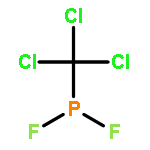
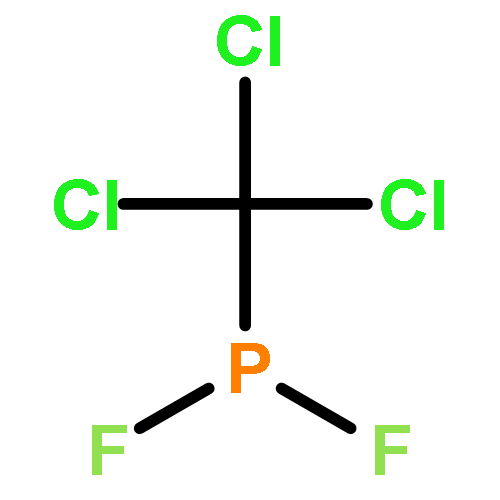
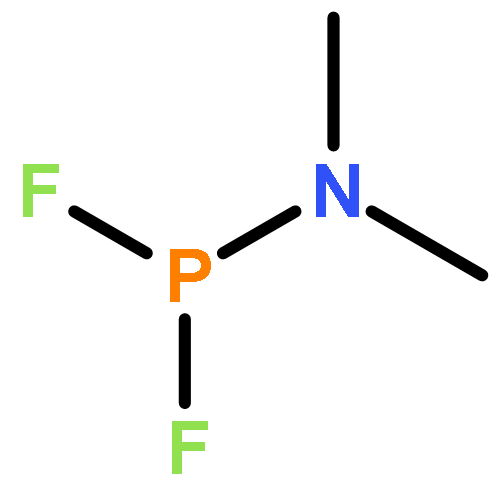
Co-reporter:John F. Nixon
Journal of Fluorine Chemistry 2017 Volume 197(Volume 197) pp:
Publication Date(Web):1 May 2017
DOI:10.1016/j.jfluchem.2017.03.011
Co-reporter:K. Harikrishna Reddy;Dr. Damudi Usharani; John F. Nixon; Eluvathingal D. Jemmis
Chemistry - A European Journal 2011 Volume 17( Issue 33) pp:9142-9152
Publication Date(Web):
DOI:10.1002/chem.201100239
Abstract
The potential energy surfaces of both neutral and dianionic SnC2P2R2 (R=H, tBu) ring systems have been explored at the B3PW91/LANL2DZ (Sn) and 6-311+G* (other atoms) level. In the neutral isomers the global minimum is a nido structure in which a 1,2-diphosphocyclobutadiene ring (1,2-DPCB) is capped by the Sn. Interestingly, the structure established by X-ray diffraction analysis, for R=tBu, is a 1,3-DPCB ring capped by Sn and it is 2.4 kcal mol−1 higher in energy than the 1,2-DPCB ring isomer. This is possibly related to the kinetic stability of the 1,3-DPCB ring, which might originate from the synthetic precursor ZrCp2tBu2C2P2. In the case of the dianionic isomers we observe only a 6π-electron aromatic structure as the global minimum, similarly to the cases of our previously reported results with other types of heterodiphospholes.1, 4, 19 The existence of large numbers of cluster-type isomers in neutral and 6π-planar structures in the dianions SnC2P2R22− (R=H, tBu) is due to 3D aromaticity in neutral clusters and to 2D π aromaticity of the dianionic rings. Relative energies of positional isomers mainly depend on: 1) the valency and coordination number of the Sn centre, 2) individual bond strengths, and 3) the steric effect of tBu groups. A comparison of neutral stannadiphospholes with other structurally related C5H5+ analogues indicates that Sn might be a better isolobal analogue to P+ than to BH or CH+. The variation in global minima in these C5H5+ analogues is due to characteristic features such as 1) the different valencies of C, B, P and Sn, 2) the electron deficiency of B, 3) weaker pπ–pπ bonding by P and Sn atoms, and 4) the tendency of electropositive elements to donate electrons to nido clusters. Unlike the C5H5+ systems, all C5H5− analogues have 6π-planar aromatic structures as global minima. The differences in the relative ordering of the positional isomers and ligating properties are significant and depend on 1) the nature of the π orbitals involved, and 2) effective overlap of orbitals.
Co-reporter:Scott B. Clendenning, Peter B. Hitchcock, Gerard A. Lawless, John F. Nixon, Christopher W. Tate
Journal of Organometallic Chemistry 2010 695(5) pp: 717-720
Publication Date(Web):
DOI:10.1016/j.jorganchem.2009.12.005
Co-reporter:Mahmoud M. Al-Ktaifani, Martyn P. Coles, Ian R. Crossley, Lloyd T.J. Evans, Peter B. Hitchcock, Gerard A. Lawless, John F. Nixon
Journal of Organometallic Chemistry 2009 694(26) pp: 4223-4229
Publication Date(Web):
DOI:10.1016/j.jorganchem.2009.09.002
Co-reporter:Nathan M. West, Peter S. White, Joseph L. Templeton and John F. Nixon
Organometallics 2009 Volume 28(Issue 5) pp:1425-1434
Publication Date(Web):February 11, 2009
DOI:10.1021/om800968v
Combining (Cl-nacnac)Pt(H)(1-pentene) (Cl-nacnac = N-p-Cl-phenyl-β-diiminate) and HSiR3 (SiR3 = SiPh3, SiEt3, SiHPh2) leads to rapid displacement of 1-pentene; oxidative addition of the Si−H bond produces a stable 5-coordinate Pt(IV) silyl dihydride complex which has been isolated and characterized crystallographically. The 5-coordinate silyl dihydride platinum(IV) complex undergoes a cycloaddition reaction with either acetylene or phosphaalkyne to form a vinyl or phosphaalkenyl Pt(IV) product, respectively. One acetylenic carbon adds to the central methine carbon of the Cl-nacnac ligand and the other alkyne carbon or phosphorus coordinates to the vacant site of the d6 metal. Phosphaalkynes are polarized such that the carbon is at the negative end of the dipole; however we observe a final product that reflects a reversal of this normal polarity. The phosphorus is nucleophilic even after the cycloaddition reaction is complete, and its lone pair can be methylated by MeOTf (OTf = trifluoromethanesulfonate) or protonated by addition of HBF4·Et2O.
Co-reporter:Scott B. Clendenning Dr.;Peter B. Hitchcock Dr.;Michael F. Lappert Dr.;Philippe G. Merle Dr.;John F. Nixon Dr.;László Nyulászi Dr.
Chemistry - A European Journal 2007 Volume 13(Issue 25) pp:
Publication Date(Web):11 JUN 2007
DOI:10.1002/chem.200601626
Treatment of the lithium amide Li[NPh(SiMe3)] with 2,4,6-tri-tert-butyl-1,3,5-triphosphabenzene, P3C3tBu3, in a 1:2 ratio afforded equimolar amounts of the lithium salt of the five-membered 2,4,5-tri-tert-butyl-1,3-diphospholide anion, LiP2C3tBu3 (isolated as its N,N,N′,N′-tetramethylethylenediamine (TMEDA) adduct), and the tricyclic compound 6-[phenyl(trimethylsilyl)amino]-3,5,7-tri-tert-butyl-1,2,4,6-tetraphosphatricyclo[3.2.0.02,7]hept-3-ene. Both compounds have been structurally characterised by single-crystal X-ray diffraction studies. The mechanism of this remarkable reaction has been elucidated by theoretical methods at the B3LYP/6-311+G** level of theory. The reaction involves a hitherto unobserved aminophosphinidene, which was formed by abstraction of a phosphorus atom from triphosphabenzene. The intermediate aminophosphinidene, which is further stabilised by the solvent THF, shows, in agreement with previous theoretical predictions, enhanced stability and reacts then with a second molecule of triphosphabenzene.
Co-reporter:F. Geoffrey, N. Cloke, Peter B. Hitchcock, John F. Nixon, D. James Wilson, László Nyulászi, Tamás Kárpáti
Journal of Organometallic Chemistry 2005 Volume 690(Issue 17) pp:3983-3989
Publication Date(Web):1 September 2005
DOI:10.1016/j.jorganchem.2005.05.046
The 1-triphenylstannyl-2, 4, 5-tritertiarybutyl-1,3-diphosphole, Ph3SnP2C3Bu3t, which has been synthesised from Ph3SnCl and KP2C3Bu3t, has been fully structurally characterised in the solid state but in solution undergoes ready intra-molecular shifts of the Ph3Sn-group around the 1,3-diphospholyl ring. Theoretical calculations concerning both the structure and the dynamic process are presented and discussed.The preparation and theoretical discussion of the first stanna-1,3-diphosphole is described.
Co-reporter:Sarah E d'Arbeloff–Wilson, Peter B Hitchcock, John F Nixon, Hiroyuki Kawaguchi, Kazuyuki Tatsumi
Journal of Organometallic Chemistry 2003 Volume 672(1–2) pp:1-10
Publication Date(Web):14 April 2003
DOI:10.1016/S0022-328X(03)00047-0
The chalcogenide complexes [Zr(η5-(C5Me5)2(E))] (E=S, Se), (generated in situ from the corresponding pyridine adducts), undergo [2+2] cyclo-addition reactions with the phospha-alkyne tBuCP to afford the structurally characterised complexes [Zr(η5-(C5Me5)2(SC(tBu)P))] and [Zr(η5-(C5Me5)2(SeC(tBu)P))]. The former complex undergoes ring expansion reactions with either selenium to afford [Zr(η5-(C5Me5)2(SC(tBu)PSe))] or with PhCN to give [Zr(η5(C5Me5)2(SC(tBu)PC(Ph)N))] which have also been characterised structurally.[2+2] Cyclo-addition reactions of chalcogenide (S, Se and Te) complexes of zirconium with the phospha-alkyne tBuCP are reported and further ring expansions with chalcogens and PhCN are described. NMR and single crystal data of the products are presented.
Co-reporter:Mahmoud M Al-Ktaifani, Peter B Hitchcock, John F Nixon
Inorganica Chimica Acta 2003 Volume 356() pp:103-108
Publication Date(Web):3 December 2003
DOI:10.1016/S0020-1693(03)00340-2
UV irradiation of the hexaphospha-pentaprismane cage, P6C4tBu4, causes a structural rearrangement affording two new isomers, one involving a different closed cage structure and the other a more open cage structure containing one PC double bond. The new closed cage isomer was also unexpectedly obtained as the sole product by protonation of P6C4tBu4 with CF3SO3H. Both new P6C4tBu4 isomers were fully characterised by multinuclear NMR spectroscopy and single crystal X-ray diffraction studies.UV irradiation of the hexaphospha-pentaprismane cage, P6C4tBu4, affords two fully structurally characterised new isomers, one involving a different closed cage structure and the other a more open cage structure containing one PC double bond. The former isomer was also unexpectedly obtained as the sole product via protonation of the hexaphospha-pentaprismane with CF3SO3H.
Co-reporter:Guy K. B. Clentsmith Dr.;F. Geoffrey N. Cloke Dr.;Jennifer C. Green Dr.;John Hanks Dr.;Peter B. Hitchcock Dr.;John F. Nixon Dr.
Angewandte Chemie International Edition 2003 Volume 42(Issue 9) pp:
Publication Date(Web):26 FEB 2003
DOI:10.1002/anie.200390267
ScIIor mixed oxidation state ScIII/ScI? Reduction of the homoleptic ScIII complex [Sc(P3C2tBu2)3] allows access to the subvalent [{Sc(P3C2tBu2)2}2] complex (see picture), in which the metal centers are formally bivalent. DFT calculations support fomulation of the reduced species as a mixed oxidation state ScIII/ScI complex.
Co-reporter:Mahmoud M. Al-Ktaifani;Walter Bauer Priv.-Doz. Dr.;Uwe Bergsträßer Dr.;Bernhard Breit Dr.;Matthew D. Francis Dr.;Frank W. Heinemann Dr.;Peter B. Hitchcock Dr.;Andreas Mack Dr.;John F. Nixon ;Hans Pritzkow Dr.;Manfred Regitz Dr.;Matthias Zeller Dr.;Ulrich Zenneck Dr.
Chemistry - A European Journal 2002 Volume 8(Issue 11) pp:
Publication Date(Web):17 MAY 2002
DOI:10.1002/1521-3765(20020603)8:11<2622::AID-CHEM2622>3.0.CO;2-#
Several independent synthetic routes are described leading to the formation of a novel unsaturated tetracyclic phosphorus carbon cage compound tBu4C4P6 (1), which undergoes a light-induced valence isomerization to produce the first hexaphosphapentaprismane cage tBu4C4P6 (2). A second unsaturated isomer tBu4C4P6 (9) of 1 and the bis-[W(CO)5] complex 13 of 1 are stable towards similar isomerization reactions. Another starting material for the synthesis of the hexaphosphapentaprismane cage tBu4C4P6 (2) is the trimeric mercury complex [(tBu4C4P6)Hg]3 (11), which undergoes elimination of mercury to afford the title compound 2. Single-crystal X-ray structural determinations have been carried out on compounds 1, 2, 9, 11, and 13.
Co-reporter:Mahmoud M Al-Ktaifani, Matthew D Francis, Peter B Hitchcock, John F Nixon
Journal of Organometallic Chemistry 2001 Volume 633(1–2) pp:143-148
Publication Date(Web):10 August 2001
DOI:10.1016/S0022-328X(01)01061-0
The reaction between zinc or cadmium dichloride and the triphospholyl anion P3C2tBu2− in each case leads to highly insoluble orange compounds, the structures of which are believed to be polymeric. Subsequent treatment with pyridine leads to the monomeric complexes [M(η1-P3C2tBu2)2(NC5H5)n] (M=Zn, n=2; M=Cd, n=3). Both complexes, which are fluxional in solution even at low temperature, have been fully characterised by multinuclear NMR spectroscopy and single crystal X-ray diffraction studies.The reaction between zinc or cadmium dichloride and the triphospholyl anion P3C2tBu2− followed by subsequent treatment with pyridine leads to the monomeric complexes [M(η1-P3C2tBu2)2(NC5H5)n] (M=Zn, n=2; M=Cd, n=3). Both complexes, which are fluxional in solution even at low temperature, have been fully characterised by multinuclear NMR spectroscopy and single crystal X-ray diffraction studies.
Co-reporter:Mahmoud M. Al-Ktaifani;Daniel P. Chapman;Matthew D. Francis Dr.;Peter B. Hitchcock Dr.;John F. Nixon Dr.;László Nyulászi Dr.
Angewandte Chemie International Edition 2001 Volume 40(Issue 18) pp:
Publication Date(Web):14 SEP 2001
DOI:10.1002/1521-3773(20010917)40:18<3474::AID-ANIE3474>3.0.CO;2-K
Exclusive opening of the unique P−P bond with formation of 2 is observed when 1 is treated with carbene analogues [Eq. (1)]! This behavior is explicable by the release of strain resulting in a low-lying (20 kcal mol−1) reactive singlet biradical. The HOMO and LUMO centred at the reactive sites and the thermodynamic stability of the products support this behavior.
Co-reporter:Mahmoud M. Al-Ktaifani;Daniel P. Chapman;Matthew D. Francis Dr.;Peter B. Hitchcock Dr.;John F. Nixon Dr.;László Nyulászi Dr.
Angewandte Chemie 2001 Volume 113(Issue 18) pp:
Publication Date(Web):14 SEP 2001
DOI:10.1002/1521-3757(20010917)113:18<3582::AID-ANGE3582>3.0.CO;2-K
Gezielt geöffnet wird nur die zentrale P-P-Bindung unter Bildung von 2, wenn 1 mit Carbenanaloga umgesetzt wird [Gl. (1)]! Dieses Verhalten kann mit dem Verlust an Ringspannung erklärt werden, der zu einem energetisch niedrig liegenden (20 kcal mol−1) reaktiven Singulett-Diradikal führt. Das HOMO und das LUMO, die sich beide an den Reaktionszentren befinden, und die thermodynamische Stabilität der Produkte unterstützen dieses Verhalten.
Co-reporter:F. Geoffrey N. Cloke, Peter B. Hitchcock, John F. Nixon and D. James Wilson
Chemical Communications 2000 (Issue 23) pp:2387-2388
Publication Date(Web):22 Nov 2000
DOI:10.1039/B007512O
Treatment of a dichloromethane solution of ButCP,
containing a catalytic amount of TiCl4, with an excess of the
primary amines RNH2 (R = Pri, But)
quantitatively affords the corresponding bis-dialkylaminophosphine
P(ButNH)2- CH2But.
P(PriNH)2CH2But is structurally
characterised by a single crystal X-ray diffraction study of its
bis-trans-PdCl2 complex.
Co-reporter:Matthew D. Francis, Peter B. Hitchcock and John F. Nixon
Chemical Communications 2000 (Issue 20) pp:2027-2028
Publication Date(Web):28 Sep 2000
DOI:10.1039/B005855F
Treatment of SrI2 with 2 equivalents of
K[P3C2But2] affords the
hexaphosphastrontocene
[{Sr(η5-P3C2But2
)2}∞] which is polymeric in the
solid state; treatment of this complex with an excess of pyrazine leads to
the corresponding Lewis base adduct
[{Sr(η5-P3C2But2
)2(μ-C4H4N2)}
∞] which is also polymeric in the solid state.
Co-reporter:Peter B Hitchcock, John F Nixon, Nurgün Sakarya
Journal of Organometallic Chemistry 2000 Volume 601(Issue 2) pp:335-340
Publication Date(Web):28 April 2000
DOI:10.1016/S0022-328X(00)00106-6
Treatment of the 1,2,4-triphosphole, P3C2But2(CH(SiMe3)2), with [Co2(CO)8] in THF under mild conditions affords the binuclear complex [Co2(CO)5(μ-P3C2But2CH(SiMe3)2)], in which the ring acts as a six-electron-donor. Under slightly more vigorous reaction conditions an H-transfer reaction occurs (presumably from the solvent) to afford the mononuclear compound [Co(CO)2(P3C2But2H(CH(SiMe3)2)] containing an η4-ligated five-electron-donor P3C2But2H(CH(SiMe3)2 ring system. The molecular structures of both compounds have been determined by single-crystal X-ray diffraction studies.
Co-reporter:Charlotte S.J Callaghan, Peter B Hitchcock, John F Nixon
Journal of Organometallic Chemistry 1999 Volume 584(Issue 1) pp:87-93
Publication Date(Web):10 July 1999
DOI:10.1016/S0022-328X(99)00097-2
Single-crystal X-ray diffraction studies on the triphosphaferrocene [Fe(η5-C5H5)(η5P3C2tBu2)] and its [Rh(η5-C5Me5)CO] complex reveal an unusual η2-ring edge coordination mode in the latter resulting in a significant elongation of the triphosphacyclopentadienyl ring P–P bond.
Co-reporter:F. Geoffrey N. Cloke;Peter B. Hitchcock;Philip Hunnable;John F. Nixon;Lászlo Nyulászi;Edgar Niecke;Vera Thelen
Angewandte Chemie 1998 Volume 110(Issue 8) pp:
Publication Date(Web):12 MAR 1999
DOI:10.1002/(SICI)1521-3757(19980420)110:8<1139::AID-ANGE1139>3.0.CO;2-N
Die sterisch anspruchsvolle Gruppe am dreifach koordinierten Phosphoratom, die π-Elektronen-Acceptorsubstituenten und die zweifach koordinierten Phosphoratome im Ring sind die wesentlichen Faktoren, die dazu beitragen, daß der 1,2,4-Triphospholring in 1 planar ist. Gemessen am Bird-Aromatizitätsindex ist 1 dasjenige unter den bekannten Phospholen, das den am stärksten ausgeprägten aromatischen Charakter aufweist.
Co-reporter:F. Geoffrey N. Cloke;Peter B. Hitchcock;Philip Hunnable;John F. Nixon;Lászlo Nyulászi;Edgar Niecke;Vera Thelen
Angewandte Chemie International Edition 1998 Volume 37(Issue 8) pp:
Publication Date(Web):17 DEC 1998
DOI:10.1002/(SICI)1521-3773(19980504)37:8<1083::AID-ANIE1083>3.0.CO;2-C
The sterically demanding groups on the tricoordinate phosphorus atom, the π-electron acceptors substituted on the ring, and the dicoordinate phosphorus atoms within the ring are the most significant factors contributing to the planarity and aromaticity of the 1,2,4-triphosphole ring in 1. The Bird aromaticity index for 1 shows that it has the most pronounced aromatic character of all known phospholes.