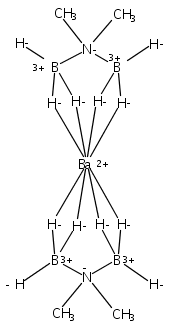
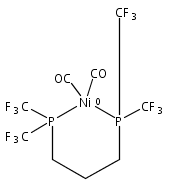
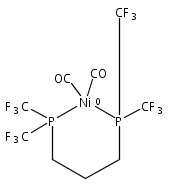
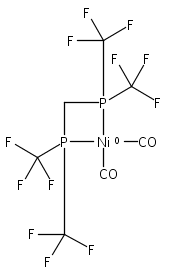
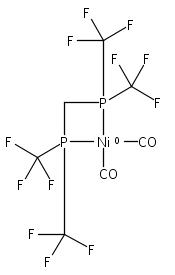
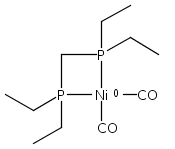
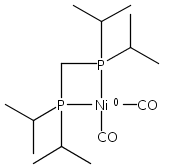
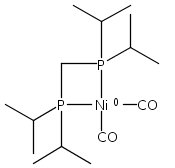
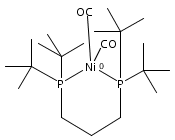
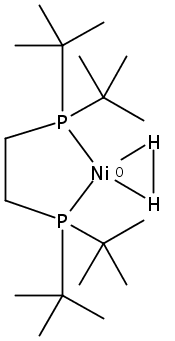
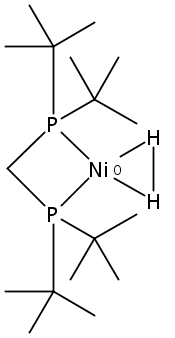
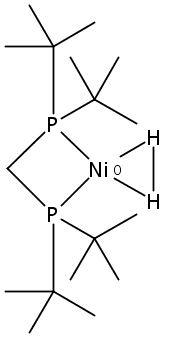
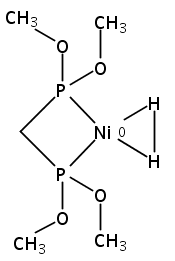
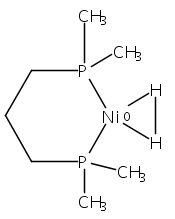
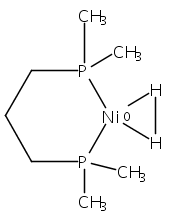
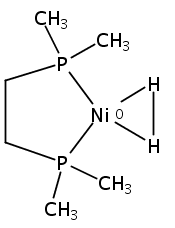
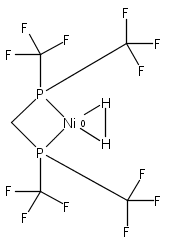
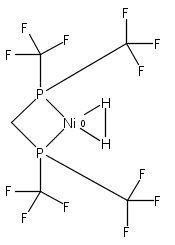
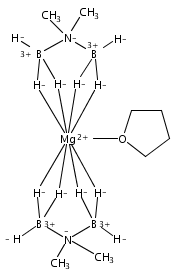
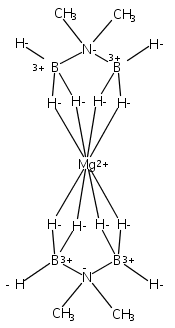
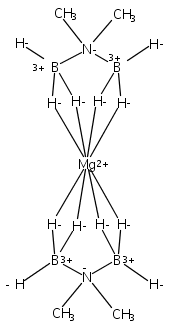
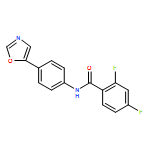
Co-reporter:Edward J. Reijerse, Cindy C. Pham, Vladimir Pelmenschikov, Ryan Gilbert-Wilson, Agnieszka Adamska-Venkatesh, Judith F. Siebel, Leland B. Gee, Yoshitaka Yoda, Kenji Tamasaku, Wolfgang Lubitz, Thomas B. Rauchfuss, and Stephen P. Cramer
Journal of the American Chemical Society March 29, 2017 Volume 139(Issue 12) pp:4306-4306
Publication Date(Web):March 14, 2017
DOI:10.1021/jacs.7b00686
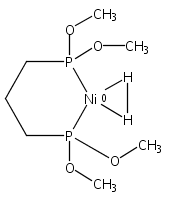
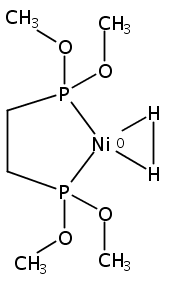
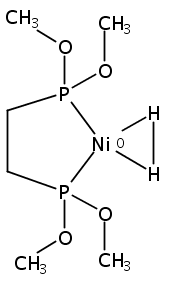
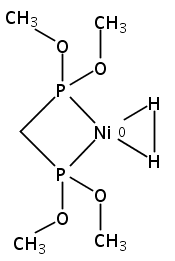
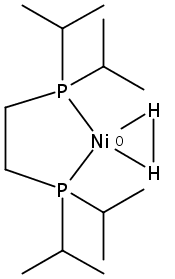
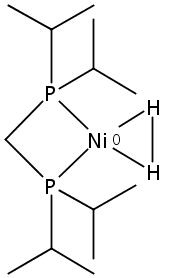
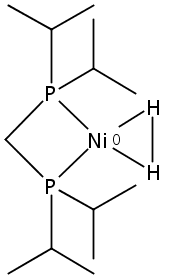
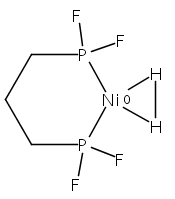
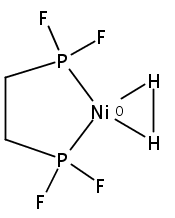
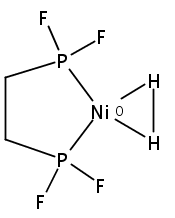
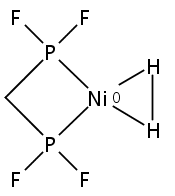
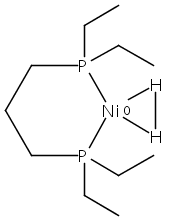
[FeFe]-hydrogenases catalyze the reversible reduction of protons to molecular hydrogen with extremely high efficiency. The active site (“H-cluster”) consists of a [4Fe–4S]H cluster linked through a bridging cysteine to a [2Fe]H subsite coordinated by CN– and CO ligands featuring a dithiol-amine moiety that serves as proton shuttle between the protein proton channel and the catalytic distal iron site (Fed). Although there is broad consensus that an iron-bound terminal hydride species must occur in the catalytic mechanism, such a species has never been directly observed experimentally. Here, we present FTIR and nuclear resonance vibrational spectroscopy (NRVS) experiments in conjunction with density functional theory (DFT) calculations on an [FeFe]-hydrogenase variant lacking the amine proton shuttle which is stabilizing a putative hydride state. The NRVS spectra unequivocally show the bending modes of the terminal Fe–H species fully consistent with widely accepted models of the catalytic cycle.
Co-reporter:Xin Yu, Chen-Ho Tung, and Wenguang Wang, Mioy T. Huynh, Danielle L. Gray, Sharon Hammes-Schiffer, and Thomas B. Rauchfuss
Organometallics June 12, 2017 Volume 36(Issue 11) pp:2245-2245
Publication Date(Web):May 18, 2017
DOI:10.1021/acs.organomet.7b00297
This study describes the structural, spectroscopic, and electrochemical properties of electronically unsymmetrical diiron hydrides. The terminal hydride Cp*Fe(pdt)Fe(dppe)(CO)H ([1(t-H)]0, Cp*– = Me5C5–, pdt2– = CH2(CH2S–)2, dppe = Ph2PC2H4PPh2) was prepared by hydride reduction of [Cp*Fe(pdt)Fe(dppe)(CO)(NCMe)]+. As established by X-ray crystallography, [1(t-H)]0 features a terminal hydride ligand. Unlike previous examples of terminal diiron hydrides, [1(t-H)]0 does not isomerize to the bridging hydride [1(μ-H)]0. Oxidation of [1(t-H)]0 gives [1(t-H)]+, which was also characterized crystallographically as its BF4– salt. Density functional theory (DFT) calculations indicate that [1(t-H)]+ is best described as containing an Cp*FeIII center. In solution, [1(t-H)]+ isomerizes to [1(μ-H)]+, as anticipated by DFT. Reduction of [1(μ-H)]+ by Cp2Co afforded the diferrous bridging hydride [1(μ-H)]0. Electrochemical measurements and DFT calculations indicate that the couples [1(t-H)]+/0 and [1(μ-H)]+/0 differ by 210 mV. Qualitative measurements indicate that [1(t-H)]0 and [1(μ-H)]0 are close in free energy. Protonation of [1(t-H)]0 in MeCN solution affords H2 even with weak acids via hydride transfer. In contrast, protonation of [1(μ-H)]0 yields 0.5 equiv of H2 by a proposed protonation-induced electron transfer process. Isotopic labeling indicates that μ-H/D ligands are inert.
Co-reporter:Noémie Lalaoui, Toby Woods, Thomas B. Rauchfuss, and Giuseppe Zampella
Organometallics June 12, 2017 Volume 36(Issue 11) pp:2054-2054
Publication Date(Web):May 16, 2017
DOI:10.1021/acs.organomet.7b00236
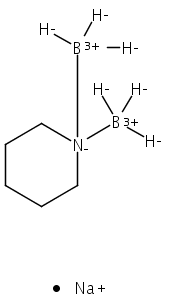
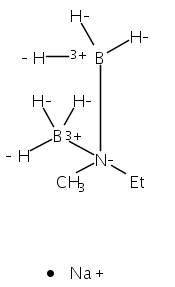
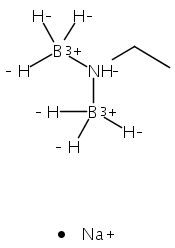
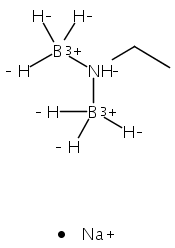
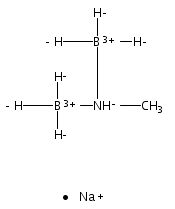
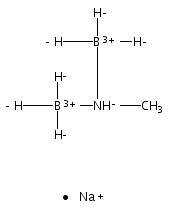
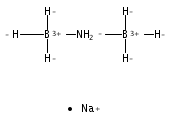
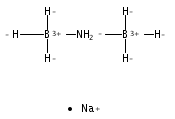
The azadithiolate complex Fe2[(SCH2)2NMe](CO)6 reacts with borane to give an initial 1:1 adduct, which spontaneously decarbonylates to give Fe2[(SCH2)2NMeBH3](CO)5. Featuring a Fe–H–B three-center, two-electron interaction, the pentacarbonyl complex is a structural model for H2 complexes invoked in the [FeFe]-hydrogenases. The pentacarbonyl compound is a “σ complex”, where a B–H σ bond serves as a ligand for iron. The structure of this σ complex was characterized by variable-temperature NMR spectroscopy and X-ray crystallography. Complementary to the 1:1 borane adduct is the quaternary ammonium complex [Fe2[(SCH2)2NMe2](CO)6]+, which was also characterized. It represents a kinetically robust analogue of the N-protonated amine cofactor, as indicated by its mild reduction potential.
Co-reporter:Casseday P. Richers;Jeffery A. Bertke
Dalton Transactions 2017 vol. 46(Issue 27) pp:8756-8762
Publication Date(Web):2017/07/11
DOI:10.1039/C6DT04205H
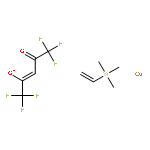
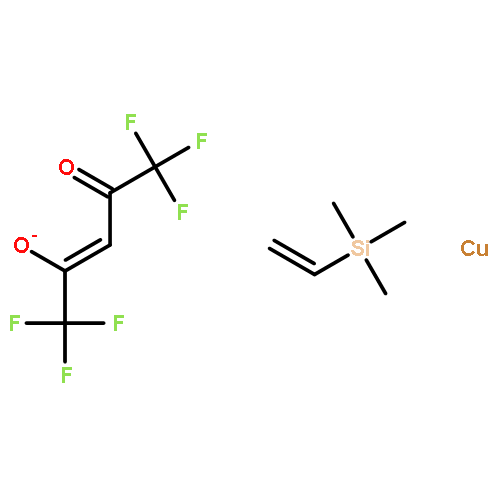
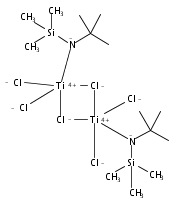
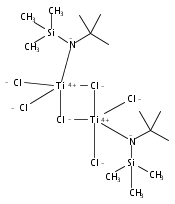
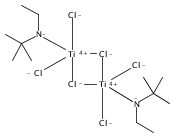
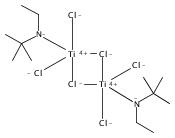
The paper describes three methods for the preparation of methoxysiloxide complexes, a rare class of complexes of relevance to room temperature vulcanization (RTV) of polysiloxanes. The salt metathesis reaction involves the use of the recently described reagent NaOSi(OMe)2Me with various metal chlorides to give Cp*2Ti[OSi(OMe)2Me](OMe), (Me,MeN2N)NiOSi(OMe)2Me, (IPr)CuOSi(OMe)2Me, and (triphos)CoOSi(OMe)2Me (Cp* = C5Me5, triphos = Me(CH2PPh2)3). Several attempted reactions gave methoxide complexes instead, a pathway that is attributed to the intermediacy of κ2-OSi(OMe)2Me species. The diol Cp*2Zr(OH)2 reacts with excess (MeO)3SiMe to give Cp*2Zr[OSi(OMe)2Me]2. In contrast the less nucleophilic Cp*2Ti(OH)2 was unreactive. The third route to methoxysiloxide complexes involves the reaction of Cp*2M(O)(py) with (MeO)3SiMe to give Cp*2M[OSi(OMe)2Me](OMe) in nearly quantitative yield (M = Ti, Zr). The structures of Cp*2Ti[OSi(OMe)2Me](OMe), Cp*2Zr[OSi(OMe)2Me](OMe), (IPr)CuOSi(OMe)2Me, and (triphos)CoOSi(OMe)2Me were confirmed by single crystal X-ray diffraction.
Co-reporter:Yulong Li and Thomas B. Rauchfuss
Chemical Reviews 2016 Volume 116(Issue 12) pp:7043
Publication Date(Web):June 3, 2016
DOI:10.1021/acs.chemrev.5b00669
Virtually all organosulfur compounds react with Fe(0) carbonyls to give the title complexes. These reactions are reviewed in light of major advances over the past few decades, spurred by interest in Fe2(μ-SR)2(CO)x centers at the active sites of the [FeFe]-hydrogenase enzymes. The most useful synthetic route to Fe2(μ-SR)2(CO)6 involves the reaction of thiols with Fe2(CO)9 and Fe3(CO)12. Such reactions can proceed via mono-, di-, and triiron intermediates. The reactivity of Fe(0) carbonyls toward thiols is highly chemoselective, and the resulting dithiolato complexes are fairly rugged. Thus, many complexes tolerate further synthetic elaboration directed at the organic substituents. A second major route involves alkylation of Fe2(μ-S2)(CO)6, Fe2(μ-SH)2(CO)6, and Li2Fe2(μ-S)2(CO)6. This approach is especially useful for azadithiolates Fe2[(μ-SCH2)2NR](CO)6. Elaborate complexes arise via addition of the FeSH group to electrophilic alkenes, alkynes, and carbonyls. Although the first example of Fe2(μ-SR)2(CO)6 was prepared from ferrous reagents, ferrous compounds are infrequently used, although the Fe(II)(SR)2 + Fe(0) condensation reaction is promising. Almost invariably low-yielding, the reaction of Fe3(CO)12, S8, and a variety of unsaturated substrates results in C–H activation, affording otherwise inaccessible derivatives. Thiones and related C═S-containing reagents are highly reactive toward Fe(0), often giving complexes derived from substituted methanedithiolates and C–H activation.
Co-reporter:Olbelina A. Ulloa; Mioy T. Huynh; Casseday P. Richers; Jeffery A. Bertke; Mark J. Nilges; Sharon Hammes-Schiffer
Journal of the American Chemical Society 2016 Volume 138(Issue 29) pp:9234-9245
Publication Date(Web):June 21, 2016
DOI:10.1021/jacs.6b04579
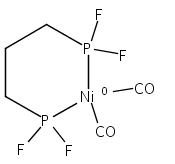
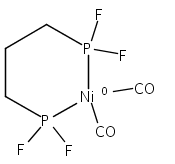
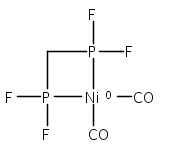
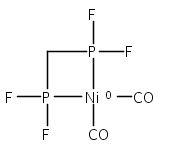
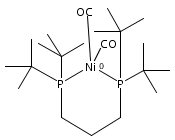
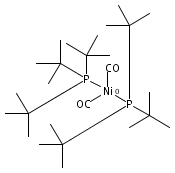
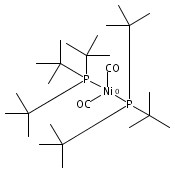
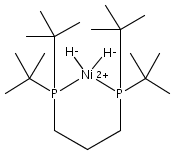
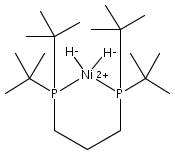
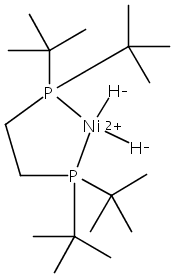
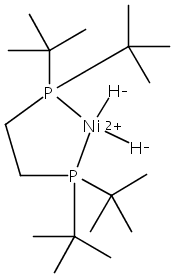
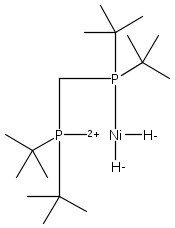
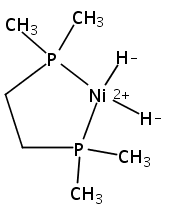
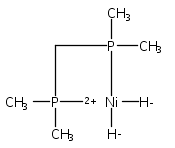
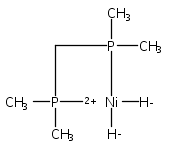
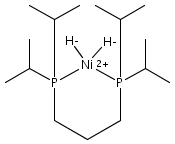
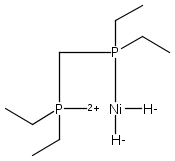
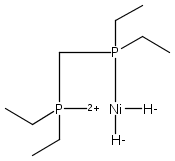
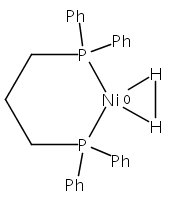
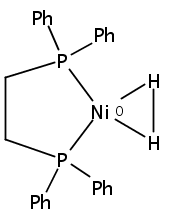
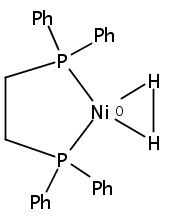
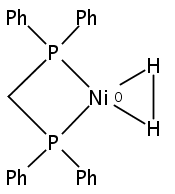
The intermediacy of a reduced nickel–iron hydride in hydrogen evolution catalyzed by Ni–Fe complexes was verified experimentally and computationally. In addition to catalyzing hydrogen evolution, the highly basic and bulky (dppv)Ni(μ-pdt)Fe(CO)(dppv) ([1]0; dppv = cis-C2H2(PPh2)2) and its hydride derivatives have yielded to detailed characterization in terms of spectroscopy, bonding, and reactivity. The protonation of [1]0 initially produces unsym-[H1]+, which converts by a first-order pathway to sym-[H1]+. These species have C1 (unsym) and Cs (sym) symmetries, respectively, depending on the stereochemistry of the octahedral Fe site. Both experimental and computational studies show that [H1]+ protonates at sulfur. The S = 1/2 hydride [H1]0 was generated by reduction of [H1]+ with Cp*2Co. Density functional theory (DFT) calculations indicate that [H1]0 is best described as a Ni(I)–Fe(II) derivative with significant spin density on Ni and some delocalization on S and Fe. EPR spectroscopy reveals both kinetic and thermodynamic isomers of [H1]0. Whereas [H1]+ does not evolve H2 upon protonation, treatment of [H1]0 with acids gives H2. The redox state of the “remote” metal (Ni) modulates the hydridic character of the Fe(II)–H center. As supported by DFT calculations, H2 evolution proceeds either directly from [H1]0 and external acid or from protonation of the Fe–H bond in [H1]0 to give a labile dihydrogen complex. Stoichiometric tests indicate that protonation-induced hydrogen evolution from [H1]0 initially produces [1]+, which is reduced by [H1]0. Our results reconcile the required reductive activation of a metal hydride and the resistance of metal hydrides toward reduction. This dichotomy is resolved by reduction of the remote (non-hydride) metal of the bimetallic unit.
Co-reporter:Casseday P. Richers, Jeffery Bertke, and Thomas B. Rauchfuss
Inorganic Chemistry 2016 Volume 55(Issue 12) pp:5744
Publication Date(Web):June 3, 2016
DOI:10.1021/acs.inorgchem.6b00874
The commercially practiced conversion of trimethoxymethylsilane (MTM) to [OSi(OMe)Me)]n polymers and resins is assumed to proceed via the silanol (MeO)2MeSiOH. Access to this crucial silanol is gained via the synthesis of (MeO)2MeSiONa, the first methoxysilanoate to be crystallographically characterized. Mild protonation of this silanoate gives (MeO)2MeSiOH, which is shown to condense with (MeO)2MeSiOH but not with MTM. Condensation generates reactive disiloxanols but does not produce symmetric disiloxanes. Parallel results were obtained with (MeO)2PhSiOH.
Co-reporter:Edward Reijerse;Wolfgang Lubitz
Inorganic Chemistry 2016 Volume 55(Issue 2) pp:419-431
Publication Date(Web):September 30, 2015
DOI:10.1021/acs.inorgchem.5b01662
A new class of synthetic models for the active site of [NiFe]-hydrogenases are described. The NiI/II(SCys)2 and FeII(CN)2CO sites are represented with (RC5H4)NiI/II and FeII(diphos)(CO) modules, where diphos = 1,2-C2H4(PPh2)2(dppe) or cis-1,2-C2H2(PPh2)2(dppv). The two bridging thiolate ligands are represented by CH2(CH2S)22– (pdt2–), Me2C(CH2S)22– (Me2pdt2–), and (C6H5S)22–. The reaction of Fe(pdt)(CO)2(dppe) and [(C5H5)3Ni2]BF4 affords [(C5H5)Ni(pdt)Fe(dppe)(CO)]BF4 ([1a]BF4). Monocarbonyl [1a]BF4 features an S = 0 NiIIFeII center with five-coordinated iron, as proposed for the Ni-SIa state of the enzyme. One-electron reduction of [1a]+ affords the S = 1/2 derivative [1a]0, which, according to density functional theory (DFT) calculations and electron paramagnetic resonance and Mössbauer spectroscopies, is best described as a NiIFeII compound. The NiIFeII assignment matches that for the Ni-L state in [NiFe]-hydrogenase, unlike recently reported NiIIFeI-based models. Compound [1a]0 reacts with strong acids to liberate 0.5 equiv of H2 and regenerate [1a]+, indicating that H2 evolution is catalyzed by [1a]0. DFT calculations were used to investigate the pathway for H2 evolution and revealed that the mechanism can proceed through two isomers of [1a]0 that differ in the stereochemistry of the Fe(dppe)CO center. Calculations suggest that protonation of [1a]0 (both isomers) affords NiIII–H–FeII intermediates, which represent mimics of the Ni-C state of the enzyme.
Co-reporter:Xiaoyuan Zhou, Bryan E. Barton, Geoffrey M. Chambers, and Thomas B. Rauchfuss , Federica Arrigoni and Giuseppe Zampella
Inorganic Chemistry 2016 Volume 55(Issue 7) pp:3401-3412
Publication Date(Web):March 21, 2016
DOI:10.1021/acs.inorgchem.5b02789
The complexes Fe2(pdt)(CNR)6 (pdt2– = CH2(CH2S–)2) were prepared by thermal substitution of the hexacarbonyl complex with the isocyanides RNC for R = C6H4-4-OMe (1), C6H4-4-Cl (2), Me (3). These complexes represent electron-rich analogues of the parent Fe2(pdt)(CO)6. Unlike most substituted derivatives of Fe2(pdt)(CO)6, these isocyanide complexes are sterically unencumbered and have the same idealized symmetry as the parent hexacarbonyl derivatives. Like the hexacarbonyls, the stereodynamics of 1–3 involve both turnstile rotation of the Fe(CNR)3 as well as the inversion of the chair conformation of the pdt ligand. Structural studies indicate that the basal isocyanide has nonlinear CNC bonds and short Fe–C distances, indicating that they engage in stronger Fe–C π-backbonding than the apical ligands. Cyclic voltammetry reveals that these new complexes are far more reducing than the hexacarbonyls, although the redox behavior is complex. Estimated reduction potentials are E1/2 ≈ −0.6 ([2]+/0), −0.7 ([1]+/0), and −1.25 ([3]+/0). According to DFT calculations, the rotated isomer of 3 is only 2.2 kcal/mol higher in energy than the crystallographically observed unrotated structure. The effects of rotated versus unrotated structure and of solvent coordination (THF, MeCN) on redox potentials were assessed computationally. These factors shift the redox couple by as much as 0.25 V, usually less. Compounds 1 and 2 protonate with strong acids to give the expected μ-hydrides [H1]+ and [H2]+. In contrast, 3 protonates with [HNEt3]BArF4 (pKaMeCN = 18.7) to give the aminocarbyne [Fe2(pdt)(CNMe)5(μ-CN(H)Me)]+ ([3H]+). According to NMR measurements and DFT calculations, this species adopts an unsymmetrical, rotated structure. DFT calculations further indicate that the previously described carbyne complex [Fe2(SMe)2(CO)3(PMe3)2(CCF3)]+ also adopts a rotated structure with a bridging carbyne ligand. Complex [3H]+ reversibly adds MeNC to give [Fe2(pdt)(CNR)6(μ-CN(H)Me)]+ ([3H(CNMe)]+). Near room temperature, [3H]+ isomerizes to the hydride [(μ-H)Fe2(pdt)(CNMe)6]+ ([H3]+) via a first-order pathway.
Co-reporter:Wan-Yi Chu, Ryan Gilbert-Wilson, and Thomas B. Rauchfuss, Maurice van Gastel and Frank Neese
Organometallics 2016 Volume 35(Issue 17) pp:2900-2914
Publication Date(Web):August 22, 2016
DOI:10.1021/acs.organomet.6b00457
A family of CoCl2(PNpy) complexes were prepared, where PNpy = 2-iminopyridyl-phosphine ligands derived from aminoalkyl and aminoaryl phosphines and 2-keto- and 2-formylpyridines. Reduction of CoCl2(PNpy) complexes in the presence of PPh3 gave CoH(PNpy)(PPh3) and CoMe(PNpy)(PPh3), which were active for hydrofunctionalization of alkenes. According to DFT calculations, the CoMe(PNpy)(PPh3) complexes are best described as Co(II) derivatives of the anion [PNpy]−, with a labile PPh3 coligand. Metalation of Na[Ph2PC2NHpy] gave the dimers [CoCl(Ph2PC2NHpy)]2. Monomeric complexes catalyze hydrosilylation of 1-octene with Ph2SiH2, with the CoCl2(iPr2PC3NHpy)/2NaBEt3H system exhibiting the highest rate and selectivity for anti-Markovnikov product. In situ NMR studies established the following: (i) silanes protonolyze catalyst precursors to give the Co-silyl complexes Co(SiR3)(Ph2PC6H4NPhpy)(PPh3), (ii) alkenes compete with PPh3 to give Co(SiHPh2)(Ph2PC6H4NPhpy)(η2-alkene), (iii) ethylene inserts into the Co–Si bond to give Co(CH2CH2SiR3)(Ph2PC6H4NPhpy)(PPh3).
Co-reporter:Geoffrey M. Chambers and Thomas B. Rauchfuss, Federica Arrigoni and Giuseppe Zampella
Organometallics 2016 Volume 35(Issue 5) pp:836-846
Publication Date(Web):February 25, 2016
DOI:10.1021/acs.organomet.6b00068
The effects of simple thiolates vs chelating dithiolates on M–M bonding, redox potentials, and synthetic outcomes have been probed experimentally and computationally. Nickelocene (Cp2Ni) has long been known to react with simple thiols to give diamagnetic Cp2Ni2(SR)2 with planar Ni2S2 cores and long Ni- - -Ni distances. Ethane- and propanedithiol (edtH2 and pdtH2, respectively) instead give di-, tri-, and pentanickel complexes, with nonplanar Ni2S2 cores. The 36e Cp2Ni2(pdt) (1pdt) adopts a symmetrical butterfly Ni2S2 structure. Variable-temperature NMR spectra indicate that 1pdt possesses a thermally accessible triplet state (ΔG = 2.65(5) kcal/mol) in equilibrium with a diamagnetic ground state. DFT calculations indicate that the singlet–triplet gap is highly sensitive to the nonplanarity of the Ni2S2 core. The calculations further reveal that only the high-spin form of 1pdt features Ni–Ni bonding, which is unprecedented. Cp2Ni3(pdt)2 (2pdt), which derives from 1pdt, crystallized as cis and trans isomers, both with a central Ni(pdt)22– unit that is S,S-chelated to two CpNi+ centers. Reaction of Cp2Ni with 1,2-ethanedithiol (H2edt) and 1,2-benzenedithiol (bdtH2) exclusively gave the trinickel species 2edt and 2bdt, which are structurally analogous to cis-2pdt. Solutions of 2edt are unstable, depositing crystals of Cp2Ni5(edt)4 (3edt). Cyclic voltammetric studies show that the Ni2 species oxidize readily to give the mixed-valence cations [1pdt]+ and [1edt]+. Crystallographic and EPR analyses indicate that these cations are delocalized mixed-valence Ni(II)–Ni(III) species. Oxidation of Cp2Ni2(SEt)2, which features a planar Ni2S2 core, afforded a mixed-valence cation, showing the pyramidal Ni2S2 core observed in [1pdt]0/+ and [1edt]0/+. Although not obtained from the Cp2Ni/H2edt reaction, the neutral complex 1edt was obtained by reduction of [1edt]+. Variable-temperature NMR measurements and DFT calculations indicate that the triplet is further stabilized in this highly pyramidalized species.
Co-reporter:Wan-Yi Chu, Casseday P. Richers, Elizabeth R. Kahle, and Thomas B. Rauchfuss, Federica Arrigoni and Giuseppe Zampella
Organometallics 2016 Volume 35(Issue 17) pp:2782-2792
Publication Date(Web):August 19, 2016
DOI:10.1021/acs.organomet.6b00318
Fundamental reactions of imino-phosphine ligands were elucidated through studies on Ph2PC6H4CH═NC6H4-4-Cl (PCHNArCl) complexes of iron(0), iron(I), and iron(II). The reaction of PCHNArCl with Fe(bda)(CO)3 gives Fe(PCHNArCl)(CO)3 (1), featuring an η2-imine. DNMR studies, its optical properties, and DFT calculations suggest that 1 racemizes on the NMR time scale via an achiral N-bonded imine intermediate. The N-imine isomer is more stable in Fe(PCHNArOMe)(CO)3 (1OMe), which crystallized despite being the minor isomer in solution. Protonation of 1 by HBF4·Et2O gave the iminium complex [1H]BF4. The related diphosphine complex Fe(PCHNArCl)(PMe3)(CO)2 (2), which features an η2-imine, was shown to also undergo N protonation. Oxidation of 1 and 2 with FcBF4 gave the Fe(I) compounds [1]BF4 and [2]BF4. The oxidation-induced change in hapticity of the imine from η2 in [1]0 to κ1 in [1]+ was verified crystallographically. Substitution of a CO ligand in 1 with PCHNArCl gave Fe[P2(NArCl)2](CO)2 (3), which contains the tetradentate diamidodiphosphine ligand. This C–C coupling is reversed by chemical oxidation of 3 with FcOTf. The oxidized product of [Fe(PCHNArCl)2(CO)2]2+ ([4]2+) was prepared independently by the reaction of [1]+, PCHNArCl, and Fc+. The C–C scission is proposed to proceed concomitantly with the reduction of Fe(II) via an intermediate related to [2]+.
Co-reporter:Thomas B. Rauchfuss
Accounts of Chemical Research 2015 Volume 48(Issue 7) pp:2107
Publication Date(Web):June 16, 2015
DOI:10.1021/acs.accounts.5b00177
The [FeFe] hydrogenases (H2ases) catalyze the redox reaction that interconverts protons and H2. This area of biocatalysis has attracted attention because the metal-based chemistry is unusual, and the reactions have practical implications. The active site consists of a [4Fe–4S] cluster bridged to a [Fe2(μ-dithiolate)(CN)2(CO)3]z center (z = 1– and 2−). The dithiolate cofactor is [HN(CH2S)2]2–, called the azadithiolate ([adtH]2–). Although many derivatives of Fe2(SR)2(CO)6–xLx are electrocatalysts for the hydrogen evolution reaction (HER), most operate by slow nonbiomimetic pathways. Biomimetic hydrogenogenesis is thought to involve intermediates, wherein the hydride substrate is adjacent to the amine of the adtH, being bonded to only one Fe center.Formation of terminal hydride complexes is favored when the diiron carbonyl models contain azadithiolate. Although unstable in the free state, the adt cofactor is stable once it is affixed to the Fe2 center. It can be prepared by alkylation of Fe2(SH)2(CO)6 with formaldehyde in the presence of ammonia (to give adtH derivatives) or amines (to give adtR derivatives). Weak acids protonate Fe2(adtR)(CO)2(PR3)4 to give terminal hydrido (term-H) complexes. In contrast, protonation of the related 1,3-propanedithiolate (pdt2−) complexes Fe2(pdt)(CO)2(PR3)4 requires strong acids. The amine in the azadithiolate is a kinetically fast base, relaying protons to and from the iron, which is a kinetically slow base. The crystal structure of the doubly protonated model [(term-H)Fe2(HadtH)(CO)2(dppv)2]2+ confirms the presence of both ammonium and terminal hydrido centers, which interact through a dihydrogen bond (dppv = cis-C2H2(PPh2)2). DFT calculations indicate that this H---H interaction is sensitive to the counterions and is strengthened upon reduction of the diiron center. For the monoprotonated models, the hydride [(term-H)Fe2(adtH)(CO)2(dppv)2]+ exists in equilibrium with the ammonium tautomer [Fe2(HadtH)(CO)2(dppv)2]+. Both [(term-H)Fe2(HadtH)(CO)2(dppv)2]2+ and [(term-H)Fe2(adtH)(CO)2(dppv)2]+ are highly active electrocatalysts for HER. Catalysis is initiated by reduction of the diferrous center, which induces coupling of the protic ammonium center and the hydride ligand. In contrast, the propanedithiolate [(term-H)Fe2(pdt)(CO)2(dppv)2]+ is a poor electrocatalyst for HER.Oxidation of H2 has been demonstrated, starting with models for the oxidized state (“Hox”), for example, [Fe2(adtH)(CO)3(dppv)(PMe3)]+. Featuring a distorted Fe(II)Fe(I) center, this Hox model reacts slowly with high pressures of H2 to give [(μ-H)Fe2(adtH)(CO)3(dppv)(PMe3)]+. Highlighting the role of the proton relay, the propanedithiolate [Fe2(pdt)(CO)3(dppv)(PMe3)]+ is unreactive toward H2. The Hox-model + H2 reaction is accelerated in the presence of ferrocenium salts, which simulate the role of the attached [4Fe–4S] cluster. The redox-complemented complex [Fe2(adtBn)(CO)3(dppv)(FcP*)]n+ catalyzes both proton reduction and hydrogen oxidation (FcP* = (C5Me5)Fe(C5Me4CH2PEt2)).
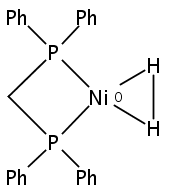
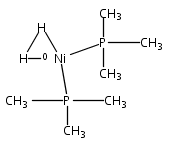
Co-reporter:Ryan Gilbert-Wilson; Judith F. Siebel; Agnieszka Adamska-Venkatesh; Cindy C. Pham; Edward Reijerse; Hongxin Wang; Stephen P. Cramer; Wolfgang Lubitz
Journal of the American Chemical Society 2015 Volume 137(Issue 28) pp:8998-9005
Publication Date(Web):June 19, 2015
DOI:10.1021/jacs.5b03270
The preparation and spectroscopic characterization of a CO-inhibited [FeFe] hydrogenase with a selectively 57Fe-labeled binuclear subsite is described. The precursor [57Fe2(adt)(CN)2(CO)4]2– was synthesized from the 57Fe metal, S8, CO, (NEt4)CN, NH4Cl, and CH2O. (Et4N)2[57Fe2(adt)(CN)2(CO)4] was then used for the maturation of the [FeFe] hydrogenase HydA1 from Chlamydomonas reinhardtii, to yield the enzyme selectively labeled at the [2Fe]H subcluster. Complementary 57Fe enrichment of the [4Fe-4S]H cluster was realized by reconstitution with 57FeCl3 and Na2S. The Hox-CO state of [257Fe]H and [457Fe-4S]H HydA1 was characterized by Mössbauer, HYSCORE, ENDOR, and nuclear resonance vibrational spectroscopy.
Co-reporter:Joyee Mitra, Xiaoyuan Zhou and Thomas Rauchfuss
Green Chemistry 2015 vol. 17(Issue 1) pp:307-313
Publication Date(Web):16 Sep 2014
DOI:10.1039/C4GC01520G
The diverse reactivity of 5-hydroxymethylfural (HMF) in Pd/C-catalyzed reactions is described with emphasis on the role of additives that affect selectivity. Three broad reactions are examined: decarbonylation, hydrogenation, and hydrogenolysis. Especially striking are the multiple roles of formic acid in hydrogenolysis/hydrogenation and in suppressing decarbonylation, as illustrated by the conversion of HMF to DMF. Hydrogenation of the furan ring is suppressed by CO2 and carboxylic acids. These results emphasize the utility of Pd/C as a convenient catalyst for upgradation of cellulosic biomass.
Co-reporter:Ryan Gilbert-Wilson, Wan-Yi Chu, and Thomas B. Rauchfuss
Inorganic Chemistry 2015 Volume 54(Issue 11) pp:5596-5603
Publication Date(Web):May 15, 2015
DOI:10.1021/acs.inorgchem.5b00692
A series of phosphine-diimine ligands were synthesized by the condensation of 2-(diphenylphosphino)aniline (PNH2) with a variety of formyl and ketopyridines. Condensation of PNH2 with acetyl- and benzoylpyridine yielded the Ph2P(C6H4)N═C(R)(C5H4N), respectively abbreviated PNMepy and PNPhpy. With ferrous halides, PNPhpy gave the complexes FeX2(PNPhpy) (X = Cl, Br). Condensation of pyridine carboxaldehyde and its 6-methyl derivatives with PNH2 was achieved using a ferrous template, affording low-spin complexes [Fe(PNHpyR)2]2+ (R = H, Me). Dicarbonyls Fe(PNRpy)(CO)2 were produced by treating PNMepy with Fe(benzylideneacetone)(CO)3 and reduction of FeX2(PNPhpy) with NaBEt3H under a CO atmosphere. Cyclic voltammetric studies show that the [FeL3(CO)2]0/– and [FeL3(CO)2]+/0 couples are similar for a range of tridentate ligands, but the PNPhpy system uniquely sustains two one-electron reductions. Treatment of Fe(PNPhpy)X2 with NaBEt3H gave active catalysts for the hydroboration of 1-octene with pinacolborane. Similarly, these catalysts proved active for the addition of diphenylsilane, but not HSiMe(OSiMe3)2, to 1-octene and vinylsilanes. Evidence is presented that catalysis occurs via iron hydride complexes of intact PNPhpy.
Co-reporter:Raja Angamuthu, Chi-Shian Chen, Tyler R. Cochrane, Danielle L. Gray, David Schilter, Olbelina A. Ulloa, and Thomas B. Rauchfuss
Inorganic Chemistry 2015 Volume 54(Issue 12) pp:5717-5724
Publication Date(Web):May 22, 2015
DOI:10.1021/acs.inorgchem.5b00290
Experiments are described that probe the stability of N-substituted derivatives of the azadithiolate cofactor recently confirmed in the [FeFe] hydrogenases (Berggren, G., et al. Nature 2013, 499, 66). Acid-catalyzed hydrolysis of bis(thioester) BnN(CH2SAc)2 gives [BnNCH2SCH2]2 rather than azadithiol BnN(CH2SH)2. Treatment of BnN(CH2SAc)2 with NaOtBu generates BnN(CH2SNa)2, which was trapped with NiCl2(diphos) (diphos = 1,2-C2H4(PR2)2; R = Ph (dppe) and Cy (dcpe)) to give fully characterized complexes Ni[(SCH2)2NBn](diphos). The related N-aryl derivative Ni[(SCH2)2NC6H4Cl](diphos) was prepared analogously from 4-ClC6H4N(CH2SAc)2, NaOtBu, and NiCl2(dppe). Crystallographic analysis confirmed that these rare nonbridging [adtR]2– complexes feature distorted square planar Ni centers. The analogue Pd[(SCH2)2NBn](dppe) was also prepared. 31P NMR analysis indicates that Ni[(SCH2)2NBn](dppe) has basicity comparable to typical amines. As shown by cyclic voltammetry, the couple [M[(SCH2)2NBn](dppe)]+/0 is reversible near −2.0 V versus Fc+/0. The wave shifts to −1.78 V upon N-protonation. In the presence of CF3CO2H, Ni[(SCH2)2NBn](dppe) catalyzes hydrogen evolution at rate of 22 s–1 in the acid-independent regime, at room temperature in CH2Cl2 solution. In contrast to the instability of RN(CH2SH)2 (R = alkyl, aryl), the dithiol of tosylamide TsN(CH2SH)2 proved sufficiently stable to allow full characterization. This dithiol reacts with Fe3(CO)12 and, in the presence of base, NiCl2(dppe) to give Fe2[(SCH2)2NTs](CO)6 and Ni[(SCH2)2NTs](dppe), respectively.
Co-reporter:Wan-Yi Chu, Xiaoyuan Zhou, and Thomas B. Rauchfuss
Organometallics 2015 Volume 34(Issue 9) pp:1619-1626
Publication Date(Web):February 6, 2015
DOI:10.1021/om501152h
This report describes examples of combined Fe- and O-centered reactivity of Fe(P2O2)(CO)2 (1), where P2O2 is the diphosphinoglycolate (Ph2PC6H4CHO)22–. This 18e low-spin ferrous dialkoxide undergoes substitution of CO to give the labile monosubstituted derivatives Fe(P2O2)(CO)(L) (L = PMe3, pyridine, MeCN). Treatment of Fe(P2O2)(CO)2 with Brønsted acids results in stepwise O-protonation, affording rare examples of low-spin Fe(II) complexes containing alcohol ligands. Substitution reactions with amides (RC(O)NH2) proceeds with binding of the carbonyl and formation of an intramolecular hydrogen bond between NH and the neighboring alkoxo ligand. This two-site binding was confirmed with crystallographic characterization of the thiourea-substituted derivative. Fe(P2O2)(CO)2 reacts with Ph2SiH2 to give the O-silylated hydrido complex, which is inactive for hydrosilylation. The monocarbonyl derivatives Fe(P2O2)(CO)(L) (L = NCMe, PMe3, acetamide) are precursors to catalysts for the hydrosilylation of benzaldehyde, acetophenone, and styrene.
Co-reporter:Mioy T. Huynh ; David Schilter ; Sharon Hammes-Schiffer
Journal of the American Chemical Society 2014 Volume 136(Issue 35) pp:12385-12395
Publication Date(Web):August 5, 2014
DOI:10.1021/ja505783z
Theory and experiment indicate that the protonation of reduced NiFe dithiolates proceeds via a previously undetected isomer with enhanced basicity. In particular, it is proposed that protonation of (OC)3Fe(pdt)Ni(dppe) (1; pdt2– = –S(CH2)3S–; dppe = Ph2P(CH2)2PPh2) occurs at the Fe site of the two-electron mixed-valence Fe(0)Ni(II) species, not the Fe(I)-Ni(I) bond for the homovalence isomer of 1. The new pathway, which may have implications for protonation of other complexes and clusters, was uncovered through studies on the homologous series L(OC)2Fe(pdt)M(dppe), where M = Ni, Pd (2), and Pt (3) and L = CO, PCy3. Similar to 1, complexes 2 and 3 undergo both protonation and 1e– oxidation to afford well-characterized hydrides ([2H]+ and [3H]+) and mixed-valence derivatives ([2]+ and [3]+), respectively. Whereas the Pd site is tetrahedral in 2, the Pt site is square-planar in 3, indicating that this complex is best described as Fe(0)Pt(II). In view of the results on 2 and 3, the potential energy surface of 1 was reinvestigated with density functional theory. These calculations revealed the existence of an energetically accessible and more basic Fe(0)Ni(II) isomer with a square-planar Ni site.
Co-reporter:Giuseppe Zampella
Journal of the American Chemical Society 2014 Volume 136(Issue 15) pp:5773-5782
Publication Date(Web):March 22, 2014
DOI:10.1021/ja501366j
Mechanisms for biological and bioinspired dihydrogen activation and production often invoke the intermediacy of diiron dithiolato dihydrides. The first example of such a Fe2(SR)2H2 species is provided by the complex [(term-H)(μ-H)Fe2(pdt)(CO)(dppv)2] ([H1H]0). Spectroscopic and computational studies indicate that [H1H]0 contains both a bridging hydride and a terminal hydride, which, notably, occupies a basal site. The synthesis begins with [(μ-H)Fe2(pdt)(CO)2(dppv)2]+ ([H1(CO)]+), which undergoes substitution to afford [(μ-H)Fe2(pdt)(CO)(NCMe)(dppv)2]+ ([H1(NCMe)]+). Upon treatment of [H1(NCMe)]+ with borohydride salts, the MeCN ligand is displaced to afford [H1H]0. DNMR (EXSY, SST) experiments on this complex show that the terminal and bridging hydride ligands interchange intramolecularly at a rate of 1 s–1 at −40 °C. The compound reacts with D2 to afford [D1D]0, but not mixed isotopomers such as [H1D]0. The dihydride undergoes oxidation with Fc+ under CO to give [1(CO)]+ and H2. Protonation in MeCN solution gives [H1(NCMe)]+ and H2. Carbonylation converts [H1H]0 into [1(CO)]0.
Co-reporter:Geoffrey M. Chambers, Joyee Mitra, Thomas B. Rauchfuss, and Matthias Stein
Inorganic Chemistry 2014 Volume 53(Issue 8) pp:4243-4249
Publication Date(Web):March 31, 2014
DOI:10.1021/ic500389p
This study describes the characterization of a mixed-valence RuII/NiI complex, a structural model for the Ni–L state of the [NiFe]hydrogenases. One-electron oxidation of (cymene)Ru(μ-pdt)Ni(diphos) ([1]0, diphos = dppe, C2H4(PPh2)2; [2]0, diphos = dcpe, C2H4(P(C6H11)2)2] affords the mixed-valence cations [(cymene)Ru(pdt)Ni(diphos)]+ ([1]+ and [2]+). Crystallographic and spectroscopic measurements indicate that these cations are described as RuII/NiI. Although [1]0 and [1]+ are very similar structurally, the following changes are notable: the Ni–P distances elongate upon oxidation, and the Ru–Ni distance changes insignificantly. The molecular and electronic structures of the Ni center in [1]+ approaches that observed in the [NiFe]hydrogenases. Density functional theory calculations indicate that [1]0 is best described as RuII/Ni0, consistent with its oxidation to RuII/NiI in [1]+. The fast electron self-exchange rate of 107 M–1 s–1 between [1]0 and [1]+ suggests minor reorganization, more consistent with a Ni0/NiI oxidation state change than a NiI/NiII couple. In solution, [1]+ slowly converts to [H1]+ and [1-H]+, with the latter being a complex of the thioaldehyde SCHCH2CH2S arising from C–H activation of the pdt backbone. Treatment of [1]+ with the H-atom abstracting reagent 2,2,6,6-tetramethylpiperidine-1-oxy also gives [1-H]+.
Co-reporter:Tai Lin;Olbelina A. Ulloa;Danielle L. Gray
European Journal of Inorganic Chemistry 2014 Volume 2014( Issue 25) pp:4109-4114
Publication Date(Web):
DOI:10.1002/ejic.201402413
Abstract
The reactions of [Fe3(CO)12] with the dithiadiazacyclooctanes [SCH2N(R)CH2]2 (R = Me, Bn) afford the diiron complexes [Fe2{SCH2N(R)CH2}(CO)6] (1Me, 1Bu). The methyl derivative 1Me was characterized crystallographically [Fe–Fe = 2.5702(5) Å]. Its low symmetry was verified by variable-temperature 13C NMR spectroscopy, which revealed that the turnstile rotation of the {S(CH2)Fe(CO)3} and {S(NMe)Fe(CO)3} centers are subject to very different energy barriers. Although 1Me resists protonation, it readily undergoes substitution by tertiary phosphines, first at the {S(CH2)Fe(CO)3} center, as verified crystallographically for [Fe2{SCH2N(Me)CH2}(CO)5(PPh3)]. Substitution by the chelating diphosphine Ph2PCH2CH2PPh2 (dppe) gave [Fe2{SCH2N(Me)CH2}(CO)4(dppe)] by substitution at both the {S(CH2)Fe(CO)3} and {S(NMe)Fe(CO)3} sites.
Co-reporter:Maria E. Carroll, Jinzhu Chen, Danielle E. Gray, James C. Lansing, Thomas B. Rauchfuss, David Schilter, Phillip I. Volkers, and Scott R. Wilson
Organometallics 2014 Volume 33(Issue 4) pp:858-867
Publication Date(Web):February 3, 2014
DOI:10.1021/om400752a
Reported are complexes of the formula Fe(dithiolate)(CO)2(diphos) and their use to prepare homo- and heterobimetallic dithiolato derivatives. The starting iron dithiolates were prepared by a one-pot reaction of FeCl2 and CO with chelating diphosphines and dithiolates, where dithiolate = S2(CH2)22– (edt2–), S2(CH2)32– (pdt2–), S2(CH2)2(C(CH3)2)2– (Me2pdt2–) and diphos = cis-C2H2(PPh2)2 (dppv), C2H4(PPh2)2 (dppe), C6H4(PPh2)2 (dppbz), C2H4[P(C6H11)2]2 (dcpe). The incorporation of 57Fe into such building block complexes commenced with the conversion of 57Fe into 57Fe2I4(iPrOH)4, which then was treated with K2pdt, CO, and dppe to give 57Fe(pdt)(CO)2(dppe). NMR and IR analyses show that these complexes exist as mixtures of all-cis and trans-CO isomers, edt2– favoring the former and pdt2– the latter. Treatment of Fe(dithiolate)(CO)2(diphos) with the Fe(0) reagent (benzylideneacetone)Fe(CO)3 gave Fe2(dithiolate)(CO)4(diphos), thereby defining a route from simple ferrous salts to models for hydrogenase active sites. Extending the building block route to heterobimetallic complexes, treatment of Fe(pdt)(CO)2(dppe) with [(acenaphthene)Mn(CO)3]+ gave [(CO)3Mn(pdt)Fe(CO)2(dppe)]+ ([3d(CO)]+). Reduction of [3d(CO)]+ with BH4– gave the Cs-symmetric μ-hydride (CO)3Mn(pdt)(H)Fe(CO)(dppe) (H3d). Complex H3d is reversibly protonated by strong acids, the proposed site of protonation being sulfur. Treatment of Fe(dithiolate)(CO)2(diphos) with CpCoI2(CO) followed by reduction by Cp2Co affords CpCo(dithiolate)Fe(CO)(diphos) (4), which can also be prepared from Fe(dithiolate)(CO)2(diphos) and CpCo(CO)2. Like the electronically related (CO)3Fe(pdt)Fe(CO)(diphos), these complexes undergo protonation to afford the μ-hydrido complexes [CpCo(dithiolate)HFe(CO)(diphos)]+. Low-temperature NMR studies indicate that Co is the kinetic site of protonation.
Co-reporter:Dr. Xiaoyuan Zhou;Dr. Joyee Mitra ; Thomas B. Rauchfuss
ChemSusChem 2014 Volume 7( Issue 6) pp:1623-1626
Publication Date(Web):
DOI:10.1002/cssc.201301253
Abstract
The cleavage of CO bonds in lignin model compounds without hydrogen was developed using the commercially available Pd/C. Hydrogen donor solvents are helpful for this reaction through transfer hydrogenation, but not necessary. A redox neutral process that utilizes the internal hydrogen source for the cleavage is also possible. An initial mechanistic study indicates that the β-benzylic-H atom in the substrate plays a critical role and that the present system undergoes a process different from previous reports.
Co-reporter:James C. Lansing, James M. Camara, Danielle E. Gray, and Thomas B. Rauchfuss
Organometallics 2014 Volume 33(Issue 20) pp:5897-5906
Publication Date(Web):July 1, 2014
DOI:10.1021/om5004013
Active site mimics of [FeFe]-hydrogenase are shown to be bidirectional catalysts, producing H2 upon treatment with protons and reducing equivalents. This reactivity complements the previously reported oxidation of H2 by these same catalysts in the presence of oxidants. The complex Fe2(adtBn)(CO)3(dppv)(PFc*Et2) ([1]0; adtBn = (SCH2)2NBn, dppv = cis-1,2-bis(diphenylphosphino)ethylene, PFc*Et2 = Et2PCH2C5Me4FeCp*) reacts with excess [H(OEt2)2]BArF4 (BArF4– = B(C6H3-3,5-(CF3)2)4–) to give ∼0.5 equiv of H2 and [Fe2(adtBnH)(CO)3(dppv)(PFc*Et2)]2+ ([1H]2+). The species [1H]2+ consists of a ferrocenium ligand, an N-protonated amine, and an FeIFeI core. In the presence of additional reducing equivalents in the form of decamethylferrocene (Fc*), hydrogen evolution is catalytic, albeit slow. The related catalyst Fe2(adtBn)(CO)3(dppv)(PMe3) (3) behaves similarly in the presence of Fc*, except that in the absence of excess reducing agent it converts to the catalytically inactive μ-hydride derivative [μ-H3]+. Replacement of the adt in [1]0 with propanedithiolate (pdt) results in a catalytically inactive complex. In the course of synthesizing [FeFe]-hydrogenase mimics, new routes to ferrocenylphosphine ligands and nonamethylferrocene were developed.
Co-reporter:Aaron M. Appel, John E. Bercaw, Andrew B. Bocarsly, Holger Dobbek, Daniel L. DuBois, Michel Dupuis, James G. Ferry, Etsuko Fujita, Russ Hille, Paul J. A. Kenis, Cheryl A. Kerfeld, Robert H. Morris, Charles H. F. Peden, Archie R. Portis, Stephen W. Ragsdale, Thomas B. Rauchfuss, Joost N. H. Reek, Lance C. Seefeldt, Rudolf K. Thauer, and Grover L. Waldrop
Chemical Reviews 2013 Volume 113(Issue 8) pp:6621
Publication Date(Web):June 14, 2013
DOI:10.1021/cr300463y
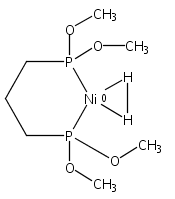
Co-reporter:Wenguang Wang ; Mark J. Nilges ; Thomas B. Rauchfuss ;Matthias Stein
Journal of the American Chemical Society 2013 Volume 135(Issue 9) pp:3633-3639
Publication Date(Web):February 5, 2013
DOI:10.1021/ja312458f
The mixed-valence diiron hydrido complex (μ-H)Fe2(pdt)(CO)2(dppv)2 ([H1]0, where pdt =1,3-propanedithiolate and dppv = cis-1,2-C2H2(PPh2)2), was generated by reduction of the differous hydride [H1]+ using decamethylcobaltocene. Crystallographic analysis shows that [H1]0 retains the stereochemistry of its precursor, where one dppv ligand spans two basal sites and the other spans apical and basal positions. The Fe---Fe bond elongates to 2.80 from 2.66 Å, but the Fe–P bonds only change subtly. Although the Fe–H distances are indistinguishable in the precursor, they differ by 0.2 Å in [H1]0. The X-band electron paramagnetic resonance (EPR) spectrum reveals the presence of two stereoisomers, the one characterized crystallographically and a contribution of about 10% from a second symmetrical (sym) isomer wherein both dppv ligands occupy apical–basal sites. The unsymmetrical (unsym) arrangement of the dppv ligands is reflected in the values of A(31P), which range from 31 MHz for the basal phosphines to 284 MHz for the apical phosphine. Density functional theory calculations were employed to rationalize the electronic structure of [H1]0 and to facilitate spectral simulation and assignment of EPR parameters including 1H and 31P hyperfine couplings. The EPR spectra of [H1]0 and [D1]0 demonstrate that the singly occupied molecular orbital is primarily localized on the Fe center with the longer bond to H, that is, FeII–H···FeI. The coupling to the hydride is A(1H) = 55 and 74 MHz for unsym- amd sym-[H1]0, respectively. Treatment of [H1]0 with H+ gives 0.5 equiv of H2 and [H1]+. Reduction of D+ affords D2, leaving the hydride ligand intact. These experiments demonstrate that the bridging hydride ligand in this complex is a spectator in the hydrogen evolution reaction.
Co-reporter:Brian C. Manor
Journal of the American Chemical Society 2013 Volume 135(Issue 32) pp:11895-11900
Publication Date(Web):July 11, 2013
DOI:10.1021/ja404580r
Described are experiments demonstrating incorporation of cyanide cofactors and hydride substrate into [NiFe]-hydrogenase (H2ase) active site models. Complexes of the type (CO)2(CN)2Fe(pdt)Ni(dxpe) (dxpe = dppe, 1; dxpe = dcpe, 2) bind the Lewis acid B(C6F5)3 (BArF3) to give the adducts (CO)2(CNBArF3)2Fe(pdt)Ni(dxpe), (1(BArF3)2, 2(BArF3)2). Upon decarbonylation using amine oxides, these adducts react with H2 to give hydrido derivatives [(CO)(CNBArF3)2Fe(H)(pdt)Ni(dxpe)]− (dxpe = dppe, [H3(BArF3)2]−; dxpe = dcpe, [H4(BArF3)2]−). Crystallographic analysis shows that Et4N[H3(BArF3)2] generally resembles the active site of the enzyme in the reduced, hydride-containing states (Ni–C/R). The Fe–H···Ni center is unsymmetrical with rFe–H = 1.51(3) Å and rNi–H = 1.71(3) Å. Both crystallographic and 19F NMR analyses show that the CNBArF3– ligands occupy basal and apical sites. Unlike cationic Ni–Fe hydrides, [H3(BArF3)2]− and [H4(BArF3)2]− oxidize at mild potentials, near the Fc+/0 couple. Electrochemical measurements indicate that in the presence of base, [H3(BArF3)2]− catalyzes the oxidation of H2. NMR evidence indicates dihydrogen bonding between these anionic hydrides and R3NH+ salts, which is relevant to the mechanism of hydrogenogenesis. In the case of Et4N[H3(BArF3)2], strong acids such as HCl induce H2 release to give the chloride Et4N[(CO)(CNBArF3)2Fe(Cl)(pdt)Ni(dppe)].
Co-reporter:Dr. Xiaoyuan Zhou ; Thomas B. Rauchfuss
ChemSusChem 2013 Volume 6( Issue 2) pp:383-388
Publication Date(Web):
DOI:10.1002/cssc.201200718
Abstract
We report the one-pot alkylation of mesitylene with carbohydrate-derived 5-(hydroxymethyl)furfural (HMF) as a step toward diesel-range liquids. Using FeCl3 as a catalyst, HMF is shown to alkylate toluene, xylene, and mesitylene in high yields in CH2Cl2 and MeNO2 solvents. Efforts to extend this reaction to greener or safer solvents showed that most ether-based solvents are unsatisfactory. Acid catalysts (e.g, p-TsOH) also proved to be ineffective. Using formic acid as a reactive solvent, mesitylene could be alkylated to give mesitylmethylfurfural (MMF) starting from fructose with yields up to approximately 70 %. The reaction of fructose with formic acid in the absence of mesitylene gave rise to low yields of the formate ester of HMF, which indicates the stabilizing effect of replacing the hydroxyl substituent with mesityl. The arene also serves as a second phase into which the product is extracted. Even by using formic acid, the mesitylation of less expensive precursors such as glucose and cellulose proceeded only in modest yields (ca. 20 %). These simpler substrates were found to undergo mesitylation by using hydrogen chloride/formic acid via the intermediate chloromethylfurfural.
Co-reporter:Geoffrey M. Chambers, Raja Angamuthu, Danielle L. Gray, and Thomas B. Rauchfuss
Organometallics 2013 Volume 32(Issue 21) pp:6324-6329
Publication Date(Web):October 24, 2013
DOI:10.1021/om4006824
Described is a new family of RuNi dithiolates featuring geometrically flexible Ni centers, which enable both acid–base and redox chemistry, behavior that is characteristic of the hydrogenases. Treatment of Ni(pdt)(dxpe) with (cymene)2Ru2Cl4 affords the salt [(cymene)Ru(Cl)(pdt)Ni(diphos)]Cl (pdt2– = 1,3-propanedithiolate, diphos = dppe = 1,2-bis(diphenylphosphino)ethane ([1Cl]Cl) and diphos = dcpe = 1,2-bis(dicyclohexylphosphino)ethane ([2Cl]Cl)). Cyclic voltammetry revealed that in CH2Cl2 solution [1Cl]Cl reduces irreversibly near −1.6 V vs Fc0/+ followed by the appearance of a reversible 1e– couple assigned to the [1]0/+ couple. Reduction of [1Cl]Cl and [2Cl]Cl with cobaltocene produced the neutral derivatives (cymene)Ru(pdt)Ni(diphos) ([1]0, [2]0). Crystallographic characterization of these compounds revealed short Ru–Ni distance of 2.5539(5) ([1]0) and 2.600(3) Å ([2]0). The 2e– reduction of these chlorides converts the Ni site from square planar to tetrahedral, highlighting the flexibility of the Ni center in these complexes. Variable-temperature NMR studies show that [1]0 and [2]0 are dynamic by virtue of ring flipping in the RuNi(pdt) core. Complexes [1]0 and [2]0 are basic: their conjugate acids, [H1]+ and [H2]+, exhibit pKaPhCN values of 18.94 and 21.65, respectively. Crystallographic characterization of [H1]+ as its aryl borate salt revealed an unsymmetrical Ru–H···Ni interaction and confirmed that the Ni center converted from tetrahedral to square planar, again demonstrating the flexibility of this site.
Co-reporter:Dr. Wenguang Wang;Dr. Thomas B. Rauchfuss;Dr. Curtis E. Moore;Dr. Arnold L. Rheingold;Dr. Luca DeGioia;Dr. Giuseppe Zampella
Chemistry - A European Journal 2013 Volume 19( Issue 46) pp:15476-15479
Publication Date(Web):
DOI:10.1002/chem.201303351
Co-reporter:Riccardo Zaffaroni, Thomas B. Rauchfuss, and Amy Fuller, Luca De Gioia and Giuseppe Zampella
Organometallics 2013 Volume 32(Issue 1) pp:232-238
Publication Date(Web):December 27, 2012
DOI:10.1021/om300997s
This paper reports on the protonation of phosphine-substituted diiron diphosphido carbonyls, analogues of diiron dithiolato centers at the active sites of hydrogenase enzymes. Reaction of the diphosphines (CH2)n(PPhH)2 (n = 2 (edpH2) and n = 3 (pdpH2)) with Fe3(CO)12 gave excellent yields of Fe2(edp)(CO)6 (1) and Fe2(pdp)(CO)6 (2). Substitution of Fe2(edp)(CO)6 with PMe3 afforded Fe2(edp)(CO)2(PMe3)4 (3; νCO 1855 and 1836 cm–1). Crystallographic analysis showed that 3 adopts an idealized C2 symmetry, with pairs of phosphine ligands occupying apical–basal sites on each Fe center. Relative to that in the dithiolato complex, the Fe–Fe bond (2.7786(8) Å) is elongated by 0.15 Å. Treatment of 3 with H(OEt2)2BArF4 (ArF = C6H3-3,5-(CF3)2) gave exclusively the C2-symmetric μ-hydride complex [HFe2(edp)(CO)2(PMe3)4]+. This result contrasts with the behavior of the analogous ethanedithiolate Fe2(edt)(CO)2(PMe3)4 (edt = 1,2-C2H4S2), protonation of which gives both the bridging and terminal hydride complexes. This difference points to the participation of the sulfur centers in the formation of terminal hydrides. The absence of terminal hydride intermediates was also revealed in the protonation of the diphosphine diphosphido complexes Fe2(pdp)(CO)4(dppv) (4; dppv = cis-1,2-C2H2(PPh2)2) and Fe2(edp)(CO)4(dppbz) (5; dppbz = 1,2-C6H4(PPh2)2). Protonation of these diphosphine complexes afforded μ-hydrido cations with apical–basal diphosphine ligands, which convert to the isomer where the diphosphine is dibasal. In contrast, protonation of the dithiolato complex Fe2(pdt)(CO)4(dppv) gave terminal hydrides, which isomerize to μ-hydrides. In a competition experiment, 4 was shown to protonate faster than Fe2(pdt)(CO)4(dppv).
Co-reporter:Dr. David Schilter;Dr. Thomas B. Rauchfuss
Angewandte Chemie International Edition 2013 Volume 52( Issue 51) pp:13518-13520
Publication Date(Web):
DOI:10.1002/anie.201307132
Co-reporter:Maria E. Carroll, Bryan E. Barton, Thomas B. Rauchfuss, and Patrick J. Carroll
Journal of the American Chemical Society 2012 Volume 134(Issue 45) pp:18843-18852
Publication Date(Web):November 5, 2012
DOI:10.1021/ja309216v
This report compares biomimetic hydrogen evolution reaction catalysts with and without the amine cofactor (adtNH): Fe2(adtNH)(CO)2(dppv)2 (1NH) and Fe2(pdt)(CO)2(dppv)2 (2) [(adtNH)2– = HN(CH2S)22–, pdt2– = 1,3-(CH2)3S22–, and dppv = cis-C2H2(PPh2)2]. These compounds are spectroscopically, structurally, and stereodynamically very similar but exhibit very different catalytic properties. Protonation of 1NH and 2 gives three isomeric hydrides each, beginning with the kinetically favored terminal hydride, which converts sequentially to sym and unsym isomers of the bridging hydrides. In the case of 1NH, the corresponding ammonium hydrides are also observed. In the case of the terminal amine hydride [t-H1NH]BF4, the ammonium/amine hydride equilibrium is sensitive to counteranions and solvent. The species [t-H1NH2](BF4)2 represents the first example of a crystallographically characterized terminal hydride produced by protonation. The NH---HFe distance of 1.88(7) Å indicates dihydrogen-bonding. The bridging hydrides [μ-H1NH]+ and [μ-H2]+ reduce near −1.8 V, about 150 mV more negative than the reductions of the terminal hydride [t-H1NH]+ and [t-H2]+ at −1.65 V. Reductions of the amine hydrides [t-H1NH]+ and [t-H1NH2]2+ are irreversible. For the pdt analogue, the [t-H2]+/0 couple is unaffected by weak acids (pKaMeCN = 15.3) but exhibits catalysis with HBF4·Et2O, albeit with a turnover frequency (TOF) around 4 s–1 and an overpotential greater than 1 V. The voltammetry of [t-H1NH]+ is strongly affected by relatively weak acids and proceeds at 5000 s–1 with an overpotential of 0.7 V. The ammonium hydride [t-H1NH2]2+ is a faster catalyst, with an estimated TOF of 58 000 s–1 and an overpotential of 0.5 V.
Co-reporter:Riccardo Zaffaroni ; Thomas B. Rauchfuss ; Danielle L. Gray ; Luca De Gioia ;Giuseppe Zampella
Journal of the American Chemical Society 2012 Volume 134(Issue 46) pp:19260-19269
Publication Date(Web):October 24, 2012
DOI:10.1021/ja3094394
This investigation examines the protonation of diiron dithiolates, exploiting the new family of exceptionally electron-rich complexes Fe2(xdt)(CO)2(PMe3)4, where xdt is edt (ethanedithiolate, 1), pdt (propanedithiolate, 2), and adt (2-aza-1,3-propanedithiolate, 3), prepared by the photochemical substitution of the corresponding hexacarbonyls. Compounds 1–3 oxidize near −950 mV vs Fc+/0. Crystallographic analyses confirm that 1 and 2 adopt C2-symmetric structures (Fe–Fe = 2.616 and 2.625 Å, respectively). Low-temperature protonation of 1 afforded exclusively [μ-H1]+, establishing the non-intermediacy of the terminal hydride ([t-H1]+). At higher temperatures, protonation afforded mainly [t-H1]+. The temperature dependence of the ratio [t-H1]+/[μ-H1]+ indicates that the barriers for the two protonation pathways differ by ∼4 kcal/mol. Low-temperature 31P{1H} NMR measurements indicate that the protonation of 2 proceeds by an intermediate, proposed to be the S-protonated dithiolate [Fe2(Hpdt)(CO)2(PMe3)4]+ ([S-H2]+). This intermediate converts to [t-H2]+ and [μ-H2]+ by first-order and second-order processes, respectively. DFT calculations support transient protonation at sulfur and the proposal that the S-protonated species (e.g., [S-H2]+) rearranges to the terminal hydride intramolecularly via a low-energy pathway. Protonation of 3 affords exclusively terminal hydrides, regardless of the acid or conditions, to give [t-H3]+, which isomerizes to [t-H3′]+, wherein all PMe3 ligands are basal.
Co-reporter:Wenguang Wang and Thomas B. Rauchfuss , Luca Bertini and Giuseppe Zampella
Journal of the American Chemical Society 2012 Volume 134(Issue 10) pp:4525-4528
Publication Date(Web):February 27, 2012
DOI:10.1021/ja211778j
The diiron hydride [(μ-H)Fe2(pdt)(CO)4(dppv)]+ ([H2]+, dppv = cis-1,2-C2H2(PPh2)2) is shown to be an effective photocatalyst for the H2 evolution reaction (HER). These experiments establish the role of hydrides in photocatalysis by biomimetic diiron complexes. Trends in redox potentials suggests that other unsymmetrically substituted diiron hydrides are promising catalysts. Unlike previous catalysts for photo-HER, [H2]+ functions without sensitizers: irradiation of [H2]+ in the presence of triflic acid (HOTf) efficiently affords H2. Instead of sacrificial electron donors, ferrocenes can be used as recyclable electron donors for the photocatalyzed HER, resulting in 4 turnovers.
Co-reporter:Christopher S. Letko ; Thomas B. Rauchfuss ; Xiaoyuan Zhou ;Danielle L. Gray
Inorganic Chemistry 2012 Volume 51(Issue 8) pp:4511-4520
Publication Date(Web):March 27, 2012
DOI:10.1021/ic202207e
We report the enhanced reactivity of hydroxyl substituted CuN3+ derivatives, where N3 = tris(picolinyl)methane (tripic) and related derivatives, upon deprotonation of the O–H functionality. The work capitalizes on new methodology for incorporating hydroxyl groups into the second coordination sphere of copper centers. The key synthetic methodology relies on Pd-catalyzed coupling reactions of dilithiated 6-methyl-2-pyridone with bromopyridyl derivatives. These building blocks allow the preparation of tridentate N3 ligands with OH and OMe substituents flanking the fourth coordination site of a tetrahedral complex. Coupling of these tridendate ligands gives the corresponding hydroxy- and methoxy-functionalized bistripodal ligands. [Cu[bis(2-methylpyrid-6-yl)(2-hydroxypyrid-6-yl)methane](NCMe)]+ ([Cu(2H)(NCMe)]+) oxidizes readily in air to afford the mixed valence Cu1.5 dimer ([Cu2(2)2]+). Formation of [Cu2(2)2]+ is accelerated in the presence of base and can be reversed with a combination of decamethylferrocene and acid. The reactivity of [Cu(2H)(NCMe)]+ with dioxygen requires deprotonation of the hydroxyl substituent: neither [Cu(tripic)(NCMe)]+ nor the methoxy-derivatives displayed comparable reactivity. A related mixed valence dimer formed upon oxidation of the dicopper(I) complex of a tetrahydroxy bis(tridentate) ligand, [Cu2(6H4)(NCMe)2]2+. The dicopper(I) complex of the analogous tetramethoxy N6-ligand, [Cu2(5)(NCMe)2]2+, instead reversibly binds O2. Deprotonation of [Cu(2H)(CO)]+ and [Cu(2H)(NCMe)]+ afforded the neutral derivatives Cu(2)(CO) and Cu2(2)2, respectively. The dicopper(I) derivative Cu2(2)2 can be reoxidized, reprotonated, and carbonylated. The silver(I) complex, [Ag(2H)(NCMe)]BF4, forms an analogous neutral dimer (Ag2(2)2) upon deprotonation of the hydroxyl group. The structures of ligand 2H, [Cu2(5)(NCMe)2]+, [Cu2(2)2]+, [Cu2(6H2)]+, [Ag(2H)(NCMe)]BF4, and Ag2(2)2 were confirmed by single crystal X-ray diffraction.
Co-reporter:David Schilter, Thomas B. Rauchfuss, and Matthias Stein
Inorganic Chemistry 2012 Volume 51(Issue 16) pp:8931-8941
Publication Date(Web):July 27, 2012
DOI:10.1021/ic300910r
New mixed-valence iron–nickel dithiolates are described that exhibit structures similar to those of mixed-valence diiron dithiolates. The interaction of tricarbonyl salt [(dppe)Ni(pdt)Fe(CO)3]BF4 ([1]BF4, where dppe = Ph2PCH2CH2PPh2 and pdt2– = −SCH2CH2CH2S−) with P-donor ligands (L) afforded the substituted derivatives [(dppe)Ni(pdt)Fe(CO)2L]BF4 incorporating L = PHCy2 ([1a]BF4), PPh(NEt2)2 ([1b]BF4), P(NMe2)3 ([1c]BF4), P(i-Pr)3 ([1d]BF4), and PCy3 ([1e]BF4). The related precursor [(dcpe)Ni(pdt)Fe(CO)3]BF4 ([2]BF4, where dcpe = Cy2PCH2CH2PCy2) gave the more electron-rich family of compounds [(dcpe)Ni(pdt)Fe(CO)2L]BF4 for L = PPh2(2-pyridyl) ([2a]BF4), PPh3 ([2b]BF4), and PCy3 ([2c]BF4). For bulky and strongly basic monophosphorus ligands, the salts feature distorted coordination geometries at iron: crystallographic analyses of [1e]BF4 and [2c]BF4 showed that they adopt “rotated” FeI centers, in which PCy3 occupies a basal site and one CO ligand partially bridges the Ni and Fe centers. Like the undistorted mixed-valence derivatives, members of the new class of complexes are described as NiIIFeI (S = 1/2) systems according to electron paramagnetic resonance spectroscopy, although with attenuated 31P hyperfine interactions. Density functional theory calculations using the BP86, B3LYP, and PBE0 exchange-correlation functionals agree with the structural and spectroscopic data, suggesting that the spin for [1e]+ is mostly localized in a FeI-centered d(z2) orbital, orthogonal to the Fe–P bond. The PCy3 complexes, rare examples of species featuring “rotated” Fe centers, both structurally and spectroscopically incorporate features from homobimetallic mixed-valence diiron dithiolates. Also, when the NiS2Fe core of the [NiFe]-hydrogenase active site is reproduced, the “hybrid models” incorporate key features of the two major classes of hydrogenase. Furthermore, cyclic voltammetry experiments suggest that the highly basic phosphine ligands enable a second oxidation corresponding to the couple [(dxpe)Ni(pdt)Fe(CO)2L]+/2+. The resulting unsaturated 32e– dications represent the closest approach to modeling the highly electrophilic Ni–SIa state. In the case of L = PPh2 (2-pyridyl), chelation of this ligand accompanies the second oxidation.
Co-reporter:David Schilter and Thomas B. Rauchfuss
Dalton Transactions 2012 vol. 41(Issue 43) pp:13324-13329
Publication Date(Web):20 Sep 2012
DOI:10.1039/C2DT31895D
Described herein are preparations of synthetic models for the deactivated Ni(II)Fe(II) states of the [NiFe]-hydrogenases. Iodination of the S = ½ species [(dppe)Ni(pdt)Fe(CO)3]+ afforded the diamagnetic iodo complex [(dppe)Ni(pdt)IFe(CO)3]+. Crystallographic analysis of this species confirmed the presence of square-pyramidal Ni linked to an octahedral Fe centre. The Ni⋯Fe separation of 3.018 Å indicated the absence of metal–metal bonding. This complex could be reduced to give (dppe)Ni(pdt)Fe(CO)3 and, in the presence of iodide, decarbonylated to afford (dppe)Ni(pdt)FeI2. Derivatives of the type [(diphosphine)Ni(dithiolate)XFe(CO)2L]+ (X = Cl, Br, I) were prepared by halogenation of mixed-valence precursors [(diphosphine)Ni(dithiolate)Fe(CO)2L]+ (diphosphine = dppe, dcpe; L = tertiary phosphine or CO). The Fe(CO)2(PR3)-containing derivatives are more robust than the related tricarbonyl derivatives. Exploiting this greater stability, we characterised examples of chloride and bromide derivatives. Related fluorides could be prepared by F− abstraction from BF4−. Spectroscopic evidence is presented for the hydroperoxide [(dppe)Ni(pdt)(OOH)Fe(CO)2L]+, which represents a model for the Ni-SU state.
Co-reporter:Mark R. Ringenberg
European Journal of Inorganic Chemistry 2012 Volume 2012( Issue 3) pp:490-495
Publication Date(Web):
DOI:10.1002/ejic.201101011
Abstract
The effect of protonation of metal complexes of noninnocent ligands is investigated. The anilidophenolate complexes Cp*IrtBAtBu (1) and Cp*IrtBAFPh (2) were examined [H2tBAtBu = 2-tert-butylamino-4,6-di-tert-butylphenol, H2tBAFPh = 2-(2-trifluoromethyl)amino-4,6-di-tert-butylphenol]. Protonation of 1 with HBF4 gave the anilinophenolate [1H]+. Crystallographic characterization of this complex revealed an elongatated Ir–N bond, but the IrIII center remains pentacoordinate. The anilinophenolate hydride 1H(H) was prepared by treatment of [1H]+ with boron hydride reagents and with formic acid. X-ray crystallography confirmed the structure and revealed that the hydride and ammonium hydrons occupy sites on opposite sides of the IrONC6 ring. Complex 2 is protonated only by strong acids, and the resulting derivative is unstable in the absence of ligands such as CO and PMe3. Crystallographic characterization of [2(PMe3)]+ revealed that protonation occurs at O not N. The carbonylation of [2H]+ reveals the intermediacy of kinetic adducts, which are proposed to be the anilino tautomer.
Co-reporter:Hao Lei, Aaron M. Royer, Thomas B. Rauchfuss, and Danielle Gray
Organometallics 2012 Volume 31(Issue 17) pp:6408-6414
Publication Date(Web):August 15, 2012
DOI:10.1021/om300631a
Reaction of Fe(bda)(CO)3 (bda = benzylideneacetone) and Ph2P-2-C6H4CHO (PCHO) affords the bisphosphine bisalkoxide complex Fe[(Ph2PC6H4)2C2H2O2](CO)2 (1) arising from the head-to-head coupling of two formyl groups concomitant with oxidation of Fe(0) to Fe(II). Crystallographic studies show that 1 features cis alkoxide ligands that are trans to CO; the two phosphine groups are mutually trans with a P–Fe–P angle of 167.44(4)°. The pathway leading to 1 was examined, starting with the adduct Fe(PCHO)(CO)4 (2), which was obtained by addition of PCHO to Fe2(CO)9. Compound 2 decarbonylates to give tricarbonyl Fe(κ1,η2-PCHO)(CO)3 (3), which features a π-bonded aldehyde. Photolysis of 2 gives a mixture of 3 and isomeric hydride HFe(κ2-PCO)(CO)3. Complex 3 reacts with an additional equivalent of PCHO to afford 1, whereas treatment with PPh3 afforded the substituted product Fe(κ1,η2-PCHO)(PPh3)(CO)2 (4). In 4, the phosphine ligands are trans and the aldehyde is π-bonded. The geometry around Fe is pseudo trigonal bipyramidal. To gain insights into the mechanism and scope of the C–C coupling reaction, complexes were prepared with the imine Ph2PC6H4CH═NC6H4Cl (abbreviated as PCHNAr), derived by condensation of 4-chloroaniline and PCHO. PCHNAr reacts with Fe2(CO)9 and with Fe(bda)(CO)3 to afford the tetra- and tricarbonyl compounds Fe(PCHNAr)(CO)4 (5) and Fe(PCHNAr)(CO)3 (6), respectively. Treatment of 6 with PCHO gave the unsymmetrical C–C coupling complex Fe[(Ph2PC6H4)2CH(O)CH(NAr)](CO)2 (7). Compound 7 was also prepared by the reaction of 3 and PCHNAr. The solid-state structure of 7, as established by X-ray crystallography, is similar to that of 1 but with an amido group in place of one alkoxide. The deuterium-labeled phosphine aldehyde PCDO was prepared by the reaction of ortho-lithiated phosphine Ph2PC6H4-2-Li with DMF-d7. Reaction of 6 with PCDO gave 7-d1 with no scrambling of the deuterium label. Attempted oxidation of 1 with FcBF4 (Fc+ = ferrocenium) gave the adduct Fe[(Ph2PC6H4)2C2H2O2(BF3)2](CO)2 (8). The structures of 1 and 8 are almost identical. Compound 8 was independently synthesized by treating 1 with BF3OEt2 via the intermediacy of the 1:1 adduct, which was detected spectroscopically. Qualitative tests showed that 1 also reversibly protonates with HOSO2CF3 and binds TiCl4.
Co-reporter:James M. Camara
Journal of the American Chemical Society 2011 Volume 133(Issue 21) pp:8098-8101
Publication Date(Web):May 6, 2011
DOI:10.1021/ja201731q
Mild oxidants such as [Fe(C5Me5)2]+ accelerate the activation of H2 by [Fe2[(SCH2)2NBn](CO)3(dppv)(PMe3)]+ ([1]+), despite the fact that the ferrocenium cation is incapable of oxidizing [1]+. The reaction is first-order in [1]+ and [H2] but independent of the E1/2 and concentration of the oxidant. The analogous reaction occurs with D2 and proceeds with an inverse kinetic isotope effect of 0.75(8). The activation of H2 is further enhanced with the tetracarbonyl [Fe2[(SCH2)2NBn](CO)4(dppn)]+ ([2]+), the first crystallographically characterized model for the Hox state of the active site containing an amine cofactor. These studies point to rate-determining binding of H2 followed by proton-coupled electron transfer. Relative to that by [1]+, the rate of H2 activation by [2]+/Fc+ is enhanced by a factor of 104 at 25 °C.
Co-reporter:Maria E. Carroll, Bryan E. Barton, Danielle L. Gray, Amanda E. Mack, and Thomas B. Rauchfuss
Inorganic Chemistry 2011 Volume 50(Issue 19) pp:9554-9563
Publication Date(Web):August 25, 2011
DOI:10.1021/ic2012759
Described are new derivatives of the type [HNiFe(SR)2(diphosphine)(CO)3]+, which feature a Ni(diphosphine) group linked to a Fe(CO)3 group by two bridging thiolate ligands. Previous work had described [HNiFe(pdt)(dppe)(CO)3]+ ([1H]+) and its activity as a catalyst for the reduction of protons (J. Am. Chem. Soc.2010, 132, 14877). Work described in this paper focuses on the effects on properties of NiFe model complexes of the diphosphine attached to nickel as well as the dithiolate bridge, 1,3-propanedithiolate (pdt) vs 1,2-ethanedithiolate (edt). A new synthetic route to these Ni–Fe dithiolates is described, involving reaction of Ni(SR)2(diphosphine) with FeI2(CO)4 followed by in situ reduction with cobaltocene. Evidence is presented that this route proceeds via a metastable μ-iodo derivative. Attempted isolation of such species led to the crystallization of NiFe(Me2pdt)(dppe)I2, which features tetrahedral Fe(II) and square planar Ni(II) centers (H2Me2pdt = 2,2-dimethylpropanedithiol). The new tricarbonyls prepared in this work are NiFe(pdt)(dcpe)(CO)3 (2, dcpe = 1,2-bis(dicyclohexylphosphino)ethane), NiFe(edt)(dppe)(CO)3 (3), and NiFe(edt)(dcpe)(CO)3 (4). Attempted preparation of a phenylthiolate-bridged complex via the FeI2(CO)4 + Ni(SPh)2(dppe) route gave the tetrametallic species [(CO)2Fe(SPh)2Ni(CO)]2(μ-dppe)2. Crystallographic analysis of the edt-dcpe compund [2H]BF4 and the edt-dppe compound [3H]BF4 verified their close resemblance. Each features pseudo-octahedral Fe and square pyramidal Ni centers. Starting from [3H]BF4 we prepared the PPh3 derivative [HNiFe(edt)(dppe)(PPh3)(CO)2]BF4 ([5H]BF4), which was obtained as a ∼2:1 mixture of unsymmetrical and symmetrical isomers. Acid–base measurements indicate that changing from Ni(dppe) (dppe = Ph2PCH2CH2PPh2) to Ni(dcpe) decreases the acidity of the cationic hydride complexes by 2.5 pKaPhCN units, from ∼11 to ∼13.5 (previous work showed that substitution at Fe leads to more dramatic effects). The redox potentials are more strongly affected by the change from dppe to dcpe, for example the [2]0/+ couple occurs at E1/2 = −820 for [2]0/+ vs −574 mV (vs Fc+/0) for [1]0/+. Changes in the dithiolate do not affect the acidity or the reduction potentials of the hydrides. The acid-independent rate of reduction of CH2ClCO2H by [2H]+ is about 50 s–1 (25 °C), twice that of [1H]+. The edt-dppe complex [2H]+ proved to be the most active catalyst, with an acid-independent rate of 300 s–1.
Co-reporter:Christopher S. Letko ; Zachariah M. Heiden ; Thomas B. Rauchfuss ;Scott R. Wilson
Inorganic Chemistry 2011 Volume 50(Issue 12) pp:5558-5566
Publication Date(Web):May 16, 2011
DOI:10.1021/ic200160q
The paper surveys the binding of anions to the unsaturated 16e Lewis acid [Cp*Ir(TsDPEN)]+ ([1H]+), where TsDPEN is racemic H2NCHPhCHPhNTs–. The derivatives Cp*IrX(TsDPEN) were characterized crystallographically for X– = CN–, Me(C═NH)S–, NO2–, 2-pyridonate, and 0.5 MoS42–. [(1H)2(μ-CN)]+ forms from [1H]+ and 1H(CN). Aside from 2-pyridone, amides generally add reversibly and bind to Ir through N. Thioacetamide binds irreversibly through sulfur. Compounds of the type Cp*IrX(TsDPEN) generally form diastereoselectively, although diastereomeric products were observed for the strong ligands (X = CN–, H– (introduced via BH4–), or Me(C═NH)S–). Related experiments on the reaction (p-cymene)Ru(TsDPEN-H) + BH4– gave two diastereomers of (p-cymene)RuH(TsDPEN), the known hydrogenation catalyst and a second isomer that hydrogenated acetophenone more slowly. These experiment provide new insights into the enantioselectivity of these catalysts. Diastereomerization in all cases was first order in metal with modest solvent effects. The diphenyl groups are generally diequatorial for the stable diastereomers. For the 2-pyridonate adduct, axial phenyl groups are stabilized in the solid state by puckering of the IrN2C2 ring induced by intramolecular hydrogen-bonding. Crystallographic analysis of [Cp*Ir(TsDPEN)]2(MoS4) revealed a unique example of a κ1,κ1-tetrathiometallate ligand. Cp*Ir(SC(NH)Me)TsDPEN) is the first example of a κ1-S-thioamidato complex.
Co-reporter:Matthew T. Olsen ; Thomas B. Rauchfuss ;Scott R. Wilson
Journal of the American Chemical Society 2010 Volume 132(Issue 50) pp:17733-17740
Publication Date(Web):November 29, 2010
DOI:10.1021/ja103998v
This paper summarizes studies on the redox behavior of synthetic models for the [FeFe]-hydrogenases, consisting of diiron dithiolato carbonyl complexes bearing the amine cofactor and its N-benzyl derivative. Of specific interest are the causes of the low reactivity of oxidized models toward H2, which contrasts with the high activity of these enzymes for H2 oxidation. The redox and acid−base properties of the model complexes [Fe2[(SCH2)2NR](CO)3(dppv)(PMe3)]+ ([2]+ for R = H and [2′]+ for R = CH2C6H5, dppv = cis-1,2-bis(diphenylphosphino)ethylene)) indicate that addition of H2 followed by deprotonation are (i) endothermic for the mixed valence (FeIIFeI) state and (ii) exothermic for the diferrous (FeIIFeII) state. The diferrous state is shown to be unstable with respect to coordination of the amine to Fe, a derivative of which was characterized crystallographically. The redox and acid−base properties for the mixed valence models differ strongly for those containing the amine cofactor versus those derived from propanedithiolate. Protonation of [2′]+ induces disproportionation to a 1:1 mixture of the ammonium [H2′]+ (FeIFeI) and the dication [2′]2+ (FeIIFeII). This effect is consistent with substantial enhancement of the basicity of the amine in the FeIFeI state vs the FeIIFeI state. The FeIFeI ammonium compounds are rapid and efficient H-atom donors toward the nitroxyl compound TEMPO. The atom transfer is proposed to proceed via the hydride. Collectively, the results suggest that proton-coupled electron-transfer pathways should be considered for H2 activation by the [FeFe]-hydrogenases.
Co-reporter:Dr. Todsapon Thananatthanachon ; Thomas B. Rauchfuss
ChemSusChem 2010 Volume 3( Issue 10) pp:1139-1141
Publication Date(Web):
DOI:10.1002/cssc.201000209
Co-reporter:Christopher S. Letko;Zachariah M. Heiden
European Journal of Inorganic Chemistry 2009 Volume 2009( Issue 33) pp:4927-4930
Publication Date(Web):
DOI:10.1002/ejic.200900780
Abstract
The addition of H3PO4 to Cp*Ir(TsDPEN-H), where TsDPEN = H2NCHPhCHPhNTs–, is a simple method to obtain a water-soluble hydrogenation catalyst capable of reducing aromatic ketones to their corresponding alcohols in aqueous solutions. Key to the reactivity is the low affinity of the coordinatively unsaturated [Cp*Ir(TsDPEN)]+ for H2PO4–. Catalyst degradation proceeds via the protonation of the tosylamido ligand, as was established by the crystallographic characterization of the tosylamine complex [Cp*Ir(NCMe)(HTsDPEN)]2+.(© Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, Germany, 2009)
Co-reporter:Aaron M. Royer, Thomas B. Rauchfuss and Danielle L. Gray
Organometallics 2009 Volume 28(Issue 13) pp:3618-3620
Publication Date(Web):May 28, 2009
DOI:10.1021/om9004059
The thioester Ph2PC6H4-2-C(O)SPh reacts with Fe2(CO)9 to give [Ph2PC6H4C(O)]Fe(SPh)(CO)3, a model for the CO-inhibited active site of the enzyme Hmd. This species, which reversibly decarbonylates to give a diiron derivative, reacts with cyanide to give [[Ph2PC6H4C(O)]Fe(SPh)(CN)(CO)2]−.
Co-reporter:Didier Morvan, Thomas B. Rauchfuss and Scott R. Wilson
Organometallics 2009 Volume 28(Issue 11) pp:3161-3166
Publication Date(Web):May 13, 2009
DOI:10.1021/om9001445
In view of contemporary interest in lignocellulose, we report the synthesis, structures, and preliminary reactivity of the first π-complexes of the lignin monomers (lignols). Initial studies with guaiacol demonstrated that it binds only weakly to Mo(CO)3 but [Mn(CO)3(guaiacol)]BF4 is robust. Related adducts with the cinnamyl alcohol derivatives, which are more closely related to the lignols, were unstable. From [Cp*Ru(MeCN)3]PF6, however, we generated the complete series of π-lignol complexes, including those from coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol as well as derivatives of ferulic and cinnamic acids. From a model dilignol derived from guaiacol and coniferyl alcohol was generated the expected mixture of π-complexes. Competition studies suggest that the allyl alcohol group on lignols does not influence the rate of π-complexation. Competition studies showed that [Cp*Ru(methylferulate)]+ hydrolyzes faster than the free ester.
Co-reporter:Bryan E Barton, Matthew T Olsen, Thomas B Rauchfuss
Current Opinion in Biotechnology (June 2010) Volume 21(Issue 3) pp:292-297
Publication Date(Web):1 June 2010
DOI:10.1016/j.copbio.2010.03.003
Decades of biophysical study on the hydrogenase (H2ase) enzymes have yielded sufficient information to guide the synthesis of analogs of their active sites. Three families of enzymes serve as inspiration for this work: the [FeFe]-H2ases, [NiFe]-H2ases, and [Fe]-H2ases, all of which feature iron centers bound to both CO and thiolate. Artificial H2ases affect the oxidation of H2 and the reverse reaction, the reduction of protons. These reactions occur via the intermediacy of metal hydrides. The inclusion of amine bases within the catalysts is an important design feature that is emulated in related bioinspired catalysts. Continuing challenges are the low reactivity of H2 toward biomimetic H2ases.
Co-reporter:Casseday P. Richers, Jeffery A. Bertke and Thomas B. Rauchfuss
Dalton Transactions 2017 - vol. 46(Issue 27) pp:NaN8762-8762
Publication Date(Web):2017/01/17
DOI:10.1039/C6DT04205H
The paper describes three methods for the preparation of methoxysiloxide complexes, a rare class of complexes of relevance to room temperature vulcanization (RTV) of polysiloxanes. The salt metathesis reaction involves the use of the recently described reagent NaOSi(OMe)2Me with various metal chlorides to give Cp*2Ti[OSi(OMe)2Me](OMe), (Me,MeN2N)NiOSi(OMe)2Me, (IPr)CuOSi(OMe)2Me, and (triphos)CoOSi(OMe)2Me (Cp* = C5Me5, triphos = Me(CH2PPh2)3). Several attempted reactions gave methoxide complexes instead, a pathway that is attributed to the intermediacy of κ2-OSi(OMe)2Me species. The diol Cp*2Zr(OH)2 reacts with excess (MeO)3SiMe to give Cp*2Zr[OSi(OMe)2Me]2. In contrast the less nucleophilic Cp*2Ti(OH)2 was unreactive. The third route to methoxysiloxide complexes involves the reaction of Cp*2M(O)(py) with (MeO)3SiMe to give Cp*2M[OSi(OMe)2Me](OMe) in nearly quantitative yield (M = Ti, Zr). The structures of Cp*2Ti[OSi(OMe)2Me](OMe), Cp*2Zr[OSi(OMe)2Me](OMe), (IPr)CuOSi(OMe)2Me, and (triphos)CoOSi(OMe)2Me were confirmed by single crystal X-ray diffraction.
Co-reporter:David Schilter and Thomas B. Rauchfuss
Dalton Transactions 2012 - vol. 41(Issue 43) pp:NaN13329-13329
Publication Date(Web):2012/09/20
DOI:10.1039/C2DT31895D
Described herein are preparations of synthetic models for the deactivated Ni(II)Fe(II) states of the [NiFe]-hydrogenases. Iodination of the S = ½ species [(dppe)Ni(pdt)Fe(CO)3]+ afforded the diamagnetic iodo complex [(dppe)Ni(pdt)IFe(CO)3]+. Crystallographic analysis of this species confirmed the presence of square-pyramidal Ni linked to an octahedral Fe centre. The Ni⋯Fe separation of 3.018 Å indicated the absence of metal–metal bonding. This complex could be reduced to give (dppe)Ni(pdt)Fe(CO)3 and, in the presence of iodide, decarbonylated to afford (dppe)Ni(pdt)FeI2. Derivatives of the type [(diphosphine)Ni(dithiolate)XFe(CO)2L]+ (X = Cl, Br, I) were prepared by halogenation of mixed-valence precursors [(diphosphine)Ni(dithiolate)Fe(CO)2L]+ (diphosphine = dppe, dcpe; L = tertiary phosphine or CO). The Fe(CO)2(PR3)-containing derivatives are more robust than the related tricarbonyl derivatives. Exploiting this greater stability, we characterised examples of chloride and bromide derivatives. Related fluorides could be prepared by F− abstraction from BF4−. Spectroscopic evidence is presented for the hydroperoxide [(dppe)Ni(pdt)(OOH)Fe(CO)2L]+, which represents a model for the Ni-SU state.
.png)
![Cobalt, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/1054800/17632-19-8.png)
![Cobalt, [2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphinato(2-)-κN21,κN22,κN23,κN24]-, (SP-4-1)-](/data/chemimg/1054800/17632-19-8_b.png)