescein-based dyes. Thus, irradiation of the 6-NQMP ether of 2′-hydroxymethylfluorescein with low-intensity UVA light results in a 4-fold increase in emission intensity. Photoactivation of nonfluorescent NQMP-caged 3-allyloxyfluorescein produces a highly emissive fluorescein monoether. To facilitate conjugation of the caged dye to the substrate of interest via click chemistry, the allyloxy appendage was functionalized with an azide moiety.
Co-reporter:Selvanathan Arumugam
Journal of the American Chemical Society 2012 Volume 134(Issue 20) pp:8408-8411
Publication Date(Web):May 8, 2012
DOI:10.1021/ja302970x
A very facile reaction between photochemically generated o-naphthoquinone methides (oNQMs) and thiols is employed for reversible light-directed surface derivatization and patterning. A thiol-functionalized glass slide is covered with an aqueous solution of a substrate conjugated to 3-(hydroxymethyl)-2-naphthol (NQMP). Subsequent irradiation via shadow mask results in the efficient conversion of NQMP into reactive oNQM species in the exposed areas. The latter react with thiol groups on the surface, producing thioether links between the substrate and the surface. Unreacted oNQM groups are rapidly hydrated to regenerate NQMP. The short lifetime of oNQM in aqueous solution prevents its migration from the site of irradiation, thus allowing for the spatial control of the surface derivatization. A two-step procedure was employed for protein patterning: photobiotinylation of the surface with an NQMP–biotin conjugate followed by staining with FITC–avidin. The orthogonality of oNQM–thiol and azide click chemistry allowed for the development of a sequential click strategy, which might be useful for the immobilization of light-sensitive compounds. The thioether linkage produced by the reaction of oNQM and a thiol is stable under ambient conditions but can be cleaved by UV irradiation, regenerating the free thiol. This feature allows for the removal or replacement of immobilized substrates.
Co-reporter:Christopher D. McNitt and Vladimir V. Popik
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 41) pp:8200-8202
Publication Date(Web):31 Aug 2012
DOI:10.1039/C2OB26581H
Oxa-dibenzocyclooctynes (ODIBO, 2a–c) are prepared by photochemical decarbonylation of corresponding cyclopropenones (photo-ODIBO, 1a–c). While photo-ODIBO does not react with azides, ODIBO is one of the most reactive cyclooctynes exhibiting rates of cycloaddition over 45 M−1 s−1 in aqueous solutions. ODIBO is stable under ambient conditions and has low reactivity towards thiols. Photo-ODIBO survives heating up to 160 °C and does not react with thiols.
Co-reporter:Emmanuel E. Nekongo, Pritha Bagchi, Christoph J. Fahrni and Vladimir V. Popik
Organic & Biomolecular Chemistry 2012 vol. 10(Issue 46) pp:9214-9218
Publication Date(Web):12 Oct 2012
DOI:10.1039/C2OB26715B
In aqueous and alcohol solutions, colorless and non-fluorescent derivatives of 9-aryl-9H-xanthen-9-ol equilibrate with brightly colored and fluorescent 9-arylxanthylium cations, thus offering a convenient platform for the design of dual-mode indicators for emission and absorption-based pH measurements. The position of the prototropic equilibrium depends only on the hydronium ion concentration and is not affected by general acids or other ions. Furthermore, the equilibrium equivalence point can be readily adjusted by introducing substituents in the xanthenol core. As dehydroxylation of 3,6-dialkoxy-9-(o-tolyl)-9-xanthenol occurs at pH = 6.5, indicators of this type are well suited for biological applications as illustrated by in vitro cell culture studies with NIH 3T3 cells.
Co-reporter:Sara V. Orski, Gareth R. Sheppard, Selvanathan Arumugam, Rachelle M. Arnold, Vladimir V. Popik, and Jason Locklin
Langmuir 2012 Volume 28(Issue 41) pp:14693-14702
Publication Date(Web):September 25, 2012
DOI:10.1021/la3032418
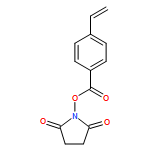
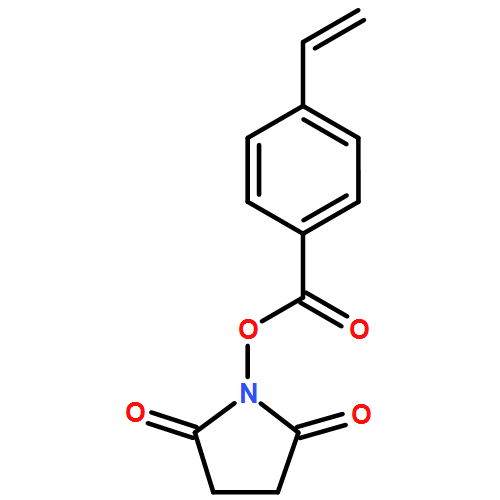
The postpolymerization functionalization of poly(N-hydroxysuccinimide 4-vinylbenzoate) brushes with reactive alkynes that differ in relative rates of activity of alkyne–azide cycloaddition reactions is described. The alkyne-derived polymer brushes undergo “click”-type cycloadditions with azido-containing compounds by two mechanisms: a strain-promoted alkyne–azide cycloaddition (SPAAC) with dibenzocyclooctyne (DIBO) and azadibenzocyclooctyne (ADIBO) or a copper-catalyzed alkyne–azide cycloaddition (CuAAC) to a propargyl group (PPG). Using a pseudo-first-order limited rate equation, rate constants for DIBO, ADIBO, and PPG-derivatized polymer brushes functionalized with an azide-functionalized dye were calculated as 7.7 × 10–4, 4.4 × 10–3, and 2.0 × 10–2 s–1, respectively. The SPAAC click reactions of the surface bound layers were determined to be slower than the equivalent reactions in solution, but the relative ratio of the reaction rates for the DIBO and ADIBO SPAAC reactions was consistent between solution and the polymer layer. The rate of functionalization was not influenced by the diffusion of azide into the polymer scaffold as long as the concentration of azide in solution was sufficiently high. The PPG functionalization by CuAAC had an extremely fast rate, which was comparable to other surface click reaction rates. Preliminary studies of dilute solution azide functionalization indicate that the diffusion-limited regime of brush functionalization impacts a 50 nm polymer brush layer and decreases the pseudo-first-order rate by a constant diffusion-limited factor of 0.233.
Co-reporter:Selvanathan Arumugam
Journal of the American Chemical Society 2011 Volume 133(Issue 39) pp:15730-15736
Publication Date(Web):August 23, 2011
DOI:10.1021/ja205652m
The utility of photochemically induced hetero-Diels–Alder reaction for the light-directed surface derivatization and patterning was demonstrated. Glass slides functionalized with vinyl ether moieties are covered with aqueous solution of substrates conjugated to 3-(hydroxymethyl)-2-naphthol (NQMP). Subsequent irradiation via shadow mask results in an efficient conversion of the latter functionality into reactive 2-napthoquinone-3-methide (oNQM) in the exposed areas. oNQM undergoes very facile hetero Diels–Alder addition (kD-A ≈ 4 × 104 M–1s–1) to immobilized vinyl ether molecules resulting in a photochemically stable covalent link between a substrate and a surface. Unreacted oNQM groups are rapidly hydrated to regenerate NQMP. The click chemistry based on the addition of photochemically generated oNQM to vinyl ether works well in aqueous solution, proceeds at high rate under ambient conditions, and does not require catalyst or additional reagents. This photoclick strategy represents an unusual paradigm in photopatterning: the surface itself is photochemically inert, while photoreactive component is present in the solution. The short lifetime (τ ≈ 7 ms in H2O) of the active form of a photoclick reagent in aqueous solution prevents its migration from the site of irradiation, thus allowing for the spatial control of surface derivatization. Both o-napthoquinone methide precursors and vinyl ethers are stable in dark and the reaction is orthogonal to other derivatization techniques, such as acetylene–azide click reaction.
Co-reporter:Selvanathan Arumugam ; Sara V. Orski ; Jason Locklin
Journal of the American Chemical Society 2011 Volume 134(Issue 1) pp:179-182
Publication Date(Web):December 14, 2011
DOI:10.1021/ja210350d
Reactive polymer brushes grown on silicon oxide surfaces were derivatized with photoreactive 3-(hydroxymethyl)naphthalene-2-ol (NQMP) moieties. Upon 300 or 350 nm irradiation, NQMP efficiently produces o-naphthoquinone methide (oNQM), which in turn undergoes very rapid Diels–Alder addition to vinyl ether groups attached to a substrate, resulting in the covalent immobilization of the latter. Any unreacted oNQM groups rapidly add water to regenerate NQMP. High-resolution surface patterning is achieved by irradiating NQMP-derivatized surfaces using photolithographic methods. The Diels–Alder photoclick reaction is orthogonal to azide–alkyne click chemistry, enabling sequential photoclick/azide-click derivatizations to generate complex surface functionalities.
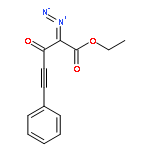
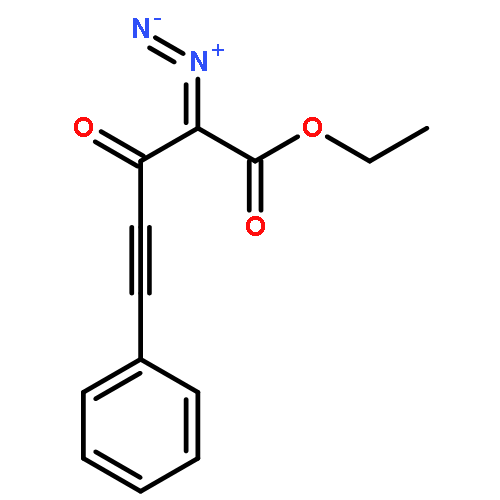
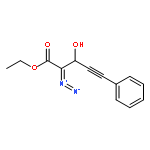
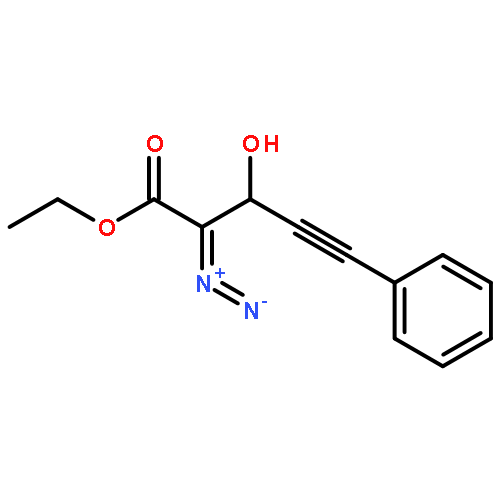
Co-reporter:Natalia G. Zhegalova
Journal of Physical Organic Chemistry 2011 Volume 24( Issue 10) pp:969-975
Publication Date(Web):
DOI:10.1002/poc.1875
Irradiation of β-phenylethynyl-α-diazo-β-ketoester with 300 or 350 nm light results in efficient and regioselective Wolff rearrangement producing only the product of alkynyl group migration. Addition of alcohols to the resulting α-oxoketene yields α-phenylethynyl-β-diester, which undergoes rapid (τ < 1 min) tautomerization to 1,1-dicarbalkoxyallene. The latter then adds second molecule of alcohol in Michael fashion to form the final product, 2-(1-alkoxy-2-phenylvinyl)malonic ester. α-Phenylethynyl-β-ketoacid produced from the ketene in aqueous solutions does not isomerize to an allene but rather undergoes decarboxylation to give β,γ-acetylenic ester. Introduction of o-(3-hydroxy-1-propynyl) fragment in the structure of the parent α-diazo-β-ketoester allowed us to achieve two goals simultaneously: ring closure by intramolecular nucleophilic attack of propargyl alcohol on photo-generated ketene and the subsequent acetylene–allene rearrangement. The resulting enyne–allene undergoes spontaneous Myers–Saito cycloaromatization generating 1,4-biradical. In alcohol solutions, however, ketene reaction with solvent outcompetes the intramolecular process. Copyright © 2011 John Wiley & Sons, Ltd.
Co-reporter:Olga V. Vinogradova, Irina A. Balova, and Vladimir V. Popik
The Journal of Organic Chemistry 2011 Volume 76(Issue 16) pp:6937-6941
Publication Date(Web):June 30, 2011
DOI:10.1021/jo201148h
A short and efficient synthesis of cinnoline-fused cyclic enediyne is reported. Richter cyclization of o-(1,3-butadiynyl)phenyltriazene produced 3-alkynyl-4-bromocinnoline. The Sonogashira coupling of the latter with 5-hexyn-1-ol was employed for the introduction of a second acetylenic moiety. The crucial cyclization step was achieved under Nozaki–Hiyama–Kishi conditions. Cinnoline-fused 10-membered ring enediyne is more reactive than corresponding carbocyclic analog and produces good yield of the Bergman cyclization product upon mild heating. This enediyne induces single-strand dDNA scissions upon incubation at 40 °C.
Co-reporter:Andrei Poloukhtine, Valentin Rassadin, Alexander Kuzmin and Vladimir V. Popik
The Journal of Organic Chemistry 2010 Volume 75(Issue 17) pp:5953-5962
Publication Date(Web):August 4, 2010
DOI:10.1021/jo101238x
Introduction of a nitrogen atom at one of the acetylenic termini of 10-, 11-, 12-, and 13-membered benzannulated cyclic enediynes results in a complete suppression of the conventional radical Bergman reaction in favor of a polar cycloaromatization. The latter reaction is catalyzed by acids and proceeds via initial protonation of an ynamide fragment. The resulting ketenimmonium cation then cyclizes to produce naphthyl cation, which rapidly reacts with nucleophiles or undergoes Friedel−Crafts addition to aromatic compounds. In alcohols, addition of the nucleophilic solvent across the activated triple bond competes with the cyclization reaction. The ratio of cyclized to solvolysis products decreases with the increase in ring size.
Co-reporter:Alexander V. Kuzmin and Vladimir V. Popik
Chemical Communications 2009 (Issue 38) pp:5707-5709
Publication Date(Web):14 Aug 2009
DOI:10.1039/B911871C
A reactive ten-membered ring enyne–allene (τ25 °C = 5–6 min) is efficiently generated (Φ300 nm = 0.57) by UV irradiation of a thermally stable precursor in which a triple bond is masked as a cyclopropenone moiety.
Co-reporter:Dewey A. Sutton, Seok-Ho Yu, Richard Steet and Vladimir V. Popik
Chemical Communications 2016 - vol. 52(Issue 3) pp:NaN556-556
Publication Date(Web):2015/11/02
DOI:10.1039/C5CC08106H
The first fully conjugated bis-cyclopropenone (photo-DIBOD), a derivative of dibenzo[a,e][8]annulene, has been synthesized. 350–420 nm irradiation of this robust compound results in the efficient formation of dibenzo [a,e] cyclooctadiyne, an unstable, but useful SPAAC cross-linking reagent. Since photo-DIBO doesn't react with organic azides, this method allows for the spatiotemporal control of the ligation of two azide-tagged substrates.
Co-reporter:Alexander V. Kuzmin and Vladimir V. Popik
Chemical Communications 2009(Issue 38) pp:NaN5709-5709
Publication Date(Web):2009/08/14
DOI:10.1039/B911871C
A reactive ten-membered ring enyne–allene (τ25 °C = 5–6 min) is efficiently generated (Φ300 nm = 0.57) by UV irradiation of a thermally stable precursor in which a triple bond is masked as a cyclopropenone moiety.
Co-reporter:Selvanathan Arumugam, Jun Guo, Ngalle Eric Mbua, Frédéric Friscourt, Nannan Lin, Emmanuel Nekongo, Geert-Jan Boons and Vladimir V. Popik
Chemical Science (2010-Present) 2014 - vol. 5(Issue 4) pp:NaN1598-1598
Publication Date(Web):2014/01/02
DOI:10.1039/C3SC51691A
The selective derivatization of solvent-exposed cysteine residues in peptides and proteins is achieved by brief irradiation of an aqueous solution containing 3-(hydroxymethyl)-2-naphthol derivatives (NQMPs) with a 350 nm fluorescent lamp. NQMP can be conjugated with various moieties, such as PEG, dyes, carbohydrates, or possess a fragment for further selective derivatization, e.g., biotin, azide, alkyne, etc. Attractive features of this labeling approach include an exceptionally fast rate of the reaction and a requirement for a low equivalence of the reagent. The NQMP-thioether linkage is stable under ambient conditions, and survives protein digestion and MS analysis. Irradiation of an NQMP-labeled protein in a dilute solution (<40 μM) or in the presence of a vinyl ether results in a traceless release of the substrate. The reversible biotinylation of bovine serum albumin, as well as the capture and release of this protein using NeutrAvidin Agarose resin beads has been demonstrated.
Co-reporter:Christopher D. McNitt and Vladimir V. Popik
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 41) pp:NaN8202-8202
Publication Date(Web):2012/08/31
DOI:10.1039/C2OB26581H
Oxa-dibenzocyclooctynes (ODIBO, 2a–c) are prepared by photochemical decarbonylation of corresponding cyclopropenones (photo-ODIBO, 1a–c). While photo-ODIBO does not react with azides, ODIBO is one of the most reactive cyclooctynes exhibiting rates of cycloaddition over 45 M−1 s−1 in aqueous solutions. ODIBO is stable under ambient conditions and has low reactivity towards thiols. Photo-ODIBO survives heating up to 160 °C and does not react with thiols.
Co-reporter:Emmanuel E. Nekongo, Pritha Bagchi, Christoph J. Fahrni and Vladimir V. Popik
Organic & Biomolecular Chemistry 2012 - vol. 10(Issue 46) pp:NaN9218-9218
Publication Date(Web):2012/10/12
DOI:10.1039/C2OB26715B
In aqueous and alcohol solutions, colorless and non-fluorescent derivatives of 9-aryl-9H-xanthen-9-ol equilibrate with brightly colored and fluorescent 9-arylxanthylium cations, thus offering a convenient platform for the design of dual-mode indicators for emission and absorption-based pH measurements. The position of the prototropic equilibrium depends only on the hydronium ion concentration and is not affected by general acids or other ions. Furthermore, the equilibrium equivalence point can be readily adjusted by introducing substituents in the xanthenol core. As dehydroxylation of 3,6-dialkoxy-9-(o-tolyl)-9-xanthenol occurs at pH = 6.5, indicators of this type are well suited for biological applications as illustrated by in vitro cell culture studies with NIH 3T3 cells.
Co-reporter:Mariia V. Sutton, Matthew McKinley, Revathy Kulasekharan and Vladimir V. Popik
Chemical Communications 2017 - vol. 53(Issue 41) pp:NaN5601-5601
Publication Date(Web):2017/04/05
DOI:10.1039/C7CC02056B
A new photocleavable analog of BAPTA chelating ligand has a high affinity towards Ca2+ ions (K = 2.5 × 106 M−1). The use of photolabile 3-(hydroxymethyl)-2-naphthol core in the design of photo-BAPTA allows for the efficient (Φ = 0. 63) and very fast (τ < 12 μs) release of Ca2+ ions upon 300 or 350 nm irradiation.
.jpg)
![Benzaldehyde, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/96100/96013-95-5.png)
![Benzaldehyde, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/96100/96013-95-5_b.png)
![Benzenemethanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/96100/96013-77-3.png)
![Benzenemethanol, 3-[[(1,1-dimethylethyl)dimethylsilyl]oxy]-](http://img.cochemist.com/ccimg/96100/96013-77-3_b.png)










![BENZALDEHYDE, 3-[2-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]ETHOXY]-](http://img.cochemist.com/ccimg/828300/828242-83-7.png)
![BENZALDEHYDE, 3-[2-[[(1,1-DIMETHYLETHYL)DIMETHYLSILYL]OXY]ETHOXY]-](http://img.cochemist.com/ccimg/828300/828242-83-7_b.png)



![4,7,10,13-Tetraoxa-16-azaheneicosanoicacid, 21-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4-yl]-17-oxo-](http://img.cochemist.com/ccimg/721500/721431-18-1.png)
![4,7,10,13-Tetraoxa-16-azaheneicosanoicacid, 21-[(3aS,4S,6aR)-hexahydro-2-oxo-1H-thieno[3,4-d]imidazol-4-yl]-17-oxo-](http://img.cochemist.com/ccimg/721500/721431-18-1_b.png)



![DIBENZO[A,E]CYCLOOCTENE, 5,11-BIS(PHENYLSULFONYL)-](http://img.cochemist.com/ccimg/468800/468751-39-5.png)
![DIBENZO[A,E]CYCLOOCTENE, 5,11-BIS(PHENYLSULFONYL)-](http://img.cochemist.com/ccimg/468800/468751-39-5_b.png)









![2-[2-[2-(2-Azidoethoxy)ethoxy]ethoxy]-1-(p-toluenesulfonyl)-ethanol](http://img.cochemist.com/ccimg/168700/168640-82-2.png)
![2-[2-[2-(2-Azidoethoxy)ethoxy]ethoxy]-1-(p-toluenesulfonyl)-ethanol](http://img.cochemist.com/ccimg/168700/168640-82-2_b.png)




![PROPANOYL CHLORIDE, 3-[(TRIFLUOROACETYL)AMINO]-](http://img.cochemist.com/ccimg/87700/87639-76-7.png)
![PROPANOYL CHLORIDE, 3-[(TRIFLUOROACETYL)AMINO]-](http://img.cochemist.com/ccimg/87700/87639-76-7_b.png)




![6-[TERT-BUTYL(DIMETHYL)SILYL]OXYHEXANOIC ACID](http://img.cochemist.com/ccimg/77800/77744-44-6.png)
![6-[TERT-BUTYL(DIMETHYL)SILYL]OXYHEXANOIC ACID](http://img.cochemist.com/ccimg/77800/77744-44-6_b.png)


![Phosphonium, [(3-methoxyphenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/72400/72311-12-7.png)
![Phosphonium, [(3-methoxyphenyl)methyl]triphenyl-, bromide](http://img.cochemist.com/ccimg/72400/72311-12-7_b.png)


![5-(2-Oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoic acid](http://img.cochemist.com/ccimg/57400/57378-70-8.png)
![5-(2-Oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanoic acid](http://img.cochemist.com/ccimg/57400/57378-70-8_b.png)
![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4.png)
![11-oxo-2,3,5,6,7,11-Hexahydro-1H-pyrano[2,3-f]pyrido[3,2,1-ij]quinoline-10-carboxylic acid](/data/chemimg/86700/55804-65-4_b.png)
![5,6,11,12-tetradehydrodibenzo[a,e][8]annulene](http://img.cochemist.com/ccimg/53400/53397-65-2.png)
![5,6,11,12-tetradehydrodibenzo[a,e][8]annulene](http://img.cochemist.com/ccimg/53400/53397-65-2_b.png)


![N-[(4-BROMOPHENYL)METHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/34200/34158-73-1.png)
![N-[(4-BROMOPHENYL)METHYLIDENE]HYDROXYLAMINE](http://img.cochemist.com/ccimg/34200/34158-73-1_b.png)







![(5E)-5-(3-iodo-5-methoxy-4-{2-[2-(prop-2-en-1-yl)phenoxy]ethoxy}benzylidene)-3-methyl-2-thioxo-1,3-thiazolidin-4-one](http://img.cochemist.com/ccimg/6200/6188-74-5.png)
![(5E)-5-(3-iodo-5-methoxy-4-{2-[2-(prop-2-en-1-yl)phenoxy]ethoxy}benzylidene)-3-methyl-2-thioxo-1,3-thiazolidin-4-one](http://img.cochemist.com/ccimg/6200/6188-74-5_b.png)















![Ethanol, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/976600/86770-67-4.png)
![Ethanol, 2-[2-[2-(2-azidoethoxy)ethoxy]ethoxy]-](/data/chemimg/976600/86770-67-4_b.png)






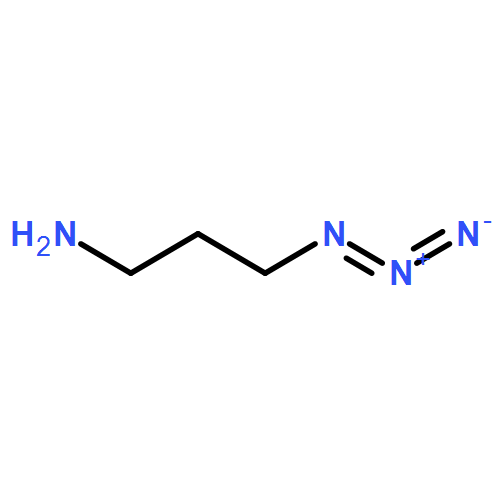
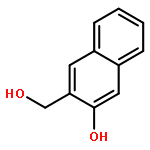
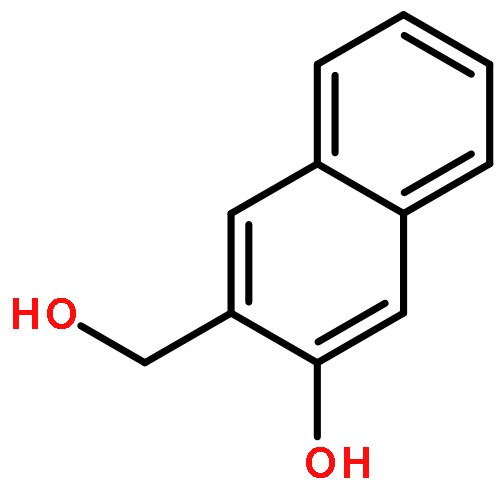


![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5.png)
![3',6'-Dihydroxy-3H-spiro[isobenzofuran-1,9'-xanthen]-3-one](http://img.cochemist.com/ccimg/2400/2321-07-5_b.png)