Co-reporter:Yong Liang, Sazzad H. Suzol, Zhiwei Wen, Alain G. Artiles, Logesh Mathivathanan, Raphael G. Raptis, and Stanislaw F. Wnuk
Organic Letters 2016 Volume 18(Issue 6) pp:1418-1421
Publication Date(Web):March 2, 2016
DOI:10.1021/acs.orglett.6b00346
The transition-metal-catalyzed or radical-mediated halosulfonylation of 5-ethynyluridine provided (E)-(1-halo-2-tosylvinyl)uridines. These (β-halo)vinyl sulfones undergo efficient stereoselective addition–elimination with amines or thiols to provide Z-β-aminovinyl or E-β-thiovinyl sulfones tethered to the C5 position of the uracil ring.
Co-reporter:Sam Kavoosi, Ramanjaneyulu Rayala, Brenna Walsh, Maria Barrios, Walter G. Gonzalez, Jaroslava Miksovska, Logesh Mathivathanan, Raphael G. Raptis, Stanislaw F. Wnuk
Tetrahedron Letters 2016 Volume 57(Issue 39) pp:4364-4367
Publication Date(Web):28 September 2016
DOI:10.1016/j.tetlet.2016.08.053
•Efficient bromination of 7-deazapurine nucleosides with DBH in MeOH.•Click reactions of 8-azidotoyocamycin with alkynes gave fluorescent triazoles.•Direct C8H bond functionalization of tubercidin with benzotriazoles.Treatment of toyocamycin or sangivamycin with 1,3-dibromo-5,5-dimethylhydantoin in MeOH (rt/30 min) gave 8-bromotoyocamycin and 8-bromosangivamycin in good yields. Nucleophilic aromatic substitution of 8-bromotoyocamycin with sodium azide provided novel 8-azidotoyocamycin. Strain promoted click reactions of the latter with cyclooctynes resulted in the formation of the 1,2,3-triazole products. Iodine-mediated direct C8H bond functionalization of tubercidin with benzotriazoles in the presence of tert-butyl hydroperoxide gave the corresponding 8-benzotriazolyltubercidin derivatives. The 8-(1,2,3-triazol-1-yl)-7-deazapurine derivatives showed moderate quantum yields and a large Stokes shifts of ∼100 nm.
Co-reporter:Cesar Gonzalez, Sam Kavoosi, Andersson Sanchez, Stanislaw F. Wnuk
Carbohydrate Research 2016 Volume 432() pp:17-22
Publication Date(Web):2 September 2016
DOI:10.1016/j.carres.2016.06.002
•Lithium triethylborohydride reduces lactones to pentose or hexose hemiacetals.•Reduction is higher yielding when lactone has a larger number of hydroxyl groups.•Reduction is more efficient in CH2Cl2 solution.•Reduction is compatible with protecting groups commonly used in sugar chemistry.Reduction of ribono-1,4-lactones and gulono-1,4-lactone as well as ribono-1,5-lactone and glucono-1,5-lactones with LTBH (1.2 equiv.) in CH2Cl2 at 0 °C for 30 min provided the corresponding pentose or hexose hemiacetals in high yields. Commonly used in carbohydrate chemistry protecting groups such as trityl, benzyl, silyl, acetals and to some extent acyls are compatible with this reduction.
Co-reporter:Ramanjaneyulu Rayala, Alain Giuglio-Tonolo, Julie Broggi, Thierry Terme, Patrice Vanelle, Patricia Theard, Maurice Médebielle, Stanislaw F. Wnuk
Tetrahedron 2016 Volume 72(Issue 16) pp:1969-1977
Publication Date(Web):21 April 2016
DOI:10.1016/j.tet.2016.02.063
Studies directed toward the oxidative and reductive desulfurization of readily available 2′-S-aryl-2′-thiouridine derivatives were investigated with the prospect to functionalize the C2′-position of nucleosides. The oxidative desulfurization-difluorination strategy was successful on 2-(arylthio)alkanoate surrogates, while extension of the combination of oxidants and fluoride sources was not an efficient fluorination protocol when applied to 2′-S-aryl-2′-thiouridine derivatives, resulting mainly in C5-halogenation of the pyrimidine ring and C2′-monofluorination without desulfurization. Cyclic voltammetry of 2′-arylsulfonyl-2′-deoxyuridines and their 2′-fluorinated analogues showed that cleavage of the arylsulfone moiety could occur, although at relatively high cathodic potentials. While reductive-desulfonylation of 2′-arylsulfonyl-2′-deoxyuridines with organic electron donors (OEDs) gave predominantly base-induced furan type products, chemical (OED) and electrochemical reductive-desulfonylation of the α-fluorosulfone derivatives yielded the 2′-deoxy-2′-fluorouridine and 2′,3′-didehydro-2′,3′-dideoxy-2′-fluorouridine derivatives. These results provided good evidence of the generation of a C2′-anion through carbon-sulfur bond cleavage, opening new horizons for the reductive-functionalization approaches in nucleosides.New strategies for the modification of nucleosides at the C2ʹ-position via oxidative and reductive activation of carbon–sulfur bonds in readily available 2ʹ-thionucleosides were studied. Oxidative fluorination of A gave α-fluorothioether B. Reductive desulfonylation of α-fluorosulfone E yielded fluoro nucleosides D and/or C providing evidence of the generation of a C2ʹ-anion through carbon-sulfur bond cleavage.
Co-reporter:Qing He, Liwen Wang, Yong Liang, Zunting Zhang, and Stanislaw F. Wnuk
The Journal of Organic Chemistry 2016 Volume 81(Issue 19) pp:9422-9427
Publication Date(Web):August 29, 2016
DOI:10.1021/acs.joc.6b01648
Transition-metal-free LiCl-promoted cross-coupling reactions of tetraphenyltin, trichlorophenyl-, dichlorodiphenyl-, and chlorotriphenylstannanes with aryl halides in DMF provided access to biaryls in good to high yields. Up to four phenyl groups were transferred from the organostannanes substrates. The aryls bearing electron-withdrawing groups in either halides or organotin substrates gave coupling products in higher yields. The methodology has been applied for the efficient synthesis of ipriflavones.
Co-reporter:Jessica Zayas, Marie Annoual, Jayanta Kumar Das, Quentin Felty, Walter G. Gonzalez, Jaroslava Miksovska, Nima Sharifai, Akira Chiba, and Stanislaw F. Wnuk
Bioconjugate Chemistry 2015 Volume 26(Issue 8) pp:1519
Publication Date(Web):June 18, 2015
DOI:10.1021/acs.bioconjchem.5b00300
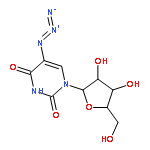
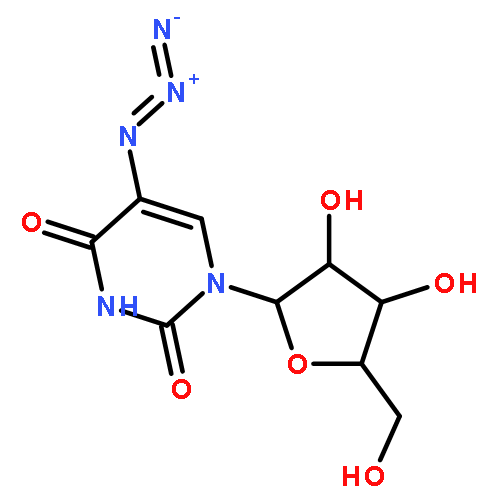
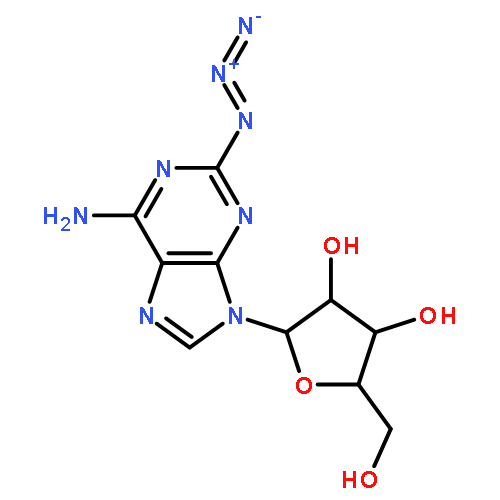
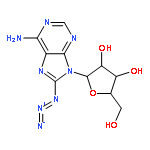
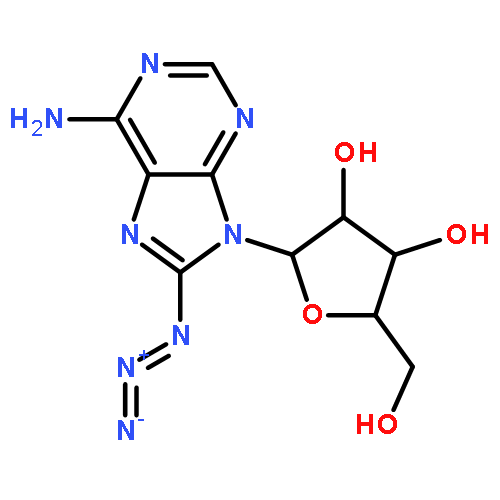
Strain-promoted click chemistry of nucleosides and nucleotides with an azido group directly attached to the purine and pyrimidine rings with various cyclooctynes in aqueous solution at ambient temperature resulted in efficient formation (3 min to 3 h) of fluorescent, light-up, triazole products. The 2- and 8-azidoadenine nucleosides reacted with fused cyclopropyl cyclooctyne, dibenzylcyclooctyne, or monofluorocyclooctyne to produce click products functionalized with hydroxyl, amino, N-hydroxysuccinimide, or biotin moieties. The 5-azidouridine and 5-azido-2′-deoxyuridine were similarly converted to the analogous triazole products in quantitative yields in less than 5 min. The 8-azido-ATP quantitatively afforded the triazole product with fused cyclopropyl cyclooctyne in aqueous acetonitrile (3 h). The novel triazole adducts at the 2- or 8-position of adenine or 5-position of uracil rings induce fluorescence properties which were used for direct imaging in MCF-7 cancer cells without the need for traditional fluorogenic reporters. FLIM of the triazole click adducts demonstrated their potential utility for dynamic measuring and tracking of signaling events inside single living cancer cells.
Co-reporter:Amitava Adhikary ; Anil Kumar ; Ramanjaneyulu Rayala ; Ragda M. Hindi ; Ananya Adhikary ; Stanislaw F. Wnuk ;Michael D. Sevilla
Journal of the American Chemical Society 2014 Volume 136(Issue 44) pp:15646-15653
Publication Date(Web):October 8, 2014
DOI:10.1021/ja5083156
Gemcitabine is a modified cytidine analog having two fluorine atoms at the 2′-position of the ribose ring. It has been proposed that gemcitabine inhibits RNR activity by producing a C3′• intermediate via direct H3′-atom abstraction followed by loss of HF to yield a C2′• with 3′-keto moiety. Direct detection of C3′• and C2′• during RNR inactivation by gemcitabine still remains elusive. To test the influence of 2′- substitution on radical site formation, electron spin resonance (ESR) studies are carried out on one-electron oxidized gemcitabine and other 2′-modified analogs, i.e., 2′-deoxy-2′-fluoro-2′-C-methylcytidine (MeFdC) and 2′-fluoro-2′-deoxycytidine (2′-FdC). ESR line components from two anisotropic β-2′-F-atom hyperfine couplings identify the C3′• formation in one-electron oxidized gemcitabine, but no further reaction to C2′• is found. One-electron oxidized 2′-FdC is unreactive toward C3′• or C2′• formation. In one-electron oxidized MeFdC, ESR studies show C2′• production presumably from a very unstable C3′• precursor. The experimentally observed hyperfine couplings for C2′• and C3′• match well with the theoretically predicted ones. C3′• to C2′• conversion in one-electron oxidized gemcitabine and MeFdC has theoretically been modeled by first considering the C3′• and H3O+ formation via H3′-proton deprotonation and the subsequent C2′• formation via HF loss induced by this proximate H3O+. Theoretical calculations show that in gemcitabine, C3′• to C2′• conversion in the presence of a proximate H3O+ has a barrier in agreement with the experimentally observed lack of C3′• to C2′• conversion. In contrast, in MeFdC, the loss of HF from C3′• in the presence of a proximate H3O+ is barrierless resulting in C2′• formation which agrees with the experimentally observed rapid C2′• formation.
Co-reporter:Jesse Pulido ; Adam J. Sobczak ; Jan Balzarini
Journal of Medicinal Chemistry 2014 Volume 57(Issue 1) pp:191-203
Publication Date(Web):December 16, 2013
DOI:10.1021/jm401586a
The coupling of gemcitabine with functionalized carboxylic acids (C9–C13) or reactions of 4-N-tosylgemcitabine with the corresponding alkyl amines afforded 4-N-alkanoyl and 4-N-alkyl gemcitabine derivatives. The analogues with a terminal hydroxyl group on the alkyl chain were efficiently fluorinated under conditions that are compatible with protocols for 18F labeling. The 4-N-alkanoylgemcitabines showed potent cytostatic activities in the low nanomolar range against a panel of tumor cell lines, whereas cytotoxicity of the 4-N-alkylgemcitabines were in the low micromolar range. The cytotoxicity for the 4-N-alkanoylgemcitabine analogues was reduced approximately by 2 orders of magnitude in the 2′-deoxycytidine kinase (dCK)-deficient CEM/dCK– cell line, whereas cytotoxicity of the 4-N-alkylgemcitabines was only 2–5 times lower. None of the compounds acted as efficient substrates for cytosolic dCK; therefore, the 4-N-alkanoyl analogues need to be converted first to gemcitabine to display a significant cytostatic potential, whereas 4-N-alkyl derivatives attain modest activity without measurable conversion to gemcitabine.
Co-reporter:Mu-Lin Zhu;Zun-Ting Zhang;Dong Xue;Hui-Liang Hua;Yong Liang
Helvetica Chimica Acta 2014 Volume 97( Issue 4) pp:561-568
Publication Date(Web):
DOI:10.1002/hlca.201300246
Abstract
The one-step cyclocondensation of substituted isoflavones (=3-phenyl-4H-1-benzopyran-4-ones) with cyanoacetohydrazide in the presence of KOH afforded a mixture of 1-amino-5,6-diaryl-3-cyano-1H-2-pyridin-2-ones and 6,7-diaryl-4-cyano-3-hydroxy-1H-[1,2]diazepines.
Co-reporter:Ramanjaneyulu Rayala;Patricia Theard;Heysell Ortiz;Dr. Sylvia Yao; James D. Young; Jan Balzarini; Morris J. Robins; Stanislaw F. Wnuk
ChemMedChem 2014 Volume 9( Issue 9) pp:2186-2192
Publication Date(Web):
DOI:10.1002/cmdc.201402047
Abstract
Human equilibrative nucleoside transporter 1 (hENT1) is a prototypical nucleoside transporter protein ubiquitously expressed on the cell surface of almost all human tissue. Given the role of hENT1 in the transport of nucleoside drugs, an important class of therapeutics in the treatment of various cancers and viral infections, efforts have been made to better understand the mechanisms by which hENT1 modulates nucleoside transport. To that end, we report here the design and synthesis of novel tool compounds for the further study of hENT1. The 7-deazapurine nucleoside antibiotic tubercidin was converted into its 4-N-benzyl and 4-N-(4-nitrobenzyl) derivatives by alkylation at N3 followed by a Dimroth rearrangement to the 4-N-isomer or by fluoro-diazotization followed by SNAr displacement of the 4-fluoro group by a benzylamine. The 4-N-(4-nitrobenzyl) derivatives of sangivamycin and toyocamycin antibiotics were prepared by the alkylation approach. Cross-membrane transport of labeled uridine by hENT1 was inhibited to a weaker extent by the 4-nitrobenzylated tubercidin and sangivamycin analogues than was observed with 6-N-(4-nitrobenzyl)adenosine. Type-specific inhibition of cancer cell proliferation was observed at micromolar concentrations with the 4-N-(4-nitrobenzyl) derivatives of sangivamycin and toyocamycin, and also with 4-N-benzyltubercidin. Treatment of 2′,3′,5′-O-acetyladenosine with aryl isocyanates gave the 6-ureido derivatives but none of them exhibited inhibitory activity against cancer cell proliferation or hENT1.
Co-reporter:Yong Liang, Jennifer Gloudeman, and Stanislaw F. Wnuk
The Journal of Organic Chemistry 2014 Volume 79(Issue 9) pp:4094-4103
Publication Date(Web):April 11, 2014
DOI:10.1021/jo500602p
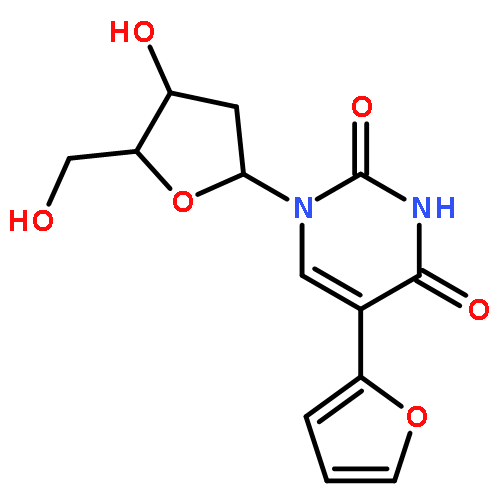
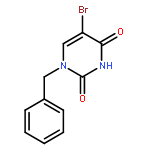
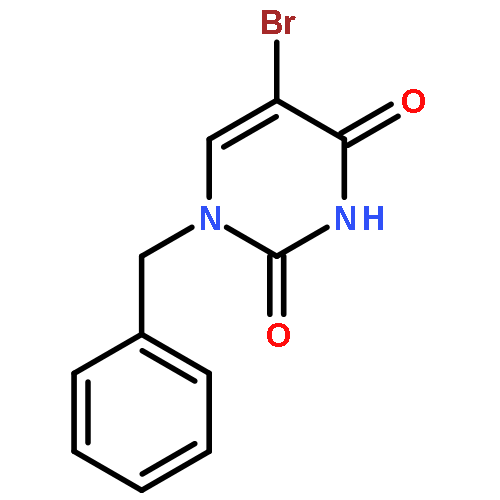
The 1-N-benzyl-5-iodo(or bromo)uracil undergoes Pd-catalyzed [Pd2(dba)3] direct arylation with benzene and other simple arenes in the presence of TBAF in DMF without the necessity of adding any ligands or additives to give 5-arylated uracil analogues. The TBAF-promoted coupling also occurs efficiently with electron rich heteroarenes at 100 °C (1 h) even with only small excess of heteroarenes. The protocol avoids usage of the arylboronic acid or stannane precursors for the synthesis of 5-(2-furyl, or 2-thienyl, or 2-pyrrolyl)uracil nucleosides, which are used as important RNA and DNA fluorescent probes. The fact that 1-N-benzyl-3-N-methyl-5-iodouracil did not undergo the TBAF-promoted couplings with arenes or heteroarenes suggests that the C4-alkoxide (enol form of uracil) facilitates coupling by participation in the intramolecular processes of hydrogen abstraction from arenes. TBAF-promoted arylation was extended into the other enolizable heterocyclic systems such as 3-bromo-2-pyridone. The π-excessive heteroarenes also coupled with 5-halouracils in the presence of Pd(OAc)2/Cs2CO3/PivOH combination in DMF (100 °C, 2 h) to yield 5-arylated uracils.
Co-reporter:Yong Liang, Jean-Philippe Pitteloud, and Stanislaw F. Wnuk
The Journal of Organic Chemistry 2013 Volume 78(Issue 11) pp:5761-5767
Publication Date(Web):April 30, 2013
DOI:10.1021/jo400590z
A stereoselective radical-mediated hydrogermylation of the protected 5-ethynyluracil nucleosides with trialkyl-, triaryl,- or tris(trimethylsilyl)germanes gave (Z)-5-(2-germylvinyl)uridine, 2′-deoxyuridine, or ara-uridine as major products. Reaction of the β-triphenylgermyl vinyl radical intermediate with oxygen and fragmentation of the resulting peroxyradical provided also 5-[2-(triphenylgermyl)acetyl]pyrimidine nucleosides in low to moderate yields. Thermal isomerization of the latter in MeOH occurred via a four-centered activated complex, and subsequent hydrolysis of the resulting O-germyl substituted enol yielded 5-acetyluracil nucleosides in quantitative yield.
Co-reporter:Ramanjaneyulu Rayala, Stanislaw F. Wnuk
Tetrahedron Letters 2012 Volume 53(Issue 26) pp:3333-3336
Publication Date(Web):27 June 2012
DOI:10.1016/j.tetlet.2012.04.081
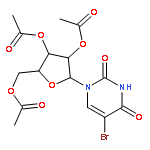
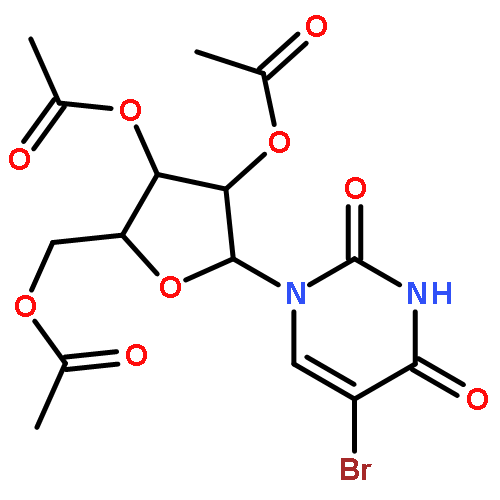
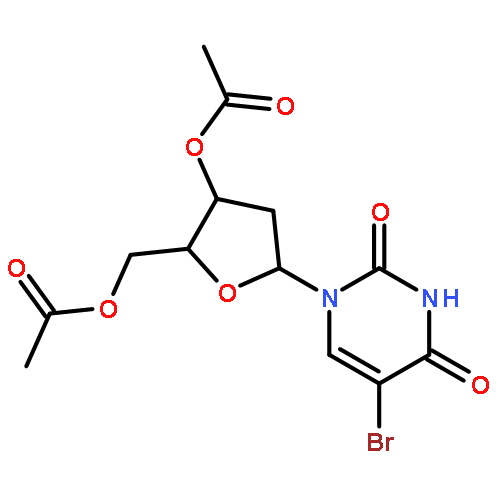
Treatment of the protected and unprotected nucleosides with 1,3-dibromo-5,5-dimethylhydantoin in aprotic solvents such as CH2Cl2, CH3CN, or DMF effected smooth bromination of uridine and cytidine derivatives at C-5 of pyrimidine rings as well as adenosine and guanosine derivatives at C-8 of purine rings. Addition of Lewis acids such as trimethylsilyl trifluoromethanesulfonate enhanced the efficiency of bromination.Bromination of pyrimidine nucleosides at C-5 position and purine nucleosides at C-8 position can be conveniently carried out with 1,3-dibromo-5,5-dimethylhydantoin in aprotic solvents (48–98% yield).
Co-reporter:Thao P. Dang, Adam J. Sobczak, Alexander M. Mebel, Chryssostomos Chatgilialoglu, Stanislaw F. Wnuk
Tetrahedron 2012 68(27–28) pp: 5655-5667
Publication Date(Web):
DOI:10.1016/j.tet.2012.04.050
Co-reporter:Venkata L.A. Malladi, Adam J. Sobczak, Natalie Maricic, Senthil Kumar Murugapiran, Lisa Schneper, John Makemson, Kalai Mathee, Stanislaw F. Wnuk
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 18) pp:5500-5506
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmc.2011.07.044
Quorum sensing (QS) is a population-dependent signaling process bacteria use to control multiple processes including virulence that is critical for establishing infection. The most common QS signaling molecule used by Gram-negative bacteria are acylhomoserine lactones. The development of non-native acylhomoserine lactone (AHL) ligands has emerged as a promising new strategy to inhibit QS in Gram-negative bacteria. In this work, we have synthesized a set of optically pure γ-lactams and their reduced cyclic azahemiacetal analogues, bearing the additional alkylthiomethyl substituent, and evaluated their effect on the AHL-dependent Pseudomonas aeruginosa las and rhl QS pathways. The concentration of these ligands and the simple structural modification such as the length of the alkylthio substituent has notable effect on activity. The γ-lactam derivatives with nonylthio or dodecylthio chains acted as inhibitors of las signaling with moderate potency. The cyclic azahemiacetal with shorter propylthio or hexylthio substituent was found to strongly inhibit both las and rhl signaling at higher concentrations while the propylthio analogue strongly stimulated the las QS system at lower concentrations.The cyclic azahemiacetals with propylthio or hexylthio substituent strongly inhibit both las and rhl signaling at higher concentrations while the propylthio analogue stimulates the las QS system at lower concentrations.
Co-reporter:Venkata L.A. Malladi, Adam J. Sobczak, Tiffany M. Meyer, Dehua Pei, Stanislaw F. Wnuk
Bioorganic & Medicinal Chemistry 2011 Volume 19(Issue 18) pp:5507-5519
Publication Date(Web):15 September 2011
DOI:10.1016/j.bmc.2011.07.043
LuxS (S-ribosylhomocysteinase) catalyzes the cleavage of the thioether linkage of S-ribosylhomocysteine (SRH) to produce homocysteine and 4,5-dihydroxy-2,3-pentanedione (DPD), the precursor to a small signaling molecule that mediates interspecies bacterial communication called autoinducer 2 (AI-2). Inhibitors of LuxS should interfere with bacterial interspecies communication and potentially provide a novel class of antibacterial agents. In this work, SRH analogues containing substitution of a nitrogen atom for the endocyclic oxygen as well as various deoxyriboses were synthesized and evaluated for LuxS inhibition. Two of the [4-aza]SRH analogues showed modest competitive inhibition (KI ∼40 μM), while most of the others were inactive. One compound that contains a hemiaminal moiety exhibited time-dependent inhibition, consistent with enzyme-catalyzed ring opening and conversion into a more potent species (KI∗ = 3.5 μM). The structure–activity relationship of the designed inhibitors highlights the importance of both the homocysteine and ribose moieties for high-affinity binding to LuxS active site.The [4-aza] S-ribosylhomocysteine analogue exhibited time-dependent inhibition of LuxS consistent with the enzyme-catalyzed ring opening and generation of 2- and/or 3-ketone intermediates.
Co-reporter:Zun-Ting Zhang, Jean-Philippe Pitteloud, Laura Cabrera, Yong Liang, Myrdich Toribio and Stanislaw F. Wnuk
Organic Letters 2010 Volume 12(Issue 4) pp:816-819
Publication Date(Web):January 21, 2010
DOI:10.1021/ol9028918
The trichlorophenyl,- dichlorodiphenyl,- and chlorotriphenylgermanes undergo Pd-catalyzed cross-couplings with aryl bromides and iodides in the presence of TBAF in toluene with addition of the measured amount of water. One chloride ligand on the Ge center allows efficient activation by fluoride to promote transfer of one, two, or three phenyl groups from the organogermane precursors.
Co-reporter:Stanislaw F. Wnuk, Jenay Robert, Adam J. Sobczak, Brandon P. Meyers, Venkata L.A. Malladi, Jinge Zhu, Bhaskar Gopishetty, Dehua Pei
Bioorganic & Medicinal Chemistry 2009 Volume 17(Issue 18) pp:6699-6706
Publication Date(Web):15 September 2009
DOI:10.1016/j.bmc.2009.07.057
S-Ribosylhomocysteinase (LuxS) catalyzes the cleavage of the thioether bond of S -ribosylhomocysteine (SRH) to produce homocysteine and 4,5-dihydroxy-2,3-pentanedione (DPD), which is the precursor of type 2 autoinducer for bacterial cell–cell communication. In this work, we have synthesized several SRH analogues modified at the ribose C3 position as potential inhibitors of LuxS. While removal or methylation of the C3–OH resulted in simple competitive inhibitors of moderate potency, inversion of the C3 stereochemistry or substitution of fluorine for C3–OH resulted in slow-binding inhibitors of improved potency. The most potent inhibitor showed a KI∗ value of 0.43 μM.S-Ribosylhomocysteine analogues that lack an enolizable hydroxyl group at the carbon-3 position act as competitive inhibitors of the LuxS enzyme.
Co-reporter:Magdalena Rapp, Xiaohong Cai, Wei Xu, William R. Dolbier Jr., Stanislaw F. Wnuk
Journal of Fluorine Chemistry 2009 Volume 130(Issue 3) pp:321-328
Publication Date(Web):March 2009
DOI:10.1016/j.jfluchem.2008.12.004
Difluorocarbene, generated from trimethylsilyl fluorosulfonyldifluoroacetate (TFDA), reacts with the uridine and adenosine substrates preferentially at the enolizable amide moiety of the uracil ring and the 6-amino group of the purine ring. 2′,3′-Di-O-benzoyl-3′-deoxy-3′-methyleneuridine reacts with TFDA to produce 4-O-difluoromethyl product derived from an insertion of difluorocarbene into the 4-hydroxyl group of the enolizable uracil ring. Reaction of the difluorocarbene with the adenosine substrates having the unprotected 6-amino group in the purine ring produced the 6-N-difluoromethyl derivative, while reaction with 6-N-benzoyl protected adenosine analogues gave the difluoromethyl ether product derived from the insertion of difluorocarbene into the enol form of the 6-benzamido group. Treatment of the 6-N-phthaloyl protected adenosine analogues with TFDA resulted in the unexpected one-pot conversion of the imidazole ring of the purine into the corresponding N-difluoromethylthiourea derivatives. Treatment of the suitably protected pyrimidine and purine nucleosides bearing an exomethylene group at carbons 2′, 3′ or 4′ of the sugar rings with TFDA afforded the corresponding spirodifluorocyclopropyl analogues but in low yields.Difluorocarbene, generated from TFDA, reacts with the uridine and adenosine substrates preferentially at the enolizable amide moieties of the heterocyclic bases. It also adds to the exomethylene double bonds of sugar rings.
Co-reporter:Pablo R. Sacasa, Jessica Zayas, Stanislaw F. Wnuk
Tetrahedron Letters 2009 50(38) pp: 5424-5427
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.07.063
Co-reporter:Stanislaw F. Wnuk, Pablo R. Sacasa, Elzbieta Lewandowska, Daniela Andrei, Sumin Cai, Ronald T. Borchardt
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 10) pp:5424-5433
Publication Date(Web):15 May 2008
DOI:10.1016/j.bmc.2008.04.017
Adenosine and uridine analogues functionalized with alkenyl or fluoroalkenyl chain at C5′ were prepared employing cross-metathesis, Negishi couplings, and Wittig reactions. Metathesis of the protected 5′-deoxy-5′-methyleneadenosine or uridine analogues with six-carbon amino acids (homoallylglycines) in the presence of Grubbs catalysts gave nucleoside analogues with the C5′–C6′ double bond. Alternatively, the Pd-catalyzed cross-coupling between the protected 5′-deoxy-5′-(iodomethylene) nucleosides and suitable alkylzinc bromides also provided analogues with alkenyl unit. Stereoselective Pd-catalyzed monoalkylation of 5′-(bromofluoromethylene)-5′-deoxyadenosine with alkylzinc bromides afforded adenosylhomocysteine analogues with a 6′-(fluoro)vinyl motif. The vinylic adenine nucleosides produced time-dependent inactivation of the S-adenosyl-l-homocysteine hydrolases.
Co-reporter:Stanislaw F. Wnuk, Jennifer Lalama, Craig A. Garmendia, Jenay Robert, Jinge Zhu, Dehua Pei
Bioorganic & Medicinal Chemistry 2008 Volume 16(Issue 9) pp:5090-5102
Publication Date(Web):1 May 2008
DOI:10.1016/j.bmc.2008.03.028
Treatment of the protected ribose or xylose 5-aldehyde with sulfonyl-stabilized fluorophosphonate gave (fluoro)vinyl sulfones. Stannyldesulfonylation followed by iododestannylation afforded 5,6-dideoxy-6-fluoro-6-iodo-d-ribo or xylo-hex-5-enofuranoses. Coupling of the hexenofuranoses with alkylzinc bromides gave 10-carbon ribosyl- and xylosylhomocysteine analogues incorporating a fluoroalkene. The fluoroalkenyl and alkenyl analogues were evaluated for inhibition of Bacillus subtilis S-ribosylhomocysteinase (LuxS). One of the compounds, 3,5,6-trideoxy-6-fluoro-d-erythro-hex-5-enofuranose, acted as a competitive inhibitor of moderate potency (KI = 96 μM).
Co-reporter:Stanislaw F. Wnuk, Elzbieta Lewandowska, Dania R. Companioni, Pedro I. Garcia Jr and John A. Secrist III
Organic & Biomolecular Chemistry 2004 vol. 2(Issue 1) pp:120-126
Publication Date(Web):20 Nov 2003
DOI:10.1039/B311504F
A series of 2′-thionucleosides, as potential inhibitors of ribonucleotide reductases, has been synthesized. Treatment of the 3′,5′-O-TPDS-2′-O-(trifluoromethanesulfonyl)adenosine with potassium thioacetate gave the arabino epimer of 2′-S-acetyl-2′-thioadenosine which was deacetylated to give 9-(3,5-O-TPDS-2-thio-β-D-arabinofuranosyl)adenine in high yield. Treatment of the latter with diethyl azodicarboxylate–C3H7SH–THF gave 2′-propyl disulfide which was desilylated to give 9-(2-deoxy-2-propyldithio-β-D-arabinofuranosyl)adenine. Subsequent tosylation (O5′) and displacement of the tosylate with pyrophosphate afforded the 5′-O-diphosphate in a stable form as propyl mixed-disulfide, which upon treatment with dithiothreitol releases 9-(2-thio-β-D-arabinofuranosyl)adenine 5′-diphosphate. The arabino 2′-mercapto group might interact with the crucial thiyl radical at cysteine 439 leading to the inhibition of ribonucleotide reductases via formation of a Cys439–2′-mercapto disulfide bridge. The 2,6-diamino-, 2-amino-6-chloro- and 2-amino-6-methoxypurine ribosides were also converted to the corresponding 2′-deoxy-2′-propyldithio-β-D-arabinofuranosyl nucleosides, which might serve as convenient precursors to the arabino epimer of 2′-thioguanosine. Analogously, 2′-deoxy-2′-propyldithioadenosine was prepared from 9-(β-D-arabinofuranosyl)adenine. The nucleoside disulfides show modest cytotoxicity in a panel of human tumor cell lines.
.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-(4-phenyl-1H-1,2,3-triazol-1-yl)-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-19-8.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-(4-phenyl-1H-1,2,3-triazol-1-yl)-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-19-8_b.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-[8-(3-amino-1-oxopropyl)-8,9-dihydro-3H-dibenzo[b,f]-1,2,3-triazolo[4,5-d]azocin-3-yl]-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-17-6.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-[8-(3-amino-1-oxopropyl)-8,9-dihydro-3H-dibenzo[b,f]-1,2,3-triazolo[4,5-d]azocin-3-yl]-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-17-6_b.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-[8-(3-amino-1-oxopropyl)-8,9-dihydro-1H-benzo[c]-1,2,3-triazolo[4,5-e][1]benzazocin-1-yl]-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-15-4.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-[8-(3-amino-1-oxopropyl)-8,9-dihydro-1H-benzo[c]-1,2,3-triazolo[4,5-e][1]benzazocin-1-yl]-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-15-4_b.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-iodo-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-25-6.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-iodo-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-25-6_b.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-azido-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-11-0.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile, 4-amino-6-azido-7-β-D-ribofuranosyl-](/data/chemimg/3723600/1996647-11-0_b.png)


![HEXANOIC ACID, 2-[(4-CHLOROPHENYL)THIO]-, ETHYL ESTER](http://img.cochemist.com/ccimg/557800/557797-65-6.png)
![HEXANOIC ACID, 2-[(4-CHLOROPHENYL)THIO]-, ETHYL ESTER](http://img.cochemist.com/ccimg/557800/557797-65-6_b.png)
![Hexanoic acid, 2-[(4-methoxyphenyl)thio]-, ethyl ester](http://img.cochemist.com/ccimg/212800/212768-75-7.png)
![Hexanoic acid, 2-[(4-methoxyphenyl)thio]-, ethyl ester](http://img.cochemist.com/ccimg/212800/212768-75-7_b.png)
![2-Furanol, tetrahydro-5-[(phenylmethoxy)methyl]-, (2S-trans)-](http://img.cochemist.com/ccimg/95800/95721-16-7.png)
![2-Furanol, tetrahydro-5-[(phenylmethoxy)methyl]-, (2S-trans)-](http://img.cochemist.com/ccimg/95800/95721-16-7_b.png)
![(3aR,6R,6aR)-6-[(Benzyloxy)methyl]-2,2-dimethyldihydrofuro[3,4-d] [1,3]dioxol-4(3aH)-one (non-preferred name)](http://img.cochemist.com/ccimg/85900/85846-80-6.png)
![(3aR,6R,6aR)-6-[(Benzyloxy)methyl]-2,2-dimethyldihydrofuro[3,4-d] [1,3]dioxol-4(3aH)-one (non-preferred name)](http://img.cochemist.com/ccimg/85900/85846-80-6_b.png)
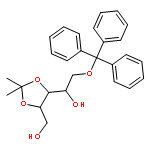
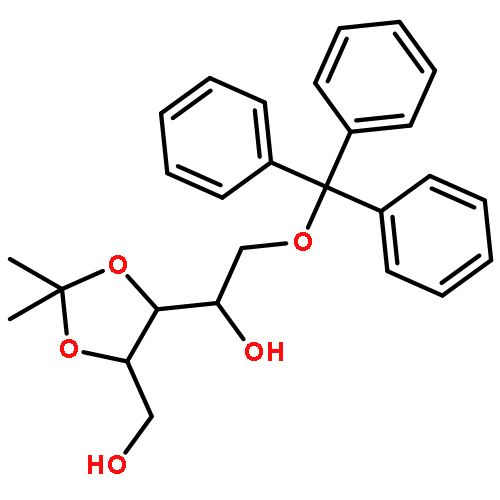
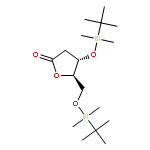
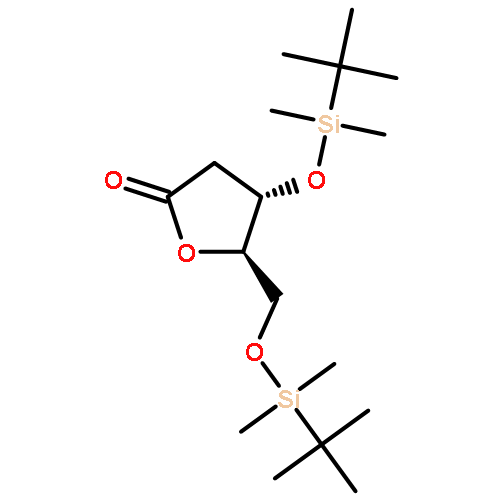
![5,6-dibromo-7-(beta-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/78100/78000-55-2.png)
![5,6-dibromo-7-(beta-D-ribofuranosyl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine](http://img.cochemist.com/ccimg/78100/78000-55-2_b.png)
![(3aR,6R,6aR)-6-(Hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-ol](http://img.cochemist.com/ccimg/67900/67814-68-0.png)
![(3aR,6R,6aR)-6-(Hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-ol](http://img.cochemist.com/ccimg/67900/67814-68-0_b.png)
![(3aR,4R,6R,6aR)-2,2-Dimethyl-6-((trityloxy)methyl)tetrahydrofuro[3,4-d][1,3]dioxol-4-ol](http://img.cochemist.com/ccimg/54600/54503-65-0.png)
![(3aR,4R,6R,6aR)-2,2-Dimethyl-6-((trityloxy)methyl)tetrahydrofuro[3,4-d][1,3]dioxol-4-ol](http://img.cochemist.com/ccimg/54600/54503-65-0_b.png)
![2(3H)-Furanone, dihydro-5-[(phenylmethoxy)methyl]-, (5S)-](http://img.cochemist.com/ccimg/32800/32780-08-8.png)
![2(3H)-Furanone, dihydro-5-[(phenylmethoxy)methyl]-, (5S)-](http://img.cochemist.com/ccimg/32800/32780-08-8_b.png)
![4-amino-6-bromo-7-[3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrrolo[2,3-d]pyrimidine-5-carboxamide](http://img.cochemist.com/ccimg/20300/20201-56-3.png)
![4-amino-6-bromo-7-[3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrrolo[2,3-d]pyrimidine-5-carboxamide](http://img.cochemist.com/ccimg/20300/20201-56-3_b.png)
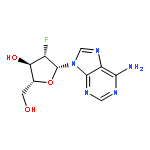
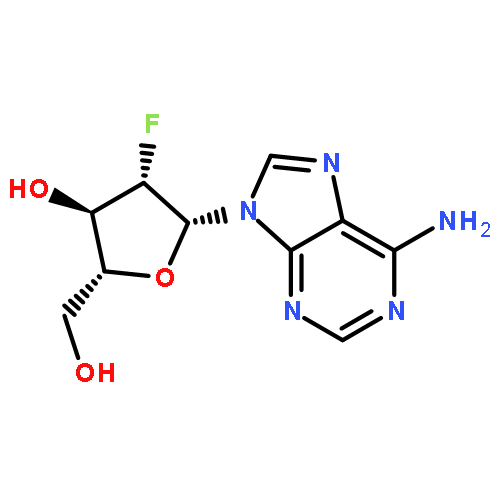
![N-[1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-oxopyrimidin-4-yl]-2-propylpentanamide](http://img.cochemist.com/ccimg/892200/892128-60-8.png)
![N-[1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-oxopyrimidin-4-yl]-2-propylpentanamide](http://img.cochemist.com/ccimg/892200/892128-60-8_b.png)
















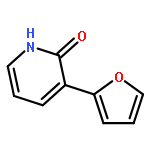
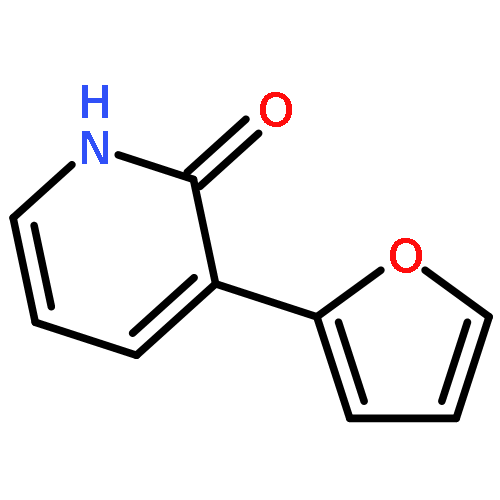


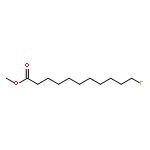
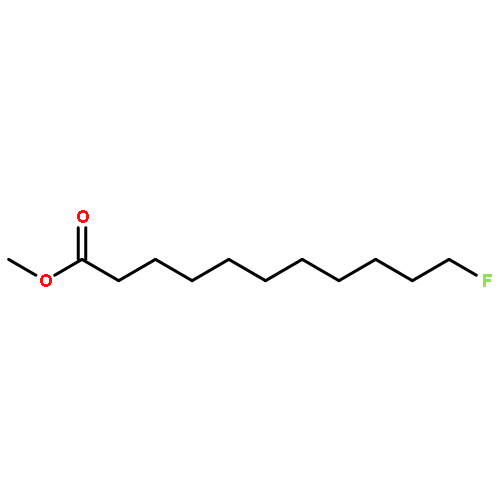








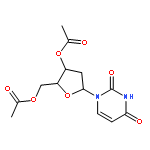
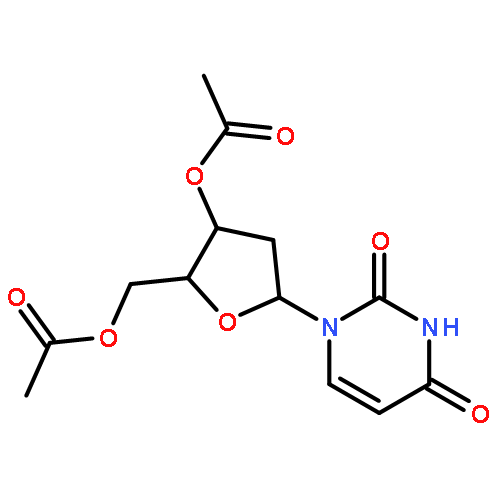





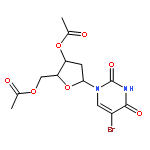



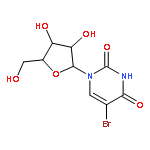
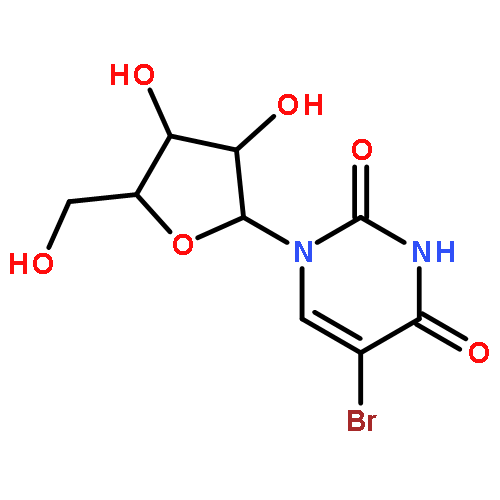
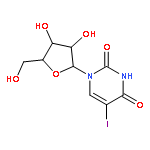
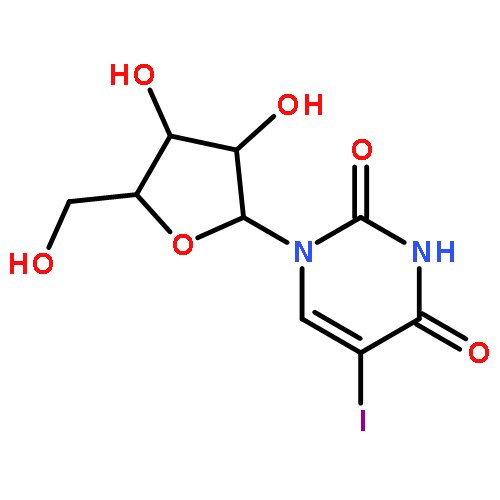














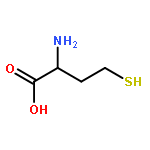
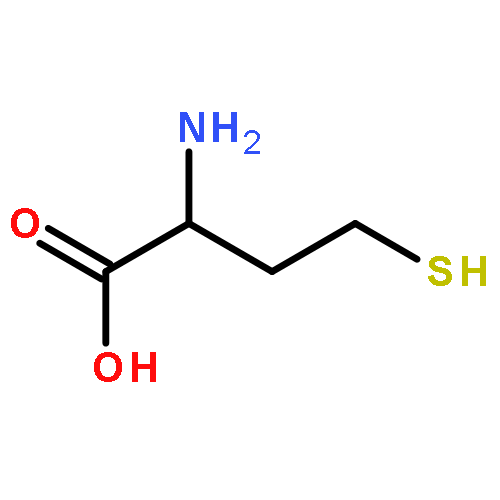




![Tert-butyl 2-[(2-methylpropan-2-yl)oxycarbonylamino]-4-sulfanylbutanoate](http://img.cochemist.com/ccimg/630200/630108-94-0.png)
![Tert-butyl 2-[(2-methylpropan-2-yl)oxycarbonylamino]-4-sulfanylbutanoate](http://img.cochemist.com/ccimg/630200/630108-94-0_b.png)
![(3AR,4R,6aS)-tert-Butyl 4-(hydroxymethyl)-2,2-dimethyldihydro-3aH-[1,3]dioxolo[4,5-c]pyrrole-5(4H)-carboxylate](http://img.cochemist.com/ccimg/155000/154905-24-5.png)
![(3AR,4R,6aS)-tert-Butyl 4-(hydroxymethyl)-2,2-dimethyldihydro-3aH-[1,3]dioxolo[4,5-c]pyrrole-5(4H)-carboxylate](http://img.cochemist.com/ccimg/155000/154905-24-5_b.png)



![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile,4-amino-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/700/606-58-6.png)
![7H-Pyrrolo[2,3-d]pyrimidine-5-carbonitrile,4-amino-7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/700/606-58-6_b.png)
![Benzene, [(1E)-2-bromoethenyl]-](/data/chemimg/1476700/588-72-7.png)
![Benzene, [(1E)-2-bromoethenyl]-](/data/chemimg/1476700/588-72-7_b.png)



![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0.png)
![7H-Pyrrolo[2,3-d]pyrimidin-4-amine,7-b-D-ribofuranosyl-](http://img.cochemist.com/ccimg/100/69-33-0_b.png)

