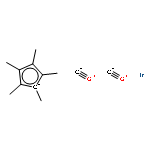
Co-reporter:Kajari Bera, Christopher J. Douglas, and Renee R. Frontiera
The Journal of Physical Chemistry Letters December 7, 2017 Volume 8(Issue 23) pp:5929-5929
Publication Date(Web):November 22, 2017
DOI:10.1021/acs.jpclett.7b02769
Singlet fission generates multiple excitons from a single photon, which in theory can result in solar cell efficiencies with values above the Shockley–Queisser limit. Understanding the molecular structural dynamics during singlet fission will help to fabricate efficient organic photovoltaic devices. Here we use femtosecond stimulated Raman spectroscopy to reveal the structural evolution during the triplet separation in rubrene. We observe vibrational signatures of the correlated triplet pair, as well as shifting of the vibrational frequencies of the 1430 and 1542 cm–1 excited state modes, which increase by more than 25 cm–1 in 5 ps. Our results indicate that the correlated pair separation into two individual triplets occurs concurrently with the loss of electron density from the tetracene backbone in rubrene. This study provides new insights into the triplet separation process and proves the utility of structurally sensitive ultrafast vibrational techniques to understand the mechanism of singlet fission.
Co-reporter:W. Ruchira Silva and Renee R. Frontiera
Physical Chemistry Chemical Physics 2016 vol. 18(Issue 30) pp:20290-20297
Publication Date(Web):04 Jan 2016
DOI:10.1039/C5CP06195D
Ultrafast photo-induced charge-transfer reactions are fundamental to a number of photovoltaic and photocatalytic devices, yet the multidimensional nature of the reaction coordinate makes these processes difficult to model theoretically. Here we use femtosecond stimulated Raman spectroscopy to probe experimentally the structural changes occurring following photoexcitation in betaine-30, a canonical intramolecular charge-transfer complex. We observe changes in vibrational mode frequencies and amplitudes on the femtosecond timescale, which for some modes results in frequency shifts of over 20 cm−1 during the first 200 fs following photoexcitation. These rapid mode-specific frequency changes track the planarization of the molecule on the 400 ± 100 fs timescale. Oscillatory amplitude modulations of the observed high frequency Raman modes indicate coupling between specific high frequency and low frequency vibrational motions, which we quantify for 6 low frequency modes and 4 high frequency modes. Analysis of the mode-specific kinetics is suggestive of the existence of a newly discovered electronic state involved in a relaxation pathway, which may be a low-lying triplet state. These results directly track the multiple nuclear coordinates involved in betaine-30's reactive pathway, and should be of use in rationally designing molecular systems with rapid electron transfer processes.
Co-reporter:Nathaniel C. Brandt, Emily L. Keller, and Renee R. Frontiera
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 16) pp:3179-3185
Publication Date(Web):August 4, 2016
DOI:10.1021/acs.jpclett.6b01453
Hot electrons generated through plasmonic excitations in metal nanostructures show great promise for efficiently driving chemical reactions with light. However, the lifetime, yield, and mechanism of action of plasmon-generated hot electrons involved in a given photocatalytic process are not well understood. Here, we develop ultrafast surface-enhanced Raman scattering (SERS) as a direct probe of plasmon–molecule interactions in the plasmon-catalyzed dimerization of 4-nitrobenzenethiol to p,p′-dimercaptoazobenzene. Ultrafast SERS probing of these molecular reporters in plasmonic hot spots reveals transient Fano resonances, which we attribute to near-field coupling of Stokes-shifted photons to hot electron-driven metal photoluminescence. Surprisingly, we find that hot spots that yield more photoluminescence are much more likely to drive the reaction, which indirectly proves that plasmon-generated hot electrons induce the photochemistry. These ultrafast SERS results provide insight into the relative reactivity of different plasmonic hot spot environments and quantify the ultrafast lifetime of hot electrons involved in plasmon-driven chemistry.
Co-reporter:James L. Brooks and Renee R. Frontiera
The Journal of Physical Chemistry C 2016 Volume 120(Issue 37) pp:20869-20876
Publication Date(Web):May 3, 2016
DOI:10.1021/acs.jpcc.6b02314
Plasmonic materials are exciting candidates for driving photochemical reactions, as they couple strongly with light across a wide range of the electromagnetic spectrum and can dramatically impact the photophysical properties of proximal molecular species. Plasmons have been shown to drive a number of photochemical reactions, but a detailed understanding of the mechanism is lacking in many cases. Here we investigate the effects of plasmonic field enhancement of the plasmon-driven conversion of 4-nitrobenzenethiol to 4,4′-dimercaptoazobenzene. By tuning the ensemble-averaged field enhancement of a plasmonic substrate, we quantify how the reaction yield and rate depend on the magnitude of the electric field. Surprisingly, we find no correlation of increased reaction rate or yield with greater field enhancement. Kinetic analysis of the reaction rate constants reveals a wide range of values, indicating that plasmonic excitation is not the rate-limiting step in this system. Additionally, we identify a competing degradation pathway that significantly contributes to the loss of reactant. This work identifies several factors that are critical in determining the overall efficiency of a plasmon-driven process and should help to lead to optimally designed plasmonic photocatalytic systems.
Co-reporter:W. Ruchira Silva, Christian T. Graefe, and Renee R. Frontiera
ACS Photonics 2016 Volume 3(Issue 1) pp:
Publication Date(Web):December 28, 2015
DOI:10.1021/acsphotonics.5b00467
We propose and implement a far-field spectroscopic system for imaging below the diffraction limit without the need for fluorescence labeling. Our technique combines concepts from Stimulated Emission Depletion (STED) microscopy and Femtosecond Stimulated Raman Spectroscopy (FSRS). The FSRS process generates signal through the creation of vibrational coherences, and here we use a toroidal-shaped decoherence pulse to eliminate vibrational signal from the edges of the focal spot. The nonlinear dependence on decoherence pulse power enables subdiffraction imaging. As in STED, the resolution is in theory infinitely small given infinite decoherence pulse power. Here, we first experimentally demonstrate that the photophysical principles behind our super-resolution Raman imaging method are sound. We then prove that addition of the decoherence pulse significantly improves the spatial resolution of our microscope, achieving values beyond the diffraction limit. We discuss future directions for this technique, including methods to reach resolution on the order of ten nanometers.
Co-reporter:Emily L. Keller, Nathaniel C. Brandt, Alyssa A. Cassabaum and Renee R. Frontiera
Analyst 2015 vol. 140(Issue 15) pp:4922-4931
Publication Date(Web):28 May 2015
DOI:10.1039/C5AN00869G
Ultrafast surface-enhanced Raman spectroscopy (SERS) with pico- and femtosecond time resolution has the ability to elucidate the mechanisms by which plasmons mediate chemical reactions. Here we review three important technological advances in these new methodologies, and discuss their prospects for applications in areas including plasmon-induced chemistry and sensing at very low limits of detection. Surface enhancement, arising from plasmonic materials, has been successfully incorporated with stimulated Raman techniques such as femtosecond stimulated Raman spectroscopy (FSRS) and coherent anti-Stokes Raman spectroscopy (CARS). These techniques are capable of time-resolved measurement on the femtosecond and picosecond time scale and can be used to follow the dynamics of molecules reacting near plasmonic surfaces. We discuss the potential application of ultrafast SERS techniques to probe plasmon-mediated processes, such as H2 dissociation and solar steam production. Additionally, we discuss the possibilities for high sensitivity SERS sensing using these stimulated Raman spectroscopies.
Co-reporter:W. Ruchira Silva, Emily L. Keller, and Renee R. Frontiera
Analytical Chemistry 2014 Volume 86(Issue 15) pp:7782
Publication Date(Web):June 30, 2014
DOI:10.1021/ac501701h
Surface-enhanced Raman spectroscopy (SERS) is a promising technique for in vivo bioanalyte detection, but accurate characterization of SERS biosensors can be challenging due to difficulties in differentiating resonance and surface enhancement contributions to the Raman signal. Here, we quantitate the resonance Raman cross-sections for a commonly used near-infrared SERS dye, 3,3′-diethylthiatricarbocyanine (DTTC). It is typically challenging to measure resonance Raman cross-sections for fluorescent dye molecules due to the overwhelming isoenergetic fluorescence signal. To overcome this issue, we used etalon-based femtosecond stimulated Raman spectroscopy, which is intrinsically designed to acquire a stimulated Raman signal without strong fluorescence or interference from signals resulting from other four-wave mixing pathways. Using this technique, we found that the cross-sections for most of the resonantly enhanced modes in DTTC exceed 10–25 cm2/molecule. These cross-sections lead to high signal magnitude SERS signals from even weakly enhancing SERS substrates, as much of what appears to be a SERS signal is actually coming from the intrinsically strong resonance Raman signal. Our work will lead to a more accurate determination of SERS enhancement factors and SERS substrate characterization in the biologically relevant near-infrared region, ultimately leading to a more widespread use of SERS for biosensing and bioimaging applications.
Co-reporter:W. Ruchira Silva and Renee R. Frontiera
Physical Chemistry Chemical Physics 2016 - vol. 18(Issue 30) pp:NaN20297-20297
Publication Date(Web):2016/01/04
DOI:10.1039/C5CP06195D
Ultrafast photo-induced charge-transfer reactions are fundamental to a number of photovoltaic and photocatalytic devices, yet the multidimensional nature of the reaction coordinate makes these processes difficult to model theoretically. Here we use femtosecond stimulated Raman spectroscopy to probe experimentally the structural changes occurring following photoexcitation in betaine-30, a canonical intramolecular charge-transfer complex. We observe changes in vibrational mode frequencies and amplitudes on the femtosecond timescale, which for some modes results in frequency shifts of over 20 cm−1 during the first 200 fs following photoexcitation. These rapid mode-specific frequency changes track the planarization of the molecule on the 400 ± 100 fs timescale. Oscillatory amplitude modulations of the observed high frequency Raman modes indicate coupling between specific high frequency and low frequency vibrational motions, which we quantify for 6 low frequency modes and 4 high frequency modes. Analysis of the mode-specific kinetics is suggestive of the existence of a newly discovered electronic state involved in a relaxation pathway, which may be a low-lying triplet state. These results directly track the multiple nuclear coordinates involved in betaine-30's reactive pathway, and should be of use in rationally designing molecular systems with rapid electron transfer processes.
.jpg)