Co-reporter:Senchuan Song, Jianghan Chen, Wenlong Pan, Hucacan Song, Huahong Shi, Yuliang Mai, Wu Wen
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2017 170() pp: 157-166
Publication Date(Web):
DOI:10.1016/j.saa.2016.07.008
Co-reporter:Ao You, Jie Zhou, Senchuan Song, Guoxun Zhu, Huacan Song, Wei Yi
European Journal of Medicinal Chemistry 2015 Volume 93() pp:255-262
Publication Date(Web):26 March 2015
DOI:10.1016/j.ejmech.2015.02.013
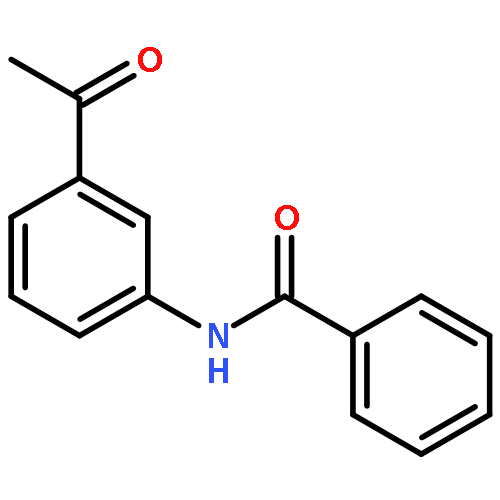
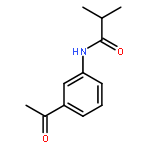
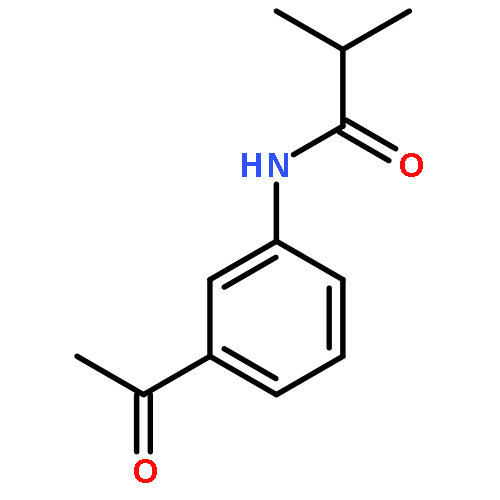
•Parent compounds were found to be the potent tyrosinase activators.•Structure-based modification toward parent compounds resulted in a remarkable change of the potency on tyrosinase.•All the designed compounds exhibited remarkable tyrosinase inhibitory activities.•SARs were discussed.•The inhibition mechanism and the inhibition kinetics of selected compounds on tyrosinase were investigated.In this study, we developed 3-/4-aminoacetophenones and their structure-based 3-/4-aminophenylethylidenethiosemicarbazide derivatives, respectively, as novel tyrosinase activators and inhibitors. Notably, all the obtained thiosemicarbazones displayed more potent tyrosinase inhibitory activities than kojic acid. Especially, compound 7k was found to be the most active tyrosinase inhibitor with IC50 value of 0.291 μM. The structure-activity relationships (SARs) analysis showed that: (1) the amine group was absolutely necessarily for determining the tyrosinase activation activity; (2) the introduction of thiosemicarbazide group played a very vital role in transforming tyrosinase activators into tyrosinase inhibitors; (3) the phenylethylenethiosemicarbazide moiety was crucial for determining the tyrosinase inhibitory activity; (4) the type of acyl group had no obvious effect on the inhibitory activity; (5) the position of amide substituent on the phenyl ring influenced the tyrosinase inhibitory potency. Moreover, the inhibition mechanism and inhibition kinetics study revealed that compound 7k was reversible and non-competitive inhibitor, and compound 8h was reversible and competitive-uncompetitive mixed-II type inhibitor.
Co-reporter:Jijun Huang, Jie Zhou, Senchuan Song, Huacan Song, Zhiyong Chen, Wei Yi
Tetrahedron 2015 Volume 71(Issue 45) pp:8628-8636
Publication Date(Web):11 November 2015
DOI:10.1016/j.tet.2015.09.018
Here a new and efficient method via ZnCl2-catalyzed direct cyclization of diverse benzylidenemalononitriles and arylamines has been developed. With this method, a variety of novel 2-amino-3,5-dicyano-4-aryl-6-aryl-aminopyridines (2a–2v) could be easily prepared under the mild conditions with board substrate/functional group tolerance and decent product yields. The biological activities of the selected compounds (2c, 2e, 2g, 2i, 2k, 2m, 2n and 2o) has also been evaluated as new antibacterial agents. The results demonstrated that almost all of the compounds had more potent antibacterial activities against HP than that of clinically used drugs, Ornidazole, Metronidazole, Nitrimidazine and Clarithromycin, suggesting that further development of such compounds might be of great interest.Herein a new and efficient method via ZnCl2-catalyzed direct cyclization of diverse benzylidenemalononitriles and arylamines for one-pot synthesis of novel 2-amino-3,5-dicyano-4-aryl-6-aryl-aminopyridines as potent antibacterial agents is described.
Co-reporter:Ao You, Jie Zhou, Senchuan Song, Guoxun Zhu, Huacan Song, Wei Yi
Bioorganic & Medicinal Chemistry 2015 23(5) pp: 924-931
Publication Date(Web):
DOI:10.1016/j.bmc.2015.01.024
Co-reporter:Zhiyong Chen, Dachuan Cai, Dehai Mou, Qin Yan, Yifeng Sun, Wenlong Pan, Yiqian Wan, Huacan Song, Wei Yi
Bioorganic & Medicinal Chemistry 2014 22(13) pp: 3279-3284
Publication Date(Web):
DOI:10.1016/j.bmc.2014.04.060
Co-reporter:Zhiyong Chen, Rihui Cao, Buxi Shi, Liang Guo, Jie Sun, Qin Ma, Wenxi Fan, Huacan Song
European Journal of Medicinal Chemistry 2011 Volume 46(Issue 10) pp:5127-5137
Publication Date(Web):October 2011
DOI:10.1016/j.ejmech.2011.08.027
A series of novel 1,9-disubstituted β-carbolines was designed, synthesized and evaluated as cytotoxic and DNA intercalating agents. Compounds 7b, 7c, 8b and 8c exhibited the most potent cytotoxic activities with IC50 values of lower than 20 μM against ten human tumor cell lines. The results indicated that (1) the 3-chlorobenzyl and 3-phenylpropyl substituents in position-9 of β-carboline nucleus were the suitable pharmacophoric group giving rise to significant antitumor agents; (2) the length of the alkylamino side chain moiety affected their cytotoxic potencies, and three CH2 units were more favorable. In addition, these compounds were found to exhibit remarkable DNA intercalating effects.A series of 1,9-disubstituted β-carbolines was synthesized and evaluated as potential cytotoxic and DNA intercalating agents.Highlights► 1,9-Disubstituted β-carbolines was synthesized and evaluated as antitumor agents. ► The 3-phenylpropyl substituents in position-9 were the suitable pharmacophoric group. ► The length of the alkylamino side chain moiety affected their cytotoxic potencies. ► These compounds were found to exhibit remarkable DNA intercalating effects.
Co-reporter:Chunming Ma, Rihui Cao, Buxi Shi, Shaoxue Li, Zhiyong Chen, Wei Yi, Wenlie Peng, Zhenhua Ren, Huacan Song
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 4) pp:1515-1523
Publication Date(Web):April 2010
DOI:10.1016/j.ejmech.2009.12.060
The β-carboline alkaloids have been characterized as a class of potential antitumor agents. To further enhance the cytotoxic potency and improve water solubility of β-carboline, a series of new β-carboline amino acid ester, β-carboline amino acid and N2-benzylated quaternary β-carboline amino acid ester conjugates were designed and synthesized, and the cytotoxic activities of these compounds were evaluated using a panel of human tumor cell lines. The N2-benzylated quaternary β-carboline amino acid ester conjugates represented the most interesting cytotoxic activities. Particularly, compounds 8b and 8g were found to be the most potent compounds with IC50 values lower than 20 μM against all human tumor cell lines investigated. These results confirmed that the N2-benzyl substituent on the β-carboline ring played an important role in the modulation of the cytotoxic potencies.A series of N2-benzylated quaternary β-carboline amino acid ester conjugates was synthesized and evaluated as new antitumor agents. Compounds 8b and 8g were found to be the most potent compounds with IC50 values lower than 20 μM against all tumor cell lines investigated.
Co-reporter:Hui Wang, Yang-Ji Du, Hua-Can Song
Food Chemistry 2010 Volume 123(Issue 1) pp:6-13
Publication Date(Web):1 November 2010
DOI:10.1016/j.foodchem.2010.03.088
The 75% ethanol extract from guava (Psidium guajava Linn.) leaves was extracted further, in turn, with CH2Cl2, EtOAc and n-BuOH to afford four fractions, CH2Cl2-soluble, EtOAc-soluble, n-BuOH-soluble and residual extract fractions. Both the n-BuOH-soluble and EtOAc-soluble fractions showed high inhibitory activity against α-glucosidase and α-amylase. Seven pure flavonoid compounds, quercetin (1), kaempferol (2), guaijaverin (3), avicularin (4), myricetin (5), hyperin (6) and apigenin (7), were isolated (using enzyme assay-guide fractionation method) from the n-BuOH-soluble and EtOAc-soluble fractions. The structures of these pure compounds were determined on the basis of MS and NMR data and the activities of these compounds were evaluated. Compounds 1, 2 and 5 showed high inhibitory activities, with IC50 values of 3.5 mM, 5.2 mM and 3.0 mM against sucrase, with IC50 values of 4.8 mM, 5.6 mM and 4.1 mM against maltase and with IC50 values of 4.8 mM, 5.3 mM and 4.3 mM against α-amylase, respectively. We found that myricetin showed the most powerful activity among these compounds with a 70% inhibition against sucrase at a concentration of 1.5 mg/ml. The hydroxyl group at the 3-position on the A-ring and a number of hydroxyl groups attached to the C-ring played important roles in the inhibition activity. There was an obvious synergistic effect (the mixing action of two compounds) against α-glucosidase, but against α-amylase this was not found. This is the first study of the active compositions of guava leaves and the biological activity of the active compositions against α-glucosidase and α-amylase.
Co-reporter:Zhiyong Chen, Rihui Cao, Liang Yu, Buxi Shi, Jie Sun, Liang Guo, Qin Ma, Wei Yi, Xiao Song, Huacan Song
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 11) pp:4740-4745
Publication Date(Web):November 2010
DOI:10.1016/j.ejmech.2010.07.037
In a continuing effort to develop novel β-carbolines endowed with better pharmacological profile, a series of water-soluble β-carbolines bearing a flexible amino side chain was designed and synthesized, and the cytotoxic activities in vitro of these compounds were evaluated. The N9-arylated alkyl substituted β-carbolines represented the most interesting cytotoxic agents, and compounds 4c and 4d were found to be the most potent compounds with IC50 values lower than 10 μM against ten human tumor cell lines. The results confirmed that the N9-arylated alkyl substituents of β-carboline played a very important role in the modulation of the cytotoxic potencies. In addition, the interaction with DNA of these compounds was also investigated, these compounds were found to exhibit significant DNA binding affinity.A series of water-soluble β-carbolines bearing a flexible amino side chain was synthesized and evaluated. Compounds 4c and 4d were found to be the most potent compounds with IC50 values lower than 10 μM against ten tumor cell lines.
Co-reporter:Yun-Nan Yan, Wen-long Pan, Hua-Can Song
Dyes and Pigments 2010 Volume 86(Issue 3) pp:249-258
Publication Date(Web):August 2010
DOI:10.1016/j.dyepig.2010.01.011
A series of 1,3,4-oxadiazole derivatives containing an imidazole unit were synthesized and characterized using 1H NMR, 13C NMR, mass spectrometry (or high-resolution mass spectrometry) and elemental analysis. The crystal structure of 2,5-bis(4-(1-n-butyl-4,5-diphenylimidazol-2-yl)phenyl)-1,3,4-oxadiazole was determined as monoclinic, space group C2/c type, using single crystal X-ray crystallography. For nine samples, UV–visible absorption coefficient (ɛ), maximum wavelength (λmax), fluorescence excitation wavelength (λex), fluorescence emission wavelength (λem), fluorescence quantum yield (ΦF), fluorescence lifetime (T) were measured in dichloromethane and in the solid state. For these selected compounds, thermogravimetric analysis was also employed and structure:optical behaviour characteristics were discussed.
Co-reporter:Wei Yi, Rihui Cao, Wenlie Peng, Huan Wen, Qin Yan, Binhua Zhou, Lin Ma, Huacan Song
European Journal of Medicinal Chemistry 2010 Volume 45(Issue 2) pp:639-646
Publication Date(Web):February 2010
DOI:10.1016/j.ejmech.2009.11.007
A series of novel 4-hydroxybenzaldehyde derivatives were synthesized and their inhibitory effects on the diphenolase activity of mushroom tyrosinase were investigated. Most of target compounds had more potent inhibitory activities than the parent compound 4-hydroxybenzaldehyde (IC50 = 1.22 mM). Interestingly, compound 3c bearing a dimethoxyl phosphate was found to be the most potent inhibitor with IC50 value of 0.059 mM. The inhibition kinetics analyzed by Lineweaver–Burk plots revealed that compound 3c was a non-competitive inhibitor (KI = 0.0368 mM). In particular, compound 3c showed no side effects at dose of 1600 mg/kg in mice. These results suggested that such compounds might be served as lead compounds for further designing new potential tyrosinase inhibitors.A series of 4-hydroxybenzaldehyde derivatives were synthesized and evaluated as mushroom tyrosinase inhibitors. Compound 3c was found to be the most potent inhibitor .
Co-reporter:Zhiyong Chen, Rihui Cao, Buxi Shi, Wei Yi, Liang Yu, Huacan Song, Zhenhua Ren, Wenlie Peng
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 13) pp:3876-3879
Publication Date(Web):1 July 2010
DOI:10.1016/j.bmcl.2010.05.034
A series of water-soluble β-carbolines, bearing a flexible amino side chain, was prepared and evaluated in vitro against a panel of human tumor cell lines. The N9-arylated alkyl substituted β-carbolines represented the most interesting cytotoxic activities, and compound 7b was found to be the most potent antitumor agent with IC50 values lower than 10 μM against eight human tumor cell lines. The results confirmed that the N9-arylated alkyl substituents of β-carboline nucleus played an important role in the modulation of the cytotoxic potencies. In addition, these compounds were found to exhibit significant DNA-binding affinity.A series of water-soluble β-carbolines, bearing a flexible amino side chain, has been prepared and evaluated in vitro against a panel of human cell lines. Compound 7b were found to be the most potent compound with IC50 values lower than 10 μM against eight human tumor cell lines.
Co-reporter:Liang Yu, Rihui Cao, Wei Yi, Qin Yan, Zhiyong Chen, Lin Ma, Wenlie Peng, Huacan Song
Bioorganic & Medicinal Chemistry Letters 2010 Volume 20(Issue 11) pp:3254-3258
Publication Date(Web):1 June 2010
DOI:10.1016/j.bmcl.2010.04.059
A series of novel cholinesterase inhibitors, being composed of 4-[(diethylamino)methyl]-phenoxy and secondary amine which were linked with a different length alkyl chain, were designed and synthesized from the starting material p-hydroxybenzaldehyde. These compounds were evaluated as acetylcholinesterase and butyrylcholinesterase (AChE/BChE) inhibitors. Compounds 25–31 having a secondary amine moiety connected to the phenyl ring via eight CH2 units spacer were found to be the most potent inhibitors with IC50 value lower than 220 nM and 48 nM against AChE and BChE, respectively. Interestingly, these inhibitors showed a surprising selectively toward BChE, and compounds 26, 27, and 30 displayed 12.5, 18.6, and 18.8-fold higher affinity to BChE. The inhibition kinetics analyzed by Linewear–Burk plots revealed that such compounds were mix-type inhibitors.A series of 4-[(diethylamino)methyl]-phenol derivatives were designed, synthesized, and evaluated as cholinesterase inhibitors. Compound 26 was found to be the most potent inhibitor.
Co-reporter:Jinbing Liu, Rihui Cao, Qifeng Wu, Chunming Ma, Zihou Wang, Wenlie Peng, Huacan Song
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 4) pp:1737-1744
Publication Date(Web):April 2009
DOI:10.1016/j.ejmech.2008.03.010
A series of novel 4-alkylphenyl β-aldehyde ketones and their derivatives were designed and synthesized on the basis of the chemical structures of Houttuynin and β-lactam antibiotics. Antibacterial activities of these compounds were investigated. The results demonstrated that most of the compounds tested had moderate antibacterial activities against Gram-positive pathogen Staphylococcus aureus (ATTC-25923) than Houttuynin, and Gram-positive bacteria were more susceptible to the compounds than Gram-negative bacteria. Compound 23 was found to be the most potent compound with MIC of 1.0 μg/mL against S. aureus. Particularly, compounds 16, 22 and 23 showed more active antibacterial activities against the clinically important pathogenic bacteria, methicillin-resistant S. aureus (MRSA) than Houttuynin and levofloxacin. The preliminary structure–activity relationship (SAR) analysis suggested that (1) the introduction of appropriate alkyl substituents into position 4 of phenyl ring enhanced antibacterial activities of these compounds, and isopropyl substituent might be more favorable; (2) the presence of ketone carbonyl moiety might play a vital role in determining significant antibacterial activities of these compounds. A series of novel 4-alkylphenyl β-aldehyde ketones and their derivatives were designed and synthesized in order to discover and develop novel antibacterial drugs. Compound 23 was found to be the most potent compound with minimum inhibitory concentration (MIC) of 1.0 μg/mL against Staphylococcus aureus.
Co-reporter:Jinbing Liu, Rihui Cao, Wei Yi, Chunming Ma, Yiqian Wan, Binhua Zhou, Lin Ma, Huacan Song
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 4) pp:1773-1778
Publication Date(Web):April 2009
DOI:10.1016/j.ejmech.2008.04.002
A series of alkylidenethiosemicarbazide compounds were synthesized and their inhibitory effects on the diphenolase activity of mushroom tyrosinase were evaluated. The results showed that most of the synthesized compounds exhibited significant inhibitory activities. Especially, compound 1f was found to be the most potent inhibitor with IC50 value of 0.086 μM, suggesting that further development of such compounds may be of interest.A series of alkylidenethiosemicarbazide compounds were synthesized and their inhibitory activities against the diphenolase activity of mushroom tyrosinase were investigated. Compound 1f was found to be the best potent compound with IC50 value of 0.086 μM.
Co-reporter:Qin Yan, Rihui Cao, Wei Yi, Zhiyong Chen, Huan Wen, Lin Ma, Huacan Song
European Journal of Medicinal Chemistry 2009 Volume 44(Issue 10) pp:4235-4243
Publication Date(Web):October 2009
DOI:10.1016/j.ejmech.2009.05.023
A series of novel 5-benzylidene barbiturate and thiobarbiturate derivatives were synthesized and evaluated as tyrosinase inhibitors and antibacterial agents. The results demonstrated that some compounds had more potent inhibitory activities than the parent compound 4-hydroxybenzaldehyde (IC50 = 1.22 mM). Particularly, compounds 1a and 2a were found to be the most potent inhibitors with IC50 value of 13.98 μM and 14.49 μM, respectively. The inhibition mechanism study revealed that these compounds were irreversible inhibitors. The circular dichroism spectra indicated that these compounds induced conformational changes of mushroom tyrosinase upon binding. In addition, these compounds exhibited selectively antibacterial activity against Staphylococcus aureus. All these results suggested that further development of such compounds may be of interest. A series of novel 5-benzylidene barbiturate and thiobarbiturate derivatives were synthesized and evaluated as tyrosinase inhibitors. Compounds 1a and 2a were found to be the most potent inhibitors.
Co-reporter:Qin Yan, Rihui Cao, Wei Yi, Liang Yu, Zhiyong Chen, Lin Ma, Huacan Song
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 15) pp:4055-4058
Publication Date(Web):1 August 2009
DOI:10.1016/j.bmcl.2009.06.018
A series of 5-benzylidene(thio)barbiturate-β-d-glycosides were designed, synthesized and evaluated as a new class of mushroom tyrosinase inhibitors. The results demonstrated that most of compounds had more potent inhibitory activities than arbutin (IC50 8.4 mmol/L). Compound 12b was found to be the most potent inhibitor with IC50 value of 0.05 mmol/L. SARs analysis suggested that (1) 5-benzylidenethiobarbiturate substructures were efficacious for the inhibitory activity; (2) the lipophilic property of acetylated sugar moiety facilitated the inhibitory potency; (3) the hydroxyl group of 3′-configuration contributed to the increase of inhibitory effects. In addition, the inhibition mechanism study revealed that 5-benzylidene(thio)barbiturate-β-d-glycosides were irreversible inhibitors.A series of 5-benzylidene(thio)barbiturate-β-d-glycosides were designed, synthesized and evaluated as tyrosinase inhibitors. Compound 12b was found to be the most potent inhibitor.
Co-reporter:Wei Yi, Rihui Cao, Huan Wen, Qin Yan, Binhua Zhou, Lin Ma, Huacan Song
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 21) pp:6157-6160
Publication Date(Web):1 November 2009
DOI:10.1016/j.bmcl.2009.09.018
A series of 4-functionalized phenyl-O-β-d-glycosides were designed, synthesized and evaluated as a new class of mushroom tyrosinase inhibitors. The results demonstrated that compounds 6a–13a bearing a thiosemicarbazide moiety exhibited potent activities with IC50 values range from 0.31 to 52.8 μM. Particularly, compound 9a containing acetylated glucose moiety was found to be the most active molecule with an IC50 value of 0.31 μM. SARs analysis suggested that (1) the thiosemicarbazide moiety remarkably contributed to the increase of inhibitory effects on tyrosinase; (2) the configuration and bond type of sugar moiety also played a very important role in determining their inhibitory activities. The inhibition kinetics and inhibition mechanism study revealed that compound 9a was reversible and competitive type inhibitor, whereas compound 13a was reversible and competitive–uncompetitive mixed-II type inhibitor.A series of 4-functionalized phenyl-O-β-d-glycosides were designed, synthesized and evaluated as tyrosinase inhibitors. Compound 9a was found to be the most potent inhibitor.
Co-reporter:Yun-Nan Yan, Dan-Yan Lin, Wen-Long Pan, Xiu-Ling Li, Yi-Qian Wan, Yu-Liang Mai, Hua-Can Song
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 2009 Volume 74(Issue 1) pp:233-242
Publication Date(Web):15 September 2009
DOI:10.1016/j.saa.2009.06.020
Eight 2-(9-phenanthrenyl)-, 2-(9-anthryl)- and 2-(1-pyrenyl)-1-alkyl-benzimidazole compounds, three 2-(9-anthryl)-1-alkylphenanthroimidazole compounds and five 4,5-diphenyl-1-alkyl-2-(9-anthryl)imidazole compounds were synthesized by alkylation reactions of the corresponding benzimidazole, phenanthroimidazole or imidazole compounds. 2-(10-Bromo-9-anthryl)-1-alkyl-benzimidazole compounds were prepared by bromination reaction of 2-(9-anthryl)-1-alkylbenzimidazole compounds. All the synthesized compounds were characterized by elemental analysis, 1H NMR, 13C NMR, MS or HRMS; their absorption coefficients (ɛ), maximum absorption λamax, fluorescence emission maximum λem, Stokes shifts and fluorescence quantum yields (ΦF) in ethyl acetate were determined; their fluorescent lifetimes (T1 and T2) were measured in ethyl acetate and in solid state, respectively. The crystal structure of 2-(9-anthryl)-1-n-butyl-4,5-diphenylimidazole (12a) was determined to be triclinic, space group P-1 types, using single crystal X-ray crystallography technique. The results showed that these compounds exhibited moderate fluorescence-emission abilities and higher solubility in most organic solvents than their corresponding starting materials. The relationships between the optical behaviors and structures for these compounds were discussed.
Co-reporter:Huan Wen, Chonglan Lin, Ling Que, Hui Ge, Lin Ma, Rihui Cao, Yiqian Wan, Wenlie Peng, Zihou Wang, Huacan Song
European Journal of Medicinal Chemistry 2008 Volume 43(Issue 1) pp:166-173
Publication Date(Web):January 2008
DOI:10.1016/j.ejmech.2007.03.018
A series of helicid analogues were prepared and evaluated in vitro for the cholinesterase (AChE and BuChE) inhibitory activities via UV spectroscopy. The results indicated that compounds 5, 6d and 8 exhibited potent AChE inhibitory activities with IC50 values of 0.45 ± 0.02 μM, 0.49 ± 0.02 μM, and 0.20 ± 0.01 μM, respectively. High selectivity for AChE over BuChE was also observed. Kinetic study showed that the mechanism of AChE inhibition of compounds 5, 6d and 8 was all mixed-type.A series of helicid analogs were prepared and evaluated in vitro for the cholinesterase (AChE and BuChE) inhibitory activities via UV spectroscopy. The results indicated that compounds 5, 6d, and 8 exhibited potent inhibitory activities with IC50 value of 0.45 ± 0.02 μM, 0.49 ± 0.02 μM, and 0.20 ± 0.01 μM, respectively. Kinetic study showed that the mechanism of AChE inhibition of compounds 5, 6d and 8 was all mixed-type.
Co-reporter:Huan Wen, Yayao Zhou, Chonglan Lin, Hui Ge, Lin Ma, Zihou Wang, Wenlie Peng, Huacan Song
Bioorganic & Medicinal Chemistry Letters 2007 Volume 17(Issue 8) pp:2123-2125
Publication Date(Web):15 April 2007
DOI:10.1016/j.bmcl.2007.01.091
A new class of inhibitors of acetylcholinesterase (methyl 2-(2-(4-formylphenoxy)acetamido)-2-substituted acetate derivatives) is described. Compounds 4b and 4i were found to be more potent than galanthamine in inhibiting acetylcholinesterase.A new class of inhibitors of acetylcholinesterase (methyl 2-(2-(4-formylphenoxy)acetamido)-2-substituted acetate derivatives) is described. Compounds 4b and 4i were found to be more potent than galanthamine in inhibiting acetylcholinesterase.

![3,5-Pyridinedicarbonitrile, 2-amino-6-[(4-bromophenyl)amino]-4-phenyl-](http://img.cochemist.com/ccimg/921000/920983-17-1.png)
![3,5-Pyridinedicarbonitrile, 2-amino-6-[(4-bromophenyl)amino]-4-phenyl-](http://img.cochemist.com/ccimg/921000/920983-17-1_b.png)
![2-[3-(TRIFLUOROMETHYL)PHENOXY]ISOINDOLE-1,3-DIONE](http://img.cochemist.com/ccimg/328100/328081-53-4.png)
![2-[3-(TRIFLUOROMETHYL)PHENOXY]ISOINDOLE-1,3-DIONE](http://img.cochemist.com/ccimg/328100/328081-53-4_b.png)











![Hydrazinecarbothioamide, 2-[1-(3-aminophenyl)ethylidene]-](http://img.cochemist.com/ccimg/87800/87743-92-8.png)
![Hydrazinecarbothioamide, 2-[1-(3-aminophenyl)ethylidene]-](http://img.cochemist.com/ccimg/87800/87743-92-8_b.png)
![2-PROPANONE, 1-[4-(PENTYLOXY)PHENYL]-](http://img.cochemist.com/ccimg/84100/84023-39-2.png)
![2-PROPANONE, 1-[4-(PENTYLOXY)PHENYL]-](http://img.cochemist.com/ccimg/84100/84023-39-2_b.png)


![2-BUTANONE, 4-[4-(PHENYLMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/74500/74432-58-9.png)
![2-BUTANONE, 4-[4-(PHENYLMETHOXY)PHENYL]-](http://img.cochemist.com/ccimg/74500/74432-58-9_b.png)
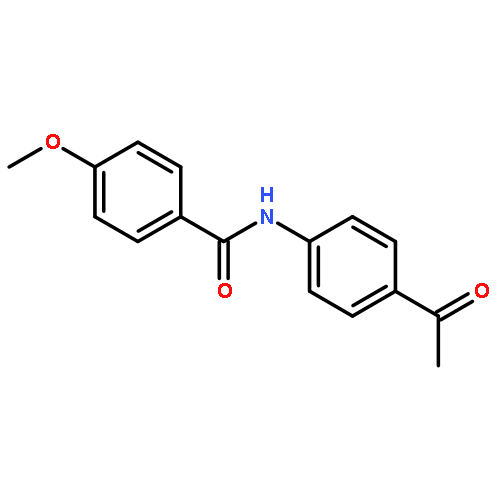
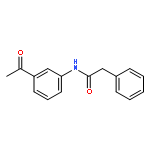
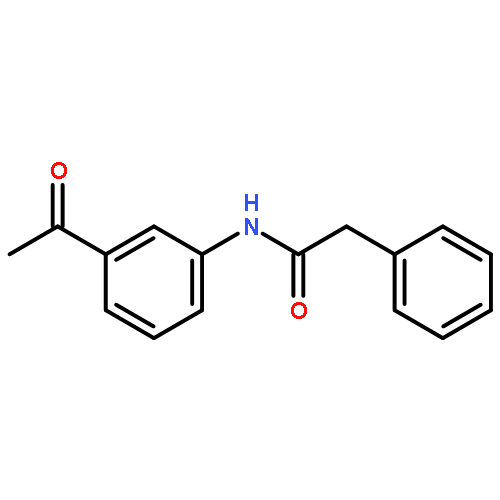


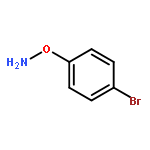





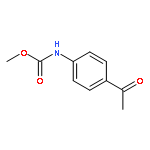
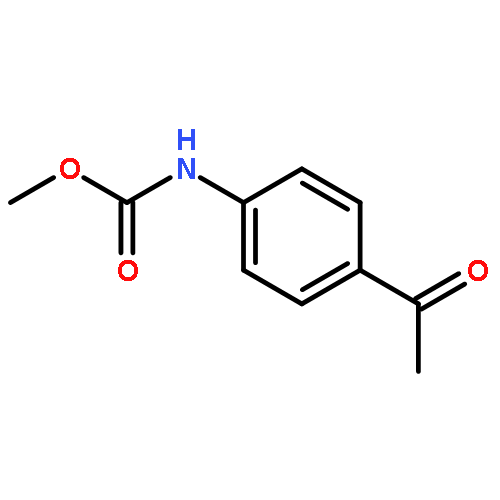
![[2-tert-butoxy-1-(tert-butoxycarbonyl)-2-oxoethylidene]diazenium](http://img.cochemist.com/ccimg/35300/35207-75-1.png)
![[2-tert-butoxy-1-(tert-butoxycarbonyl)-2-oxoethylidene]diazenium](http://img.cochemist.com/ccimg/35300/35207-75-1_b.png)
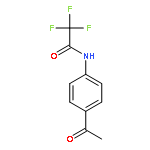
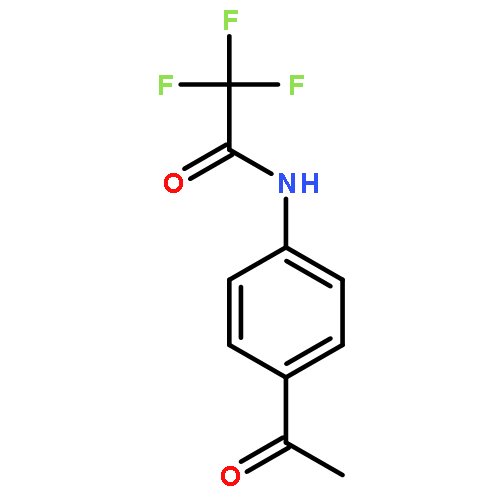
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7.png)
![Bis[(pentamethylcyclopentadienyl)dichloro-rhodium]](http://img.cochemist.com/ccimg/12400/12354-85-7_b.png)
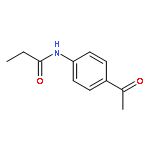
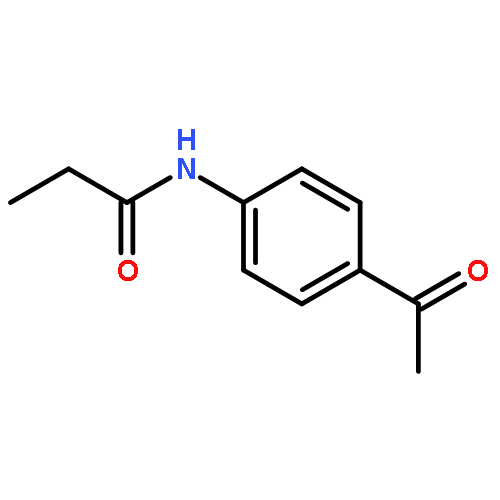
![N-{4-[(1E)-N-carbamothioylethanehydrazonoyl]phenyl}acetamide](http://img.cochemist.com/ccimg/7500/7441-55-6.png)
![N-{4-[(1E)-N-carbamothioylethanehydrazonoyl]phenyl}acetamide](http://img.cochemist.com/ccimg/7500/7441-55-6_b.png)
![[1-(4-AMINOPHENYL)ETHYLIDENEAMINO]THIOUREA](http://img.cochemist.com/ccimg/7500/7410-53-9.png)
![[1-(4-AMINOPHENYL)ETHYLIDENEAMINO]THIOUREA](http://img.cochemist.com/ccimg/7500/7410-53-9_b.png)
![Propanedinitrile,2-[(3,4,5-trimethoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/5700/5688-82-4.png)
![Propanedinitrile,2-[(3,4,5-trimethoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/5700/5688-82-4_b.png)
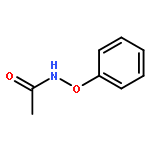
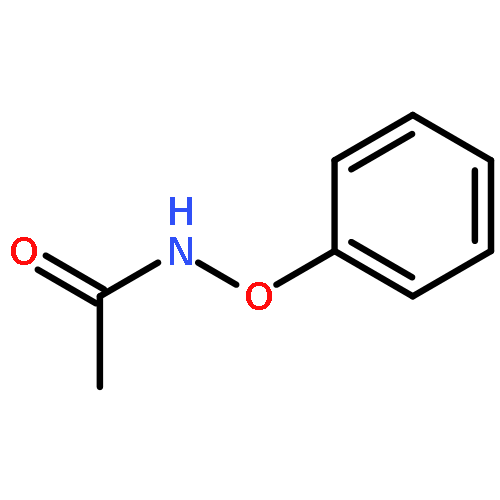







![Propanedinitrile,2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-26-8.png)
![Propanedinitrile,2-[(4-methoxyphenyl)methylene]-](http://img.cochemist.com/ccimg/2900/2826-26-8_b.png)

