Co-reporter:Matthew C. Johnson, Marta Sena-Velez, Brian K. Washburn, Georgia N. Platt, Stephen Lu, Tess E. Brewer, Jason S. Lynn, M. Elizabeth Stroupe, Kathryn M. Jones
Journal of Structural Biology 2017 Volume 200, Issue 3(Issue 3) pp:
Publication Date(Web):1 December 2017
DOI:10.1016/j.jsb.2017.08.005
Bacteriophages of nitrogen-fixing rhizobial bacteria are revealing a wealth of novel structures, diverse enzyme combinations and genomic features. Here we report the cryo-EM structure of the phage capsid at 4.9–5.7 Å-resolution, the phage particle proteome, and the genome of the Sinorhizobium meliloti-infecting Podovirus ΦM5. This is the first structure of a phage with a capsid and capsid-associated structural proteins related to those of the LUZ24-like viruses that infect Pseudomonas aeruginosa. Like many other Podoviruses, ΦM5 is a T = 7 icosahedron with a smooth capsid and short, relatively featureless tail. Nonetheless, this group is phylogenetically quite distinct from Podoviruses of the well-characterized T7, P22, and epsilon 15 supergroups. Structurally, a distinct bridge of density that appears unique to ΦM5 reaches down the body of the coat protein to the extended loop that interacts with the next monomer in a hexamer, perhaps stabilizing the mature capsid. Further, the predicted tail fibers of ΦM5 are quite different from those of enteric bacteria phages, but have domains in common with other rhizophages. Genomically, ΦM5 is highly mosaic. The ΦM5 genome is 44,005 bp with 357 bp direct terminal repeats (DTRs) and 58 unique ORFs. Surprisingly, the capsid structural module, the tail module, the DNA-packaging terminase, the DNA replication module and the integrase each appear to be from a different lineage. One of the most unusual features of ΦM5 is its terminase whose large subunit is quite different from previously-described short-DTR-generating packaging machines and does not fit into any of the established phylogenetic groups.
Co-reporter:Mykhailo Kopylov, Hank W. Bass, and M. Elizabeth Stroupe
Biochemistry 2015 Volume 54(Issue 9) pp:1743-1757
Publication Date(Web):February 13, 2015
DOI:10.1021/bi501284g
Noncanonical forms of DNA like the guanine quadruplex (G4) play important roles in regulating transcription and translation through interactions with their protein partners. Although potential G4 elements have been identified in or near genes from species diverse as bacteria, mammals, and plants, little is known about how they might function as cis-regulatory elements or as binding sites for trans-acting protein partners. In fact, until now no G4 binding partners have been identified in the plant kingdom. Here, we report on the cloning and characterization of the first plant-kingdom gene known to encode a G4-binding protein, maize (Zea mays L.) nucleoside diphosphate kinase1 (ZmNDPK1). Structural characterization by X-ray crystallography reveals that it is a homohexamer, akin to other known NDPKs like the human homologue NM23-H2. Further probing into the G4-binding properties of both NDPK homologues suggests that ZmNDPK1 possesses properties distinct from that of NM23-H2, which is known to interact with a G-rich sequence element upstream of the c-myc gene and, in doing so, modulate its expression. Indeed, ZmNDPK1 binds the folded G4 with low nanomolar affinity but corresponding unfolded G-rich DNA more weakly, whereas NM23-H2 binds both folded and unfolded G4 with low nanomolar affinities; nonetheless, both homologues appear to stabilize folded DNAs whether they were prefolded or not. We also demonstrate that the G4-binding activity of ZmNDPK1 is independent of nucleotide binding and kinase activity, suggesting that the G4-binding region and the enzyme active sites are separate. Together, these findings establish a broad evolutionary conservation of some NDPKs as G4-DNA binding enzymes, but with potentially distinct biochemical properties that may reflect divergent evolution or species-specific deployment of these elements in gene regulatory processes.
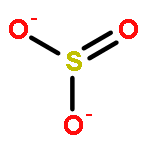
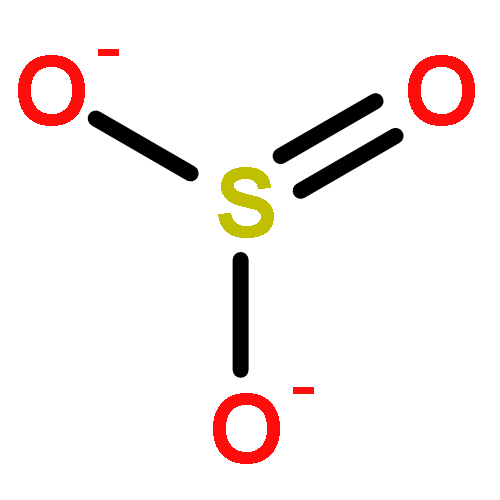
Co-reporter:Kyle W. Smith and M. Elizabeth Stroupe
Biochemistry 2012 Volume 51(Issue 49) pp:
Publication Date(Web):November 15, 2012
DOI:10.1021/bi300947a
Sulfite reductase catalyzes the six-electron reduction of sulfite to sulfide. The active site, found in the hemoprotein subunit (SiRHP), sits on the distal face of a negatively charged porphyrinoid called siroheme whose central iron atom is coupled to a proximal Fe4S4 cluster. Four positively charged amino acids are positioned around the active site cavity. Together, these two arginines (R83 and R153) and two lysines (K215 and K217) mitigate the negative charge on the siroheme macrocycle. They also serve as a cage around the distally bound anion that tightens when substrate binds and an active site loop clamps down. Structures of native SiRHP point to these amino acids as being important, but their specific roles are ill-defined. Here, we have altered those four active site amino acids and one amino acid on the flexible loop (N149) to probe their roles in SiRHP activity. None of these positively charged residues is required for electron transfer, but only R83S and N149W variants can produce a fully reduced product. By measuring the electrons used per unit of reduced sulfur released, we show that K215, R153, and K217 are responsible for intermediate and late proton transfers, whereas N149 and R153 play a role in the structure of the flexible loop that controls anion binding and release. R83 is primarily responsible for siroheme binding. Together, the activities and structures of these variants reveal specific roles for each in anion binding and in coupled proton transfer that facilitates electron transfer.
Co-reporter:Matthew C. Johnson, Homa Ghalei, Katelyn A. Doxtader, Katrin Karbstein, M. Elizabeth Stroupe
Structure (7 February 2017) Volume 25(Issue 2) pp:329-340
Publication Date(Web):7 February 2017
DOI:10.1016/j.str.2016.12.011
•A 9-Å structure of pre-40S ribosomes shows the position of seven assembly factors•Interface conformational heterogeneity suggests motions during 40S assembly•The beak architecture shows how Rps3 binds in precursor molecules•18S 3′ end formation may be regulated by Dim1 leaving the subunit interfaceLate-stage 40S ribosome assembly is a highly regulated dynamic process that occurs in the cytoplasm, alongside the full translation machinery. Seven assembly factors (AFs) regulate and facilitate maturation, but the mechanisms through which they work remain undetermined. Here, we present a series of structures of the immature small subunit (pre-40S) determined by three-dimensional (3D) cryoelectron microscopy with 3D sorting to assess the molecule's heterogeneity. These structures demonstrate an extensive structural heterogeneity of interface AFs that likely regulates subunit joining during 40S maturation. We also present structural models for the beak and the platform, two regions where the low resolution of previous studies did not allow for localization of AFs and the rRNA, respectively. These models are supported by biochemical analyses using point variants and suggest that maturation of the 18S 3′ end is regulated by dissociation of the AF Dim1 from the subunit interface, consistent with previous biochemical analyses.Download high-res image (144KB)Download full-size image
Co-reporter:Duncan R. Sousa, Scott M. Stagg, M. Elizabeth Stroupe
Journal of Molecular Biology (15 November 2013) Volume 425(Issue 22) pp:4544-4555
Publication Date(Web):15 November 2013
DOI:10.1016/j.jmb.2013.08.020
•We used cryo-3DEM to explore cardiac and gizzard muscle Tm on F-Actin.•At 8-Å resolution, we define the C-state position of Tm on F-Actin.•A model is proposed that invokes rocking of Tm to make way for myosin binding.Tropomyosin (Tm) is a key factor in the molecular mechanisms that regulate the binding of myosin motors to actin filaments (F-Actins) in most eukaryotic cells. This regulation is achieved by the azimuthal repositioning of Tm along the actin (Ac):Tm:troponin (Tn) thin filament to block or expose myosin binding sites on Ac. In striated muscle, including involuntary cardiac muscle, Tm regulates muscle contraction by coupling Ca2 + binding to Tn with myosin binding to the thin filament. In smooth muscle, the switch is the posttranslational modification of the myosin. Depending on the activation state of Tn and the binding state of myosin, Tm can occupy the blocked, closed, or open position on Ac. Using native cryogenic 3DEM (three-dimensional electron microscopy), we have directly resolved and visualized cardiac and gizzard muscle Tm on filamentous Ac in the position that corresponds to the closed state. From the 8-Å-resolution structure of the reconstituted Ac:Tm filament formed with gizzard-derived Tm, we discuss two possible mechanisms for the transition from closed to open state and describe the role Tm plays in blocking myosin tight binding in the closed-state position.Download high-res image (293KB)Download full-size image
.jpg)
![Ferrate(8-),[(12S,13S,17S,18S)-2,8,13,18-tetrakis(carboxymethyl)-12,13,17,18-tetrahydro-13,18-dimethyl-21H,22H-porphine-3,7,12,17-tetrapropanoato(10-)-kN21,kN22,kN23,kN24]-, hydrogen (1:8), (SP-4-2)-](http://img.cochemist.com/ccimg/52600/52553-42-1.png)
![Ferrate(8-),[(12S,13S,17S,18S)-2,8,13,18-tetrakis(carboxymethyl)-12,13,17,18-tetrahydro-13,18-dimethyl-21H,22H-porphine-3,7,12,17-tetrapropanoato(10-)-kN21,kN22,kN23,kN24]-, hydrogen (1:8), (SP-4-2)-](http://img.cochemist.com/ccimg/52600/52553-42-1_b.png)