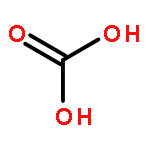
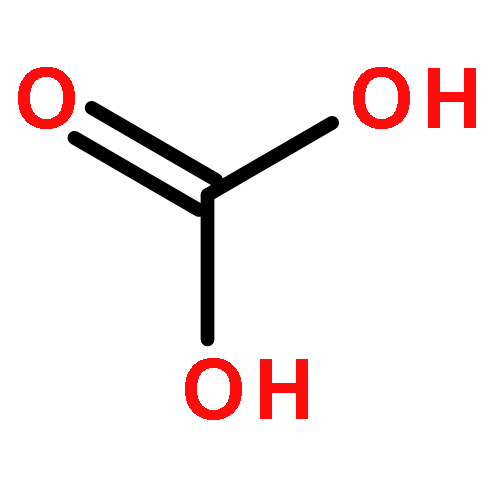
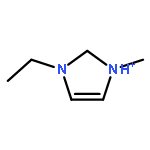
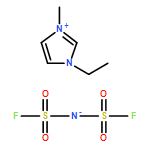
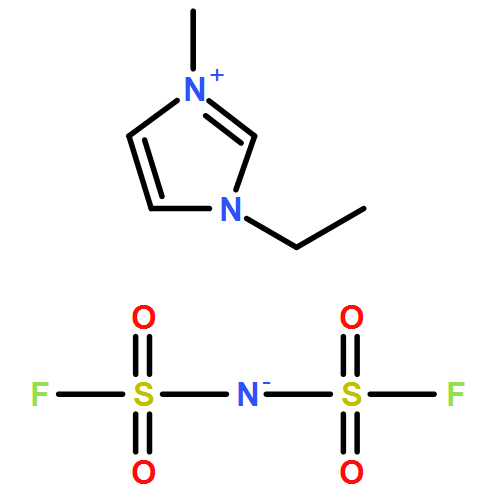
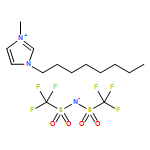
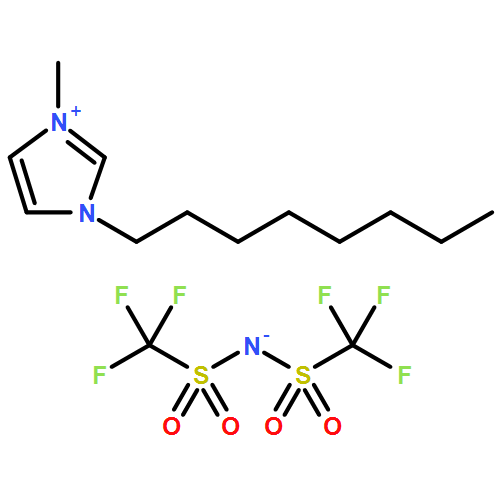
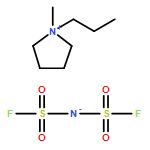
Co-reporter:Dengpan Dong, Justin B. Hooper, and Dmitry Bedrov
The Journal of Physical Chemistry B May 11, 2017 Volume 121(Issue 18) pp:4853-4853
Publication Date(Web):April 20, 2017
DOI:10.1021/acs.jpcb.7b01032
Understanding the behavior of aqueous solutions containing tetraalkylammonium (TAA) cations is of great significance in a number of applications, including polymer membranes for fuel cells. In this work, a polarizable force field has been used to perform atomistic molecular dynamics (MD) simulations of aqueous solutions containing tetramethylammonium (TMA) or tetrabutylammonium (TBA) cations and Br counterions. Extensive MD simulations of TMA-Br/water and TBA-Br/water systems were conducted as a function of solution composition (ion pair:water molar ratios of 1:10, 1:20, 1:30, 1:63, and 1:500) at atmospheric pressure and 298 K. Our simulations demonstrate excellent agreement with available experimental data for solution densities and diffusion coefficients of different species as a function of solution composition, providing us confidence in analyzed structural and dynamic correlations. Various ion–ion and ion–water spatial distributions and the extent of cation aggregation are discussed in light of changes in the structure of cations hydration shells. The delicate balance between cation ionic core interactions with water and the hydrophobic interactions of alkyl tails leads to nontrivial self-assembly of TAA cations and the formation of an interpenetrating cationic network at higher concentrations. The ions and water dynamics are strongly coupled with the observed structural correlations and are analyzed in terms of various residence time, diffusion coefficients, and ionic conductivity.
Co-reporter:Anton Iakovlev;Marcus Müller
Langmuir January 24, 2017 Volume 33(Issue 3) pp:744-754
Publication Date(Web):December 30, 2016
DOI:10.1021/acs.langmuir.6b03774
We report computer simulation of the self-assembly of alkylthiols on the surface of liquid mercury. Here we focus mainly on the alkylthiol behavior on mercury as a function of the surfactant surface coverage, which we study by means of large-scale molecular dynamics simulations of the equilibrium structure at room temperature. The majority of the presented results are obtained for octa- and dodecanethiol surfactants. This topic is particularly interesting because the properties of the alkylthiol self-assembled monolayers on liquid mercury are relevant for practical applications (e.g., in organic electronics) and can be controlled by mechanically manipulating the monolayer, i.e., by changing its structure. Our computer simulation results shed additional light on the alkylthiol self-assembly on liquid mercury by revealing the coexistence of a dense agglomerated laying-down alkylthiols with a very dilute 2D vapor on mercury surface rather than a single vapor phase in the low surface coverage regime. In the regimes of the high surface coverage we observe the coexistence of the laying-down liquid phase and crystalline phases with alkylthiols standing tilted at a sharp angle to the surface normal, which agrees with the phase behavior previously seen in X-ray and tensiometry experiments. We also discuss the influence of finite-size effects, which one inevitably encounters in molecular simulations. Our findings agree well with the general predictions of the condensation/evaporation theory for finite systems. The temperature dependence of the stability of the crystalline alkylthiol phases and details of the surfactant chemical binding to the surface are discussed. The equilibrium structure of the crystalline phase is investigated in detail for the alkylthiols of various tail lengths.
Co-reporter:Dmitry Bedrov, Oleg Borodin, and Justin B. Hooper
The Journal of Physical Chemistry C August 3, 2017 Volume 121(Issue 30) pp:16098-16098
Publication Date(Web):June 30, 2017
DOI:10.1021/acs.jpcc.7b04247
A fundamental understanding of solid electrolyte interphase (SEI) properties is critical for enabling further improvement of lithium batteries and stabilizing the anode–electrolyte interface. Mechanical and transport properties of two model SEI components were investigated using molecular dynamics (MD) simulations and a hybrid MD-Monte Carlo (MC) scheme. A many-body polarizable force field (APPLE&P) was employed in all simulations. Elastic moduli and conductivity of model SEIs comprised of dilithium ethylene dicarbonate (Li2EDC) were compared with those comprised of dilithium butylene dicarbonate (Li2BDC) over a wide temperature range. Both ordered and disordered materials were examined with the ordered materials showing higher conductivity in the conducting plane compared to conductivity of the disordered analogues. Li2BDC was found to exhibit softening and onset of anion mobility at lower temperatures compared to Li2EDC. At 120 °C and below, both SEI model compounds showed single ion conductor behavior. Ordered Li2EDC and Li2BDC phases had highly anisotropic mechanical properties, with the shear modulus of Li2BDC being systematically smaller than that for Li2EDC.
Co-reporter:Jenel Vatamanu;Oleg Borodin;Marco Olguin;Gleb Yushin
Journal of Materials Chemistry A 2017 vol. 5(Issue 40) pp:21049-21076
Publication Date(Web):2017/10/17
DOI:10.1039/C7TA05153K
Supercapacitors or electrical double layer (EDL) capacitors store charge via rearrangement of ions in electrolytes and their adsorption on electrode surfaces. They are actively researched for multiple applications requiring longer cycling life, broader operational temperature ranges, and higher power density compared to batteries. Recent developments in nanostructured carbon-based electrodes with a high specific surface area have demonstrated the potential to significantly increase the energy density of supercapacitors. Molecular modeling of electrolytes near charged electrode surfaces has provided key insights into the fundamental aspects of charge storage at the nanoscale, including an understanding of the mechanisms of ion adsorption and dynamics at flat surfaces and inside nanopores, and the influence of curvature, roughness, and electronic structure of electrode surfaces. Here we review these molecular modeling findings for EDL capacitors, dual ion batteries and pseudo-capacitors together with available experimental observations and put this analysis into the perspective of future developments in this field. Current research trends and future directions are discussed.
Co-reporter:Dmitry Bedrov and Justin B. Hooper, Matthew A. Glaser and Noel A. Clark
Langmuir 2016 Volume 32(Issue 16) pp:4004-4015
Publication Date(Web):2017-2-22
DOI:10.1021/acs.langmuir.6b00120
Extensive atomistic molecular dynamics simulations have been employed to study the structure and molecular orientational relaxation of azobenzene-based monolayers grafted to a solid substrate. Systems with surface coverage of 0.6 nm2/molecule were investigated over a wide temperature range ranging from 298 K, where the mesogens show local ordering and the monolayer dynamics was found to be glassy, up to 700 K, where the azobenzene groups have a nearly isotropic orientational distribution, with a subnanosecond characteristic orientational relaxation time scale. Biased simulations that model single-molecule thermal excitation and conformational isomerization have been conducted to obtain insight into the mechanisms for photoinduced athermal fluidization and monolayer reorganization observed experimentally in this system. Our simulations clearly indicate that trans–cis conformational isomerization transitions of azobenzene units can lead to reorientation of mesogens and to the formation of a monolayer with strong macroscopic in-plane nematic order. While local heating created by excitation process can facilitate this process, thermal excitation alone is not sufficient to induce ordering in the monolayer. Instead, the work done by a molecule undergoing cis–trans isomerization on the cage of neighboring molecules is the key mechanism for photofluidization and orientational ordering in dMR monolayers exposed to linearly polarized light leading to relaxation dynamics that can be described in terms of higher effective temperature. The obtained simulation results are discussed in light of recent experimental data reported for these systems.
Co-reporter:Anton Iakovlev; Dmitry Bedrov;Marcus Müller
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 8) pp:1546-1553
Publication Date(Web):April 5, 2016
DOI:10.1021/acs.jpclett.6b00494
Self-assembled organic films on liquid metals feature a very rich phase behavior, which qualitatively differs from the one on crystalline metals. In contrast to conventional crystalline supports, self-assembled alkylthiol monolayers on liquid metals possess a considerably higher degree of molecular order, thus enabling much more robust metal–molecule–semiconductor couplings for organic electronics applications. Yet, compared to crystalline substrates, the self-assembly of organic surfactants on liquid metals has been studied to a much lesser extent. In this Letter we report the first of its kind molecular simulation investigation of alkyl-based surfactants on a liquid mercury surface. The focus of our investigation is the surfactant conformations as a function of surface coverage and surfactant type. First, we consider normal alkanes because these systems set the basis for simulations of all other organic surfactants on liquid mercury. Subsequently, we proceed with the discussion of alkylthiols that are the most frequently used surfactants in the surface science of hybrid organometallic interfaces. Our results indicate a layering transition of normal alkanes as well as alkylthiols from an essentially bare substrate to a completely filled monolayer of laying molecules. As the surface coverage increases further, we observe a partial wetting of the laying monolayer by the bulk phase of alkanes. In the case of alkylthiols, we clearly see the coexistence of molecules in laying-down and standing-up conformations, in which the sulfur headgroups of the thiols are chemically bound to mercury. In the standing-up phase, the headgroups form an oblique lattice. For the first time we were able to explicitly characterize the molecular-scale structure and transitions between phases of alkyl-based surfactants and to demonstrate how the presence of a thiol headgroup qualitatively changes the phase equilibrium and structure in these systems. The observed phenomena are consistent with available direct and indirect experimental evidence.
Co-reporter:Dmitry Bedrov, Jenel Vatamanu, Zongzhi Hu
Journal of Non-Crystalline Solids 2015 Volume 407() pp:339-348
Publication Date(Web):1 January 2015
DOI:10.1016/j.jnoncrysol.2014.08.007
•Molecular simulations of ionic liquids (IL) were conducted on various surfaces.•On flat surfaces, all ILs show weakly changing capacitance with electrode voltage.•On atomically corrugated surfaces, capacitance strongly depends on several factors.•Surface topography can be used to increase energy density in supercapacitors.Understanding of molecular level structure and mechanisms of the formation of electric double layers in realistic ionic liquid-based electrolytes on charged electrode surfaces is one of scientifically and technologically key areas that have attracted a lot of attention over the last decade. Extensive experimental, theoretical, and modeling studies have been dedicated to this challenging topic in order to establish fundamental correlations between the details of molecular structure of electrolyte and the properties of the electric double layers (EDL) forming on various electrodes. While great progress has been made in advancing our understanding of EDL properties and their influence on the performance of supercapacitors, batteries, and other energy storage devices, there are still a number of challenges and controversies that have not been resolved. In this manuscript, we demonstrate how atomistic molecular dynamics simulations provide a powerful tool for dealing with these challenges and can facilitate the design of novel materials for advancing energy storage technologies.
Co-reporter:Jenel Vatamanu, Mihaela Vatamanu, and Dmitry Bedrov
ACS Nano 2015 Volume 9(Issue 6) pp:5999
Publication Date(Web):June 3, 2015
DOI:10.1021/acsnano.5b00945
The enhancement of non-Faradaic charge and energy density stored by ionic electrolytes in nanostructured electrodes is an intriguing issue of great practical importance for energy storage in electric double layer capacitors. On the basis of extensive molecular dynamics simulations of various carbon-based nanoporous electrodes and room temperature ionic liquid (RTIL) electrolytes, we identify atomistic mechanisms and correlations between electrode/electrolyte structures that lead to capacitance enhancement. In the symmetric electrode setup with nanopores having atomically smooth walls, most RTILs showed up to 50% capacitance increase compared to infinitely wide pore. Extensive simulations using asymmetric electrodes and pores with atomically rough surfaces demonstrated that tuning of electrode nanostructure could lead to further substantial capacitance enhancement. Therefore, the capacitance in nanoporous electrodes can be increased due to a combination of two effects: (i) the screening of ionic interactions by nanopore walls upon electrolyte nanoconfinement, and (ii) the optimization of nanopore structure (volume, surface roughness) to take into account the asymmetry between cation and anion chemical structures.Keywords: capacitance; electric double layer; energy storage; nanoconfinement; nanoporous electrodes; room temperature ionic liquids; supercapacitors;
Co-reporter:Zhe Li, Oleg Borodin, Grant D. Smith, and Dmitry Bedrov
The Journal of Physical Chemistry B 2015 Volume 119(Issue 7) pp:3085-3096
Publication Date(Web):January 16, 2015
DOI:10.1021/jp510644k
Molecular dynamics simulations of N-methyl-N-propylpyrrolidinium (pyr13) bis(trifluoromethanesulfonyl)imide (Ntf2) ionic liquid [pyr13][Ntf2] doped with [Li][Ntf2] salt and mixed with acetonitrile (AN) and ethylene carbonate (EC) organic solvents were conducted using polarizable force field. Structural and transport properties of ionic liquid electrolytes (ILEs) with 20 and 40 mol % of organic solvents have been investigated and compared to properties of neat ILEs. Addition of AN and EC solvents to ILEs resulted in the partial displacement of the Ntf2 anions from the Li+ first coordination shell by EC and AN and shifting the Li–Ntf2 coordination from bidentate to monodentate. The presence of organic solvents in ILE has increased the ion mobility, with the largest effect observed for the Li+ cation. The Li+ conductivity has doubled with addition of 40 mol % of AN. The Li+–NNtf2 residence times were dramatically reduced with addition of solvents, indicating an increasing contribution from structural diffusion of the Li+ cations.
Co-reporter:Lidan Xing, Jenel Vatamanu, Oleg Borodin, and Dmitry Bedrov
The Journal of Physical Chemistry Letters 2013 Volume 4(Issue 1) pp:132-140
Publication Date(Web):December 15, 2012
DOI:10.1021/jz301782f
The capacitance enhancement experimentally observed in electrodes with complex morphology of random subnanometer wide pores is an intriguing phenomena, yet the mechanisms for such enhancement are not completely understood. Our atomistic molecular dynamics simulations demonstrate that in subnanometer slit-geometry nanopores, a factor of 2 capacitance enhancement (compared to a flat electrode) is possible for the 1-ethyl-3-methylimidazolium (EMIM)–bis(trifluoro-methylsulfonyl)imide (TFSI) ionic liquid electrolyte. This capacitance enhancement is a result of a fast charge separation inside the nanopore due to abrupt expulsion of co-ions from the pore while maintaining an elevated counterion density due to strong screening of electrostatic repulsive interactions by the conductive pore. Importantly, we find that the capacitance enhancement can be very asymmetric. For the negatively charged 7.5 Å wide pore, the integral capacitance is 100% larger than on a flat surface; however, on the positive electrode, almost no enhancement is observed. Detailed analysis of structure and composition of electrolyte inside nanopores shows that the capacitance enhancement and the shape of differential capacitance strongly depend on the details of the ion chemical structure and a delicate balance of ion–surface and ion–ion interactions.Keywords: differential capacitance; electric double layer; electrostatic superscreening; molecular dynamics simulation; nanoporous electrode; room temperature ionic liquid; supercacitor;
Co-reporter:Birger Steinmüller, Marcus Müller, Keith R. Hambrecht, Grant D. Smith, and Dmitry Bedrov
Macromolecules 2012 Volume 45(Issue 2) pp:1107-1117
Publication Date(Web):December 21, 2011
DOI:10.1021/ma202311e
The equilibrium structure and ordering kinetics of random AB block copolymers is investigated using a Lennard-Jones bead–spring model and a soft, coarse-grained model. Upon increasing the incompatibility a disordered microemulsion-like structure is formed, whose length scale slightly increases with segregation. The structure factor of composition fluctuations, molecular conformations, single-chain dynamics and collective ordering kinetics are investigated as a function of the segregation between A and B blocks. The harsh repulsion of the Lennard-Jones potential gives rise to pronounced fluid-like packing effects that affect the liquid structure on the length scale of the bead size and, upon cooling and increase of the local density, result in an additional slowing down of the dynamics. The soft, coarse-grained model does not exhibit pronounced packing effects and the softness of the potential allows for a faster equilibration in computer simulation. The structure and dynamics of the two different models are quantitatively compared. The parameters of the soft, coarse-grained model are adjusted as to match the long-range structure of the bead–spring model, and it is demonstrated that the soft, coarse grained model can be utilized to generate starting configurations for the Lennard-Jones bead–spring model.
Co-reporter:Zhe Li, Grant D. Smith, and Dmitry Bedrov
The Journal of Physical Chemistry B 2012 Volume 116(Issue 42) pp:12801-12809
Publication Date(Web):September 14, 2012
DOI:10.1021/jp3052246
Molecular dynamics simulations of N-methyl-N-propylpyrrolidinium (pyr13) bis(trifluoromethanesulfonyl)imide (Ntf2) ionic liquid [pyr13][Ntf2] mixed with [Li][Ntf2] salt have been conducted using a polarizable force field. Mixture simulations with lithium salt mole fractions between 0% and 33% at 363 and 423 K yield densities, ion self-diffusion coefficients, and ionic conductivities in very good agreement with available experimental data. In all investigated electrolytes, each Li+ cation was found to be coordinated, on average, by 4.1 oxygen atoms from surrounding anions. At lower concentrations (x ≤ 0.20), the Li+ cation was found to be, on average, coordinated by slightly more than three Ntf2 anions with two anions contributing a single oxygen atom and one anion contributing two oxygen atoms to Li+ coordination. At the highest [Li][Ntf2] concentration, however, there were, on average, 3.5 anions coordinating each Li+ cation, corresponding to fewer bidendate and more monodentate anions in the Li+ coordination sphere. This trend is due to increased sharing of anions by Li+ at higher salt concentrations. In the [pyr13][Ntf2]/[Li][Ntf2] electrolytes, the ion diffusivity is significantly smaller than that in organic liquid electrolytes due to not only the greater viscosity of the solvent but also the formation of clusters resulting from sharing of anions by Li+ cations. The ionic conductivity of the electrolytes was found to decrease with increasing salt concentration, with the effect being greater at the higher temperature. Finally, we found that the contribution of Li+ to ionic conductivity does not increase proportionally to Li+ concentration but saturates at higher doping levels.
Co-reporter:Madhuvanthi A. Kandadai, Rajasekhar Anumolu, Xiaojun Wang, Durairaj Baskaran, Leonard F. Pease III, Dmitry Bedrov, Grant D. Smith, Jimmy W. Mays, Jules J. Magda
European Polymer Journal 2011 Volume 47(Issue 10) pp:2022-2027
Publication Date(Web):October 2011
DOI:10.1016/j.eurpolymj.2011.07.017
Rheological evidence is provided demonstrating that covalent grafting of monodisperse isotactic poly(l-leucine) branches onto linear hyaluronan (HA) polysaccharide chains yields comb-branched HA chains that self-assemble into long-lived physical networks in aqueous solutions driven by hydrophobic interactions between poly(l-leucine) chains. This is in stark contrast to native (unmodified) HA solutions which exhibit no tendency to form long-lived physical networks.Graphical abstractHighlights► Poly (l-Leucine) grafted HA chains self assemble into physical networks. ► This network has an elastic shear modulus ≈ 100 Pa and a loss tangent ≈ 0.25. ► It has a network lifetime > 20 s. ► It exhibits almost complete elastic strain recovery after removal of shear stress. ► No physical network formation seen in unmodified HA.
Co-reporter:Dmitry Bedrov, Grant D. Smith
Journal of Non-Crystalline Solids 2011 Volume 357(Issue 2) pp:258-263
Publication Date(Web):15 January 2011
DOI:10.1016/j.jnoncrysol.2010.06.043
Molecular dynamics simulations of linear polymer melts represented using simple bead-necklace models showed for the first time a distinct separation between primary α- and secondary Johari–Goldstein β-processes. The split is observed only for models where the bead diameter is much larger than the bond length connecting the beads. The overlap of neighboring (along the chain) beads results in a mismatch between local intramolecular correlations and intermolecular packing (cage size), which leads to two processes in segmental relaxation characterized by torsional autocorrelation function. The observed β-process shows all characteristics and correlations expected for the true Johari–Goldstein process.
Co-reporter:Dmitry Bedrov, Grant D. Smith, Byong-Wa Chun
European Polymer Journal 2010 Volume 46(Issue 11) pp:2129-2137
Publication Date(Web):November 2010
DOI:10.1016/j.eurpolymj.2010.08.007
A combined experimental and multiscale simulation study of the influence of polymer brush modification on interactions of colloidal particles and rheological properties of dense colloidal suspensions has been conducted. Our colloidal suspension is comprised of polydisperse MgO colloidal particles modified with poly(ethylene oxide) (PEO) brushes in water. The shear stress as a function of shear rate was determined experimentally and from multiscale simulations for a suspension of 0.48 volume fraction colloids at room temperature for both bare and PEO-modified MgO colloids. Bare MgO particles exhibited strong shear thinning behavior and a yield stress on the order of several Pascals in both experiments and simulations. In contrast, simulations of PEO-modified colloids revealed no significant yielding or shear thinning and viscosity only a few times larger than solvent viscosity. This behavior is inconsistent with results obtained from experiments where modification of colloids with PEO brushes formed by adsorption of PEO-based comb-branched chains resulted in relatively little change in suspension rheology compared to bare colloids over the range of concentration of comb-branch additives investigated. We attribute this discrepancy in rheological properties between simulation and experiment for PEO-modified colloidal suspensions to heterogeneous adsorption of the comb-branch polymers.
.jpg)