Co-reporter:Xiya Lu, Victor Ovchinnikov, Darren Demapan, Daniel Roston, and Qiang Cui
Biochemistry March 14, 2017 Volume 56(Issue 10) pp:1482-1482
Publication Date(Web):February 22, 2017
DOI:10.1021/acs.biochem.7b00016
The mechanism of ATP hydrolysis in the myosin motor domain is analyzed using a combination of DFTB3/CHARMM simulations and enhanced sampling techniques. The motor domain is modeled in the pre-powerstroke state, in the post-rigor state, and as a hybrid based on the post-rigor state with a closed nucleotide-binding pocket. The ATP hydrolysis activity is found to depend on the positioning of nearby water molecules, and a network of polar residues facilitates proton transfer and charge redistribution during hydrolysis. Comparison of the observed hydrolysis pathways and the corresponding free energy profiles leads to detailed models for the mechanism of ATP hydrolysis in the pre-powerstroke state and proposes factors that regulate the hydrolysis activity in different conformational states. In the pre-powerstroke state, the scissile Pγ–O3β bond breaks early in the reaction. Proton transfer from the lytic water to the γ-phosphate through active site residues is an important part of the kinetic bottleneck; several hydrolysis pathways that feature distinct proton transfer routes are found to have similar free energy barriers, suggesting a significant degree of plasticity in the hydrolysis mechanism. Comparison of hydrolysis in the pre-powerstroke state and the closed post-rigor model suggests that optimization of residues beyond the active site for electrostatic stabilization and preorganization is likely important to enzyme design.
Co-reporter:Jiewei Hong, Robert J. HamersJoel A. Pedersen, Qiang Cui
The Journal of Physical Chemistry C 2017 Volume 121(Issue 6) pp:
Publication Date(Web):January 27, 2017
DOI:10.1021/acs.jpcc.6b11537
The surface charge of nanomaterials determines their stability in solution and interaction with other molecules and surfaces, yet experimental determination of surface charge of complex nanomaterials is not straightforward. We propose a hybrid approach that iteratively integrates explicit solvent molecular dynamics simulations and a multiconformer continuum electrostatic model (MCCE) to efficiently sample the configurational and titration spaces of surface ligands of nanomaterials. Test calculations of model systems indicate that the iterative approach converges rapidly even for systems that contain hundreds of titratable sites, making the approach complementary to more elaborate methods such as explicit solvent-based constant-pH molecular dynamics. The hybrid method is applied to analyze the pKa distribution of alkylamines attached to a carbon-based nanoparticle as a function of ligand density, nanoparticle surface curvature, and ligand heterogeneity. The results indicate that functionalization strategies can modulate the pKa of surface ligands and therefore charge properties of nanomaterials (e.g., surface charge, charge capacitance). The hybrid computational approach makes a major step toward guiding the design of nanomaterials with desired charge properties.
Co-reporter:Anders S. Christensen, Tomáš Kubař, Qiang Cui, and Marcus Elstner
Chemical Reviews 2016 Volume 116(Issue 9) pp:5301
Publication Date(Web):April 13, 2016
DOI:10.1021/acs.chemrev.5b00584
Semiempirical (SE) methods can be derived from either Hartree–Fock or density functional theory by applying systematic approximations, leading to efficient computational schemes that are several orders of magnitude faster than ab initio calculations. Such numerical efficiency, in combination with modern computational facilities and linear scaling algorithms, allows application of SE methods to very large molecular systems with extensive conformational sampling. To reliably model the structure, dynamics, and reactivity of biological and other soft matter systems, however, good accuracy for the description of noncovalent interactions is required. In this review, we analyze popular SE approaches in terms of their ability to model noncovalent interactions, especially in the context of describing biomolecules, water solution, and organic materials. We discuss the most significant errors and proposed correction schemes, and we review their performance using standard test sets of molecular systems for quantum chemical methods and several recent applications. The general goal is to highlight both the value and limitations of SE methods and stimulate further developments that allow them to effectively complement ab initio methods in the analysis of complex molecular systems.
Co-reporter:Daniel Roston and Qiang Cui
Journal of the American Chemical Society 2016 Volume 138(Issue 36) pp:11946-11957
Publication Date(Web):August 19, 2016
DOI:10.1021/jacs.6b07347
Co-reporter:Daniel Roston; Darren Demapan
Journal of the American Chemical Society 2016 Volume 138(Issue 23) pp:7386-7394
Publication Date(Web):May 17, 2016
DOI:10.1021/jacs.6b03156
A reaction’s transition state (TS) structure plays a critical role in determining reactivity and has important implications for the design of catalysts, drugs, and other applications. Here, we explore TS structure in the enzyme alkaline phosphatase using hybrid Quantum Mechanics/Molecular Mechanics simulations. We find that minor perturbations to the substrate have major effects on TS structure and the way the enzyme stabilizes the TS. Substrates with good leaving groups (LGs) have little cleavage of the phosphorus–LG bond at the TS, while substrates with poor LGs have substantial cleavage of that bond. The results predict nonlinear free energy relationships for a single rate-determining step, and substantial differences in kinetic isotope effects for different substrates; both trends were observed in previous experimental studies, although the original interpretations differed from the present model. Moreover, due to different degrees of phosphorus–LG bond cleavage at the TS for different substrates, the LG is stabilized by different interactions at the TS: while a poor LG is directly stabilized by an active site zinc ion, a good LG is mainly stabilized by active site water molecules. Our results demonstrate the considerable plasticity of TS structure and stabilization in enzymes. Furthermore, perturbations to reactivity that probe TS structure experimentally (i.e., substituent effects) may substantially perturb the TS they aim to probe, and thus classical experimental approaches such as free energy relations should be interpreted with care.
Co-reporter:Haiyun Jin, Puja Goyal, Akshaya Kumar Das, Michael Gaus, Markus Meuwly, and Qiang Cui
The Journal of Physical Chemistry B 2016 Volume 120(Issue 8) pp:1894-1910
Publication Date(Web):December 1, 2015
DOI:10.1021/acs.jpcb.5b09656
We apply two recently developed computational methods, DFTB3 and VALBOND, to study copper oxidation/reduction processes in solution and protein. The properties of interest include the coordination structure of copper in different oxidation states in water or in a protein (plastocyanin) active site, the reduction potential of the copper ion in different environments, and the environmental response to copper oxidation. The DFTB3/MM and VALBOND simulation results are compared to DFT/MM simulations and experimental results whenever possible. For a copper ion in aqueous solution, DFTB3/MM results are generally close to B3LYP/MM with a medium basis, including both solvation structure and reduction potential for Cu(II); for Cu(I), however, DFTB3/MM finds a two-water coordination, similar to previous Born–Oppenheimer molecular dynamics simulations using BLYP and HSE, whereas B3LYP/MM leads to a tetrahedron coordination. For a tetraammonia copper complex in aqueous solution, VALBOND and DFTB3/MM are consistent in terms of both structural and dynamical properties of solvent near copper for both oxidation states. For copper reduction in plastocyanin, DFTB3/MM simulations capture the key properties of the active site, and the computed reduction potential and reorganization energy are in fair agreement with experiment, especially when the periodic boundary condition is used. Overall, the study supports the value of VALBOND and DFTB3(/MM) for the analysis of fundamental copper redox chemistry in water and protein, and the results also help highlight areas where further improvements in these methods are desirable.
Co-reporter:Qiang Cui, Rigoberto Hernandez, Sara E. Mason, Thomas Frauenheim, Joel A. Pedersen, and Franz Geiger
The Journal of Physical Chemistry B 2016 Volume 120(Issue 30) pp:7297-7306
Publication Date(Web):July 7, 2016
DOI:10.1021/acs.jpcb.6b03976
For assistance in the design of the next generation of nanomaterials that are functional and have minimal health and safety concerns, it is imperative to establish causality, rather than correlations, in how properties of nanomaterials determine biological and environmental outcomes. Due to the vast design space available and the complexity of nano/bio interfaces, theoretical and computational studies are expected to play a major role in this context. In this minireview, we highlight opportunities and pressing challenges for theoretical and computational chemistry approaches to explore the relevant physicochemical processes that span broad length and time scales. We focus discussions on a bottom-up framework that relies on the determination of correct intermolecular forces, accurate molecular dynamics, and coarse-graining procedures to systematically bridge the scales, although top-down approaches are also effective at providing insights for many problems such as the effects of nanoparticles on biological membranes.
Co-reporter:Leili Zhang, Manohary Rajendram, Douglas B. Weibel, Arun Yethiraj, and Qiang Cui
The Journal of Physical Chemistry B 2016 Volume 120(Issue 33) pp:8424-8437
Publication Date(Web):April 19, 2016
DOI:10.1021/acs.jpcb.6b02164
We describe a computational and experimental approach for probing the binding properties of the RecA protein at the surface of anionic membranes. Fluorescence measurements indicate that RecA behaves differently when bound to phosphatidylglycerol (PG)- and cardiolipin (CL)-containing liposomes. We use a multistage computational protocol that integrates an implicit membrane/solvent model, the highly mobile mimetic membrane model, and the full atomistic membrane model to study how different anionic lipids perturb RecA binding to the membrane. With anionic lipids studied here, the binding interface involves three key regions: the N-terminal helix, the DNA binding loop L2, and the M-M7 region. The nature of binding involves both electrostatic interactions between cationic protein residues and lipid polar/charged groups and insertion of hydrophobic residues. The L2 loop contributes more to membrane insertion than the N-terminal helix. More subtle aspects of RecA–membrane interaction are influenced by specific properties of anionic lipids. Ionic hydrogen bonds between the carboxylate group in phosphatidylserine and several lysine residues in the C-terminal region of RecA stabilize the parallel (∥) binding orientation, which is not locally stable on PG- and CL-containing membranes despite similarity in the overall charge density. Lipid packing defects, which are more prevalent in the presence of conical lipids, are observed to enhance the insertion depth of hydrophobic motifs. The computational finding that RecA binds in a similar orientation to PG- and CL-containing membranes is consistent with the fact that PG alone is sufficient to induce RecA polar localization, although CL might be more effective because of its tighter binding to RecA. The different fluorescence behaviors of RecA upon binding to PG- and CL-containing liposomes is likely due to the different structures and flexibility of the C-terminal region of RecA when it binds to different anionic phospholipids.
Co-reporter:Michael Gaus, Haiyun Jin, Darren Demapan, Anders S. Christensen, Puja Goyal, Marcus Elstner, and Qiang Cui
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 9) pp:4205-4219
Publication Date(Web):August 11, 2015
DOI:10.1021/acs.jctc.5b00600
We report the parametrization of a density functional tight binding method (DFTB3) for copper in a spin-polarized formulation. The parametrization is consistent with the framework of 3OB for main group elements (ONCHPS) and can be readily used for biological applications that involve copper proteins/peptides. The key to our parametrization is to introduce orbital angular momentum dependence of the Hubbard parameter and its charge derivative, thus allowing the 3d and 4s orbitals to adopt different sizes and responses to the change of charge state. The parametrization has been tested by applying to a fairly broad set of molecules of biological relevance, and the properties of interest include optimized geometries, ligand binding energies, and ligand proton affinities. Compared to the reference QM level (B3LYP/aug-cc-pVTZ, which is shown here to be similar to the B97-1 and CCSD(T) results, in terms of many properties of interest for a set of small copper containing molecules), our parametrization generally gives reliable structural properties for both Cu(I) and Cu(II) compounds, although several exceptions are also noted. For energetics, the results are more accurate for neutral ligands than for charged ligands, likely reflecting the minimal basis limitation of DFTB3; the results generally outperform NDDO based methods such as PM6 and even PBE with the 6-31+G(d,p) basis. For all ligand types, single-point B3LYP calculations at DFTB3 geometries give results very close (∼1–2 kcal/mol) to the reference B3LYP values, highlighting the consistency between DFTB3 and B3LYP structures. Possible further developments of the DFTB3 model for a better treatment of transition-metal ions are also discussed. In the current form, our first generation of DFTB3 copper model is expected to be particularly valuable as a method that drives sampling in systems that feature a dynamical copper binding site.
Co-reporter:Van Ngo, Mauricio C. da Silva, Maximilian Kubillus, Hui Li, Benoît Roux, Marcus Elstner, Qiang Cui, Dennis R. Salahub, and Sergei Yu. Noskov
Journal of Chemical Theory and Computation 2015 Volume 11(Issue 10) pp:4992-5001
Publication Date(Web):September 22, 2015
DOI:10.1021/acs.jctc.5b00524
Despite decades of investigations, the principal mechanisms responsible for the high affinity and specificity of proteins for key physiological cations K+, Na+, and Ca2+ remain a hotly debated topic. At the core of the debate is an apparent need (or lack thereof) for an accurate description of the electrostatic response of the charge distribution in a protein to the binding of an ion. These effects range from partial electronic polarization of the directly ligating atoms to long-range effects related to partial charge transfer and electronic delocalization effects. While accurate modeling of cation recognition by metalloproteins warrants the use of quantum-mechanics (QM) calculations, the most popular approximations used in major biomolecular simulation packages rely on the implicit modeling of electronic polarization effects. That is, high-level QM computations for ion binding to proteins are desirable, but they are often unfeasible, because of the large size of the reactive-site models and the need to sample conformational space exhaustively at finite temperature. Several solutions to this challenge have been proposed in the field, ranging from the recently developed Drude polarizable force-field for simulations of metalloproteins to approximate tight-binding density functional theory (DFTB). To delineate the usefulness of different approximations, we examined the accuracy of three recent and commonly used theoretical models and numerical algorithms, namely, CHARMM C36, the latest developed Drude polarizable force fields, and DFTB3 with the latest 3OB parameters. We performed MD simulations for 30 cation-selective proteins with high-resolution X-ray structures to create ensembles of structures for analysis with different levels of theory, e.g., additive and polarizable force fields, DFTB3, and DFT. The results from DFT computations were used to benchmark CHARMM C36, Drude, and DFTB3 performance. The explicit modeling of quantum effects unveils the key electrostatic properties of the protein sites and the importance of specific ion-protein interactions. One of the most interesting findings is that secondary coordination shells of proteins are noticeably perturbed in a cation-dependent manner, showing significant delocalization and long-range effects of charge transfer and polarization upon binding Ca2+.
Co-reporter:Yuqing Zheng and Qiang Cui
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 20) pp:13689-13698
Publication Date(Web):24 Apr 2015
DOI:10.1039/C5CP01858G
Histone tails are the short peptide protrusions outside of the nucleosome core particle and they play a critical role in regulating chromatin dynamics and gene activity. A histone H3 N-terminal tail, like other histone tails, can be covalently modified on different residues to activate or repress gene expression. Previous studies have indicated that, despite its intrinsically disordered nature, the histone H3 N-terminal tail has regions of notable secondary structural propensities. To further understand the structure–dynamics–function relationship in this system, we have carried out 75.6 μs long implicit solvent simulations and 29.3 μs long explicit solvent simulations. The extensive samplings allow us to better characterize not only the underlying free energy landscape but also kinetic properties through Markov state models (MSM). Dihedral principal component analysis (dPCA) and locally scaled diffusion map (LSDMap) analysis yield consistent results that indicate an overall flat free energy surface with several shallow basins that correspond to conformations with a high α-helical propensity in two regions of the peptide. Kinetic information extracted from Markov state models reveals rapid transitions between different metastable states with mean first passage times spanning from several hundreds of nanoseconds to hundreds of microseconds. These findings shed light on how the dynamical nature of the histone H3 N-terminal tail is related to its function. The complementary nature of dPCA, LSDMap and MSM for the analysis of biomolecules is also discussed.
Co-reporter:Yuqing Zheng;Elif Nihal Korkmaz;Leslie A. Leinwand;Ivan Rayment;Keenan C. Taylor;Massimo Buvoli;Ada Buvoli;Nathan T. Heinze
PNAS 2015 Volume 112 (Issue 29 ) pp:E3806-E3815
Publication Date(Web):2015-07-21
DOI:10.1073/pnas.1505813112
The rod of sarcomeric myosins directs thick filament assembly and is characterized by the insertion of four skip residues
that introduce discontinuities in the coiled-coil heptad repeats. We report here that the regions surrounding the first three
skip residues share high structural similarity despite their low sequence homology. Near each of these skip residues, the
coiled-coil transitions to a nonclose-packed structure inducing local relaxation of the superhelical pitch. Moreover, molecular
dynamics suggest that these distorted regions can assume different conformationally stable states. In contrast, the last skip
residue region constitutes a true molecular hinge, providing C-terminal rod flexibility. Assembly of myosin with mutated skip
residues in cardiomyocytes shows that the functional importance of each skip residue is associated with rod position and reveals
the unique role of the molecular hinge in promoting myosin antiparallel packing. By defining the biophysical properties of
the rod, the structures and molecular dynamic calculations presented here provide insight into thick filament formation, and
highlight the structural differences occurring between the coiled-coils of myosin and the stereotypical tropomyosin. In addition
to extending our knowledge into the conformational and biological properties of coiled-coil discontinuities, the molecular
characterization of the four myosin skip residues also provides a guide to modeling the effects of rod mutations causing cardiac
and skeletal myopathies.
Co-reporter:Xiya Lu, Michael Gaus, Marcus Elstner, and Qiang Cui
The Journal of Physical Chemistry B 2015 Volume 119(Issue 3) pp:1062-1082
Publication Date(Web):September 2, 2014
DOI:10.1021/jp506557r
We report the parametrization of the approximate density functional theory, DFTB3, for magnesium and zinc for chemical and biological applications. The parametrization strategy follows that established in previous work that parametrized several key main group elements (O, N, C, H, P, and S). This 3OB set of parameters can thus be used to study many chemical and biochemical systems. The parameters are benchmarked using both gas-phase and condensed-phase systems. The gas-phase results are compared to DFT (mostly B3LYP), ab initio (MP2 and G3B3), and PM6, as well as to a previous DFTB parametrization (MIO). The results indicate that DFTB3/3OB is particularly successful at predicting structures, including rather complex dinuclear metalloenzyme active sites, while being semiquantitative (with a typical mean absolute deviation (MAD) of ∼3–5 kcal/mol) for energetics. Single-point calculations with high-level quantum mechanics (QM) methods generally lead to very satisfying (a typical MAD of ∼1 kcal/mol) energetic properties. DFTB3/MM simulations for solution and two enzyme systems also lead to encouraging structural and energetic properties in comparison to available experimental data. The remaining limitations of DFTB3, such as the treatment of interaction between metal ions and highly charged/polarizable ligands, are also discussed.
Co-reporter:Elif Nihal Korkmaz, Brian F. Volkman, and Qiang Cui
The Journal of Physical Chemistry B 2015 Volume 119(Issue 30) pp:9547-9558
Publication Date(Web):July 2, 2015
DOI:10.1021/acs.jpcb.5b02810
The human lymphotactin (hLtn) is a protein that features two native states both of which are physiologically relevant: it is a monomer (hLtn10) at 10 °C with 200 mM salt and a dimer (hLtn40) at 40 °C and without salt. Here we focus on the networks of electrostatic and hydrophobic interactions that display substantial changes upon the conversion from hLtn10 to hLtn40 since they are expected to modulate the relative stability of the two folds. In addition to the Arg 23–Arg 43 interaction discussed in previous work, we find several other like-charge pairs that are likely important to the stability of hLtn10. Free energy perturbation calculations are carried out to explicitly evaluate the contribution of the Arg 23–Arg 43 interaction to the hLtn10 stability. hLtn40 features a larger number of salt bridges, and a set of hydrophobic residues undergo major changes in the solvent accessible surface area between hLtn10 and hLtn40, pointing to their importance to the relative stability of the two folds. We also discuss the use of explicit and implicit solvent simulations for characterizing the conformational ensembles under different solution conditions.
Co-reporter:Leili Zhang, Arun Yethiraj, and Qiang Cui
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 7) pp:2845-2859
Publication Date(Web):May 20, 2014
DOI:10.1021/ct500218p
The binding of peptides and proteins to the surface of complex lipid membranes is important in many biological processes such as cell signaling and membrane remodeling. Computational studies can aid experiments by identifying physical interactions and structural motifs that determine the binding affinity and specificity. However, previous studies focused on either qualitative behaviors of protein/membrane interactions or the binding affinity of small peptides. Motivated by this observation, we set out to develop computational protocols for bimolecular binding to charged membranes that are applicable to both peptides and large proteins. In this work, we explore a method based on an implicit membrane/solvent model (generalized Born with a simple switching in combination with the Gouy–Chapman–Stern model for a charged interface), which we expect to lead to useful results when the binding does not implicate significant membrane deformation and local demixing of lipids. We show that the binding free energy can be efficiently computed following a thermodynamic cycle similar to protein–ligand binding calculations, especially when a Bennett acceptance ratio based protocol is used to consider both the membrane bound and solution conformational ensembles. Test calculations on a series of peptides show that our computational approach leads to binding affinities in encouraging agreement with experimental data, including for the challenging example of the bringing of flexible MARCKS-ED peptides to membranes. The calculations highlight that for a membrane with a significant fraction of anionic lipids, it is essential to include the effect of ion adsorption using the Stern model, which significantly modifies the effective surface charge. This implicit membrane model based computational protocol helps lay the groundwork for more systematic analysis of protein/peptide binding to membranes of complex shape and composition.
Co-reporter:Michael Gaus, Xiya Lu, Marcus Elstner, and Qiang Cui
Journal of Chemical Theory and Computation 2014 Volume 10(Issue 4) pp:1518-1537
Publication Date(Web):March 12, 2014
DOI:10.1021/ct401002w
We report the parametrization of the approximate density functional tight binding method, DFTB3, for sulfur and phosphorus. The parametrization is done in a framework consistent with our previous 3OB set established for O, N, C, and H, thus the resulting parameters can be used to describe a broad set of organic and biologically relevant molecules. The 3d orbitals are included in the parametrization, and the electronic parameters are chosen to minimize errors in the atomization energies. The parameters are tested using a fairly diverse set of molecules of biological relevance, focusing on the geometries, reaction energies, proton affinities, and hydrogen bonding interactions of these molecules; vibrational frequencies are also examined, although less systematically. The results of DFTB3/3OB are compared to those from DFT (B3LYP and PBE), ab initio (MP2, G3B3), and several popular semiempirical methods (PM6 and PDDG), as well as predictions of DFTB3 with the older parametrization (the MIO set). In general, DFTB3/3OB is a major improvement over the previous parametrization (DFTB3/MIO), and for the majority cases tested here, it also outperforms PM6 and PDDG, especially for structural properties, vibrational frequencies, hydrogen bonding interactions, and proton affinities. For reaction energies, DFTB3/3OB exhibits major improvement over DFTB3/MIO, due mainly to significant reduction of errors in atomization energies; compared to PM6 and PDDG, DFTB3/3OB also generally performs better, although the magnitude of improvement is more modest. Compared to high-level calculations, DFTB3/3OB is most successful at predicting geometries; larger errors are found in the energies, although the results can be greatly improved by computing single point energies at a high level with DFTB3 geometries. There are several remaining issues with the DFTB3/3OB approach, most notably its difficulty in describing phosphate hydrolysis reactions involving a change in the coordination number of the phosphorus, for which a specific parametrization (3OB/OPhyd) is developed as a temporary solution; this suggests that the current DFTB3 methodology has limited transferability for complex phosphorus chemistry at the level of accuracy required for detailed mechanistic investigations. Therefore, fundamental improvements in the DFTB3 methodology are needed for a reliable method that describes phosphorus chemistry without ad hoc parameters. Nevertheless, DFTB3/3OB is expected to be a competitive QM method in QM/MM calculations for studying phosphorus/sulfur chemistry in condensed phase systems, especially as a low-level method that drives the sampling in a dual-level QM/MM framework.
Co-reporter:Qiang Cui and Marcus Elstner
Physical Chemistry Chemical Physics 2014 vol. 16(Issue 28) pp:14368-14377
Publication Date(Web):13 May 2014
DOI:10.1039/C4CP00908H
Semi-empirical (SE) methods are derived from Hartree–Fock (HF) or Density Functional Theory (DFT) by neglect and approximation of electronic integrals. Thereby, parameters are introduced which have to be determined from reference calculations and/or by fitting to available experimental data. This leads to computational methods that are about 2–3 orders of magnitude faster than the standard HF/DFT methods using medium sized basis sets while being about 3 orders of magnitude slower than empirical force field methods (Molecular Mechanics: MM). Therefore, SE methods are most appropriate for a specific range of applications. These include the study of systems that contain a large number of atoms and therefore being too large for ab initio or DFT methods and also problems where dynamic or entropic effects are particularly important. In the latter case, the errors made by considering a very limited number of molecular structures or neglecting entropic contributions can be much larger than the accuracy lost due to the use of SE methods. Another area where SE methods are attractive concerns the analysis of systems for which reliable MM models are not readily available. Therefore, even in an era when rapid progress is being made in ab initio methods, there is considerable interest in further developing SE methods. We illustrate this point by focusing on the discussion of recent development and application of the Density Functional Tight Binding method.
Co-reporter:Toshifumi Mori, Robert J. Hamers, Joel A. Pedersen, and Qiang Cui
The Journal of Physical Chemistry B 2014 Volume 118(Issue 28) pp:8210-8220
Publication Date(Web):March 18, 2014
DOI:10.1021/jp501339t
Motivated by specific applications and the recent work of Gao and co-workers on integrated tempering sampling (ITS), we have developed a novel sampling approach referred to as integrated Hamiltonian sampling (IHS). IHS is straightforward to implement and complementary to existing methods for free energy simulation and enhanced configurational sampling. The method carries out sampling using an effective Hamiltonian constructed by integrating the Boltzmann distributions of a series of Hamiltonians. By judiciously selecting the weights of the different Hamiltonians, one achieves rapid transitions among the energy landscapes that underlie different Hamiltonians and therefore an efficient sampling of important regions of the conformational space. Along this line, IHS shares similar motivations as the enveloping distribution sampling (EDS) approach of van Gunsteren and co-workers, although the ways that distributions of different Hamiltonians are integrated are rather different in IHS and EDS. Specifically, we report efficient ways for determining the weights using a combination of histogram flattening and weighted histogram analysis approaches, which make it straightforward to include many end-state and intermediate Hamiltonians in IHS so as to enhance its flexibility. Using several relatively simple condensed phase examples, we illustrate the implementation and application of IHS as well as potential developments for the near future. The relation of IHS to several related sampling methods such as Hamiltonian replica exchange molecular dynamics and λ-dynamics is also briefly discussed.
Co-reporter:Puja Goyal, Hu-Jun Qian, Stephan Irle, Xiya Lu, Daniel Roston, Toshifumi Mori, Marcus Elstner, and Qiang Cui
The Journal of Physical Chemistry B 2014 Volume 118(Issue 38) pp:11007-11027
Publication Date(Web):August 28, 2014
DOI:10.1021/jp503372v
We discuss the description of water and hydration effects that employs an approximate density functional theory, DFTB3, in either a full QM or QM/MM framework. The goal is to explore, with the current formulation of DFTB3, the performance of this method for treating water in different chemical environments, the magnitude and nature of changes required to improve its performance, and factors that dictate its applicability to reactions in the condensed phase in a QM/MM framework. A relatively minor change (on the scale of kBT) in the O–H repulsive potential is observed to substantially improve the structural properties of bulk water under ambient conditions; modest improvements are also seen in dynamic properties of bulk water. This simple change also improves the description of protonated water clusters, a solvated proton, and to a more limited degree, a solvated hydroxide. By comparing results from DFTB3 models that differ in the description of water, we confirm that proton transfer energetics are adequately described by the standard DFTB3/3OB model for meaningful mechanistic analyses. For QM/MM applications, a robust parametrization of QM-MM interactions requires an explicit consideration of condensed phase properties, for which an efficient sampling technique was developed recently and is reviewed here. The discussions help make clear the value and limitations of DFTB3 based simulations, as well as the developments needed to further improve the accuracy and transferability of the methodology.
Co-reporter:Guanhua Hou
Journal of the American Chemical Society 2013 Volume 135(Issue 28) pp:10457-10469
Publication Date(Web):June 20, 2013
DOI:10.1021/ja403293d
The first step for the hydrolysis of a phosphate monoester (pNPP2–) in enzymes of the alkaline phosphatase (AP) superfamily, R166S AP and wild-type NPP, is studied using QM/MM simulations based on an approximate density functional theory (SCC-DFTBPR) and a recently introduced QM/MM interaction Hamiltonian. The calculations suggest that similar loose transition states are involved in both enzymes, despite the fact that phosphate monoesters are the cognate substrates for AP but promiscuous substrates for NPP. The computed loose transition states are clearly different from the more synchronous ones previously calculated for diester reactions in the same AP enzymes. Therefore, our results explicitly support the proposal that AP enzymes are able to recognize and stabilize different types of transition states in a single active site. Analysis of the structural features of computed transition states indicates that the plastic nature of the bimetallic site plays a minor role in accommodating multiple types of transition states and that the high degree of solvent accessibility of the AP active site also contributes to its ability to stabilize diverse transition-state structures without the need of causing large structural distortions of the bimetallic motif. The binding mode of the leaving group in the transition state highlights that vanadate may not always be an ideal transition state analog for loose phosphoryl transfer transition states.
Co-reporter:Toshifumi Mori, Robert J. Hamers, Joel A. Pedersen, and Qiang Cui
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 11) pp:5059-5069
Publication Date(Web):October 3, 2013
DOI:10.1021/ct400487e
Identifying factors that control the strength and specificity of interactions between peptides and nanoparticles is essential for understanding the potential beneficial and deleterious effects of nanoparticles on biological systems. Computer simulations are valuable in this context, although the reliability of such calculations depends on the force field and sampling algorithm, as well as how the binding constant and binding free energy are defined; the latter must be carefully defined with a clear connection to microscopic models based on statistical mechanics. Using the example of formate binding to the rutile titanium dioxide (TiO2) (110) surface, we demonstrate that a reliable description of the binding process requires an explicit consideration of changes in the solvation state of the binding site. Specifically, we carry out metadynamics simulations in which the solvent coordination number of the binding site, s, is introduced as a collective variable in addition to the vertical distance of the adsorbate to the surface (z). The resulting two-dimensional potential of mean force (2D-PMF) clearly shows that explicitly including the local desolvation of the binding site on the TiO2 surface strongly impacts the convergence and result of the binding free energy calculations. Projecting the 2D-PMF into a one-dimensional PMF along either z or s leads to large errors in the free energy barriers. Results from metadynamics simulations are quantitatively supported by independent alchemical free energy simulations, in which the solvation state of the binding site is also carefully considered by explicitly introducing water molecules to the binding site as the adsorbate is decoupled from the system. On the other hand, preliminary committor analysis for the approximate transition state ensemble constructed based on the 2D-PMF suggests that to properly describe the binding/unbinding kinetics, variables beyond s and z, such as those describing the hydrogen bonding pattern of the adsorbate and surface water, need to be included. We expect that the insights and computational methodologies established in this work will be generally applicable to the analysis of binding interactions between highly charged adsorbates and ionic surfaces in solution, such as those implicated in peptide/nanoparticle binding and biomineralization processes.
Co-reporter:Puja Goyal;Shuo Yang;Jianxun Lu;M. R. Gunner
PNAS 2013 Volume 110 (Issue 47 ) pp:18886-18891
Publication Date(Web):2013-11-19
DOI:10.1073/pnas.1313908110
Cytochrome c oxidase contributes to the transmembrane proton gradient by removing two protons from the high-pH side of the membrane each
time the binuclear center active site is reduced. One proton goes to the binuclear center, whereas the other is pumped to
the low-pH periplasmic space. Glutamate 286 (Glu286) has been proposed to serve as a transiently deprotonated proton donor.
Using unrestrained atomistic molecular dynamics simulations, we show that the size of and water distribution in the hydrophobic
cavity that holds Glu286 is controlled by the protonation state of the propionic acid of heme a3, a group on the proton outlet pathway. Protonation of the propionate disrupts hydrogen bonding to two side chains, allowing
a loop to swing open. Continuum electrostatics and atomistic free-energy perturbation calculations show that the resultant
changes in hydration and electrostatic interactions lower the Glu proton affinity by at least 5 kcal/mol. These changes in
the internal hydration level occur in the absence of major conformational transitions and serve to stabilize needed transient
intermediates in proton transport. The trigger is not the protonation of the Glu of interest, but rather the protonation of
a residue ∼10 Å away. Thus, unlike local water penetration to stabilize a new charge, this finding represents a specific role
for water molecules in the protein interior, mediating proton transfers and facilitating ion transport.
Co-reporter:Xiya Lu and Qiang Cui
The Journal of Physical Chemistry B 2013 Volume 117(Issue 7) pp:2005-2018
Publication Date(Web):January 24, 2013
DOI:10.1021/jp309877z
Free energy simulations using a finite sphere boundary condition rather than a periodic boundary condition (PBC) are attractive in the study of very large biomolecular systems. To understand the quantitative impact of various approximations in such simulations, we compare charging free energies in both solution and protein systems calculated in a linear response framework with the Generalized Solvent Boundary Potential (GSBP) and PBC simulations. For simple ions in solution, we find good agreements between GSBP and PBC charging free energies, once the relevant correction terms are taken into consideration. For PBC simulations with the particle-mesh-Ewald for long-range electrostatics, the contribution (ΔGP–M) due to the use of a particle rather than molecule based summation scheme in real space is found to be significant, as pointed out by Hünenberger and co-workers. For GSBP, when the inner region is close to be charge neutral, the key correction is the overpolarization of water molecules at the inner/outer dielectric boundary; the magnitude of the correction (ΔGs–pol), however, is relatively small. For charging (oxidation) free energy in proteins, the situation is more complex, although good agreement between GSBP and PBC can still be obtained when care is exercised. The smooth dielectric boundary approximation inherent to GSBP tends to make significant errors when the inner region is featured with a high net charge. However, the error can be corrected with Poisson–Boltzmann calculations using snapshots from GSBP simulations in a straightforward and robust manner. Because of the more complex charge and solvent distributions, the magnitudes of ΔGP–M and ΔGs–pol in protein simulations appear to be different from those derived for solution simulations, leading to uncertainty in directly comparing absolute charging free energies from PBC and GSBP simulations for protein systems. The relative charging/oxidation free energies, however, are robust. With the linear response approximation, for the specific protein system (CueR) studied, the effect of freezing the protein structure in the outer region is found to be small, unless a very small (8 Å) inner region is used; even in the latter case, the result is substantially improved when the nearby metal binding loop is allowed to respond to metal oxidation. The implications of these results to the applicability of GSBP to complex biomolecules and in ab initio QM/MM simulations are discussed.
Co-reporter:Zhe Wu, Qiang Cui, and Arun Yethiraj
The Journal of Physical Chemistry B 2013 Volume 117(Issue 40) pp:12145-12156
Publication Date(Web):September 12, 2013
DOI:10.1021/jp4068729
An important puzzle in membrane biophysics is the difference in the behaviors of lysine (Lys) and arginine (Arg) based peptides at the membrane. For example, the translocation of poly-Arg is orders of magnitude faster than that of poly-Lys. Recent experimental work suggests that much of the difference can be inferred from the phase behavior of peptide/lipid mixtures. At similar concentrations, mixtures of phosphatidylethanolamine (PE) and phosphatidylserine (PS) lipids display different phases in the presence of these polypeptides, with a bicontinuous phase observed with poly-Arg peptides and an inverted hexagonal phase observed with poly-Lys peptides. Here we show that simulations with the coarse-grained (CG) BMW-MARTINI model reproduce the experimental results. An analysis using atomistic and CG models reveals that electrostatic and glycerol–peptide interactions play a crucial role in determining the phase behavior of peptide–lipid mixtures, with the difference between Arg and Lys arising from the stronger interactions of the former with lipid glycerols. In other words, the multivalent nature of the guanidinium group allows Arg to simultaneously interact with both phosphate and glycerol groups, while Lys engages solely with phosphate; this feature of amino acid/lipid interactions has not been emphasized in previous studies. The Arg peptides colocalize with PS in regions of high negative Gaussian curvature and stabilize the bicontinuous phase. Decreasing the strength of either the electrostatic interactions or the peptide–glycerol interactions causes the inverted hexagonal phase to become more stable. The results highlight the utility of CG models for the investigation of phase behavior but also emphasize the subtlety of the phenomena, with small changes in specific interactions leading to qualitatively different phases.
Co-reporter:Guanhua Hou, Xiao Zhu, Marcus Elstner, and Qiang Cui
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 11) pp:4293-4304
Publication Date(Web):September 21, 2012
DOI:10.1021/ct300649f
To improve the description of electrostatic interaction between QM and MM atoms when the QM is SCC-DFTB, we adopt a Klopman–Ohno (KO) functional form which considers the finite size of the QM and MM charge distributions. Compared to the original implementation that used a simple Coulombic interaction between QM Mulliken and MM point charges, the KO-based QM/MM scheme takes the charge penetration effect into consideration and therefore significantly improves the description of QM/MM interaction at short range, especially when the QM region is highly charged. To be consistent with the third-order formulation of SCC-DFTB, the Hubbard parameter in the KO functional is dependent on the QM charge. As a result, the effective size of the QM charge distribution naturally adjusts as the QM region undergoes chemical transformations, making the KO-based QM/MM scheme particularly attractive for describing chemical reactions in the condensed phase. Together with the van der Waals parameters for the QM atom, the KO-based QM/MM model introduces four parameters for each element type. They are fitted here based on microsolvation models of small solutes, focusing on negatively charged molecular ions, for elements O, C, H, and P with a specific version of SCC-DFTB (SCC-DFTBPR). Test calculations confirm that the KO-based QM/MM scheme significantly improves the interactions between QM and MM atoms over the original point charge based model, and it is transferable due to the small number of parameters. The new form of QM/MM Hamiltonian will greatly improve the applicability of SCC-DFTB based QM/MM methods to problems that involve highly charged QM regions, such as enzyme catalyzed phosphoryl transfers.
Co-reporter:Michael D. Daily, Lee Makowski, George N. Phillips Jr., Qiang Cui
Chemical Physics 2012 Volume 396() pp:84-91
Publication Date(Web):2 March 2012
DOI:10.1016/j.chemphys.2011.08.015
Abstract
While coarse-grained (CG) simulations provide an efficient approach to identify small- and large-scale motions important to protein conformational transitions, coupling with appropriate experimental validation is essential. Here, by comparing small-angle X-ray scattering (SAXS) predictions from CG simulation ensembles of adenylate kinase (AK) with a range of energetic parameters, we demonstrate that AK is flexible in solution in the absence of ligand and that a small population of the closed form exists without ligand. In addition, by analyzing variation of scattering patterns within CG simulation ensembles, we reveal that rigid-body motion of the LID domain corresponds to a dominant scattering feature. Thus, we have developed a novel approach for three-dimensional structural interpretation of SAXS data. Finally, we demonstrate that the agreement between predicted and experimental SAXS can be improved by increasing the simulation temperature or by computationally mutating selected residues to glycine, both of which perturb LID rigid-body flexibility.
Co-reporter:Jan Zienau and Qiang Cui
The Journal of Physical Chemistry B 2012 Volume 116(Issue 41) pp:12522-12534
Publication Date(Web):September 18, 2012
DOI:10.1021/jp308218m
The implementation of the solvent macromolecule boundary potential (SMBP) by Benighaus and Thiel (J. Chem. Theory Comput.2009, 5, 3114) into the program package CHARMM is presented. The SMBP allows for the efficient calculation of solvent effects for large macromolecules using irregularly shaped dielectric boundaries. In contrast to the generalized solvent boundary potential (GSBP) by Roux et al. (J. Chem. Phys.2001, 114, 2924) from which it is derived, the SMBP is targeted for quantum mechanical/molecular mechanical (QM/MM) setups using ab initio methods for the QM part. After presenting benchmark results for simple model systems, applications of the SMBP for the calculation of geometries, reaction energy barriers, and vibrational frequencies for an alkaline phosphatase (AP) enzyme are discussed. Although the effect of the boundary potential on optimized structures (including the transition state) and vibrational frequencies is relatively small, the energetics of the phosphoryl transfer catalyzed by AP depend significantly on the boundary potential. Finally, to emphasize a unique feature of our implementation, we apply both SMBP and GSBP to the calculation of the energy barrier for a proton transfer reaction in a simple model channel, where the effect of an external transmembrane potential is studied. Due to the dipolar response of the polar environment, the effective charge displacement estimated based on the effect of the membrane potential on the proton transfer energetics deviates from the net charge that passes the membrane.
Co-reporter:Demian Riccardi;Xiao Zhu;Puja Goyal;Shuo Yang;GuanHua Hou
Science China Chemistry 2012 Volume 55( Issue 1) pp:3-18
Publication Date(Web):2012 January
DOI:10.1007/s11426-011-4458-9
Motivated by several long-lasting mechanistic questions for biomolecular proton pumps, we have engaged in developing hybrid quantum mechanical/molecular mechanical (QM/MM) methods that allow an efficient and reliable description of long-range proton transport in transmembrane proteins. In this review, we briefly discuss several relevant issues: the need to develop a “multi-scale” generalized solvent boundary potential (GSBP) for the analysis of chemical events in large trans-membrane proteins, approaches to validate such a protocol, and the importance of improving the flexibility of QM/MM Hamiltonian. Several recent studies of model and realistic protein systems are also discussed to help put the discussions into context. Collectively, these studies suggest that the QM/MM-GSBP framework based on an approximate density functional theory (SCC-DFTB) as QM holds the promise to strike the proper balance between computational efficiency, accuracy and generality. With additional improvements in the methodology and recent developments by others, especially powerful sampling techniques, this “multi-scale” framework will be able to help unlock the secrets of proton pumps and other biomolecular machines.
Co-reporter:Guanhua Hou
Journal of the American Chemical Society 2011 Volume 134(Issue 1) pp:229-246
Publication Date(Web):November 18, 2011
DOI:10.1021/ja205226d
Several members of the Alkaline Phosphatase (AP) superfamily exhibit a high level of catalytic proffciency and promiscuity in structurally similar active sites. A thorough characterization of the nature of transition state for different substrates in these enzymes is crucial for understanding the molecular mechanisms that govern those remarkable catalytic properties. In this work, we study the hydrolysis of a phosphate diester, MpNPP–, in solution, two experimentally well-characterized variants of AP (R166S AP, R166S/E322Y AP) and wild type Nucleotide pyrophosphatase/phosphodiesterase (NPP) by QM/MM calculations in which the QM method is an approximate density functional theory previously parametrized for phosphate hydrolysis (SCC-DFTBPR). The general agreements found between these calculations and available experimental data for both solution and enzymes support the use of SCC-DFTBPR/MM for a semiquantitative analysis of the catalytic mechanism and nature of transition state in AP and NPP. Although phosphate diesters are cognate substrates for NPP but promiscuous substrates for AP, the calculations suggest that their hydrolysis reactions catalyzed by AP and NPP feature similar synchronous transition states that are slightly tighter in nature compared to that in solution, due in part to the geometry of the bimetallic zinc motif. Therefore, this study provides the first direct computational support to the hypothesis that enzymes in the AP superfamily catalyze cognate and promiscuous substrates via similar transition states to those in solution. Our calculations do not support the finding of recent QM/MM studies by López-Canut and co-workers, who suggested that the same diester substrate goes through a much looser transition state in NPP/AP than in solution, a result likely biased by the large structural distortion of the bimetallic zinc site in their simulations. Finally, our calculations for different phosphate diester orientations and phosphorothioate diesters highlight that the interpretation of thio-substitution experiments is not always straightforward.
Co-reporter:Puja Goyal ; Nilanjan Ghosh ; Prasad Phatak ; Maike Clemens ; Michael Gaus ; Marcus Elstner
Journal of the American Chemical Society 2011 Volume 133(Issue 38) pp:14981-14997
Publication Date(Web):July 15, 2011
DOI:10.1021/ja201568s
Identifying the group that acts as the proton storage/loading site is a challenging but important problem for understanding the mechanism of proton pumping in biomolecular proton pumps, such as bacteriorhodopsin (bR) and cytochrome c oxidase. Recent experimental studies of bR propelled the idea that the proton storage/release group (PRG) in bR is not an amino acid but a water cluster embedded in the protein. We argue that this idea is at odds with our knowledge of protein electrostatics, since invoking the water cluster as the PRG would require the protein to raise the pKa of a hydronium by almost 11 pKa units, which is difficult considering known cases of pKa shifts in proteins. Our recent quantum mechanics/molecular mechanics (QM/MM) simulations suggested an alternative “intermolecular proton bond” model in which the stored proton is shared between two conserved Glu residues (194 and 204). Here we show that this model leads to microscopic pKa values consistent with available experimental data and the functional requirement of a PRG. Extensive QM/MM simulations also show that, independent of a number of technical issues, such as the influence of QM region size, starting X-ray structure, and nuclear quantum effects, the “intermolecular proton bond” model is qualitatively consistent with available spectroscopic data. Potential of mean force calculations show explicitly that the stored proton strongly prefers the pair of Glu residues over the water cluster. The results and analyses help highlight the importance of considering protein electrostatics and provide arguments for why the “intermolecular proton bond” model is likely applicable to the PRG in biomolecular proton pumps in general.
Co-reporter:Zhe Wu, Qiang Cui, and Arun Yethiraj
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 11) pp:3793-3802
Publication Date(Web):September 20, 2011
DOI:10.1021/ct200593t
We present a new coarse-grained (CG) model for simulations of lipids and peptides. The model follows the same topology and parametrization strategy as the MARTINI force field but is based on our recently developed big multipole water (BMW) model for water (J. Phys. Chem. B2010, 114, 10524–10529). The new BMW-MARTINI force field reproduces many fundamental membrane properties and also yields improved energetics (when compared to the original MARTINI force-field) for the interactions between charged amino acids with lipid membranes, especially at the membrane–water interface. A stable attachment of cationic peptides (e.g., Arg8) to the membrane surface is predicted, consistent with experiment and in contrast to the MARTINI model. The model predicts electroporation when there is a charge imbalance across the lipid bilayer, an improvement over the original MARTINI. Moreover, the pore formed during electroporation is toroidal in nature, similar to the prediction of atomistic simulations but distinct from results of polarizable MARTINI for small charge imbalances. The simulations emphasize the importance of a reasonable description of the electrostatic properties of water in CG simulations. The BMW-MARTINI model is particularly suitable for describing interactions between highly charged peptides with lipid membranes, which is crucial to the study of antimicrobial peptides, cell penetrating peptides, and other proteins/peptides involved in the remodeling of biomembranes.
Co-reporter:L. Ma, N. K. Sundlass, R. T. Raines, and Q. Cui
Biochemistry 2011 Volume 50(Issue 2) pp:
Publication Date(Web):December 2, 2010
DOI:10.1021/bi101096k
Revealing the thermodynamic driving force of protein−DNA interactions is crucial to the understanding of factors that dictate the properties and function of protein−DNA complexes. For the binding of DNA to DNA-wrapping proteins, such as the integration host factor (IHF), Record and co-workers proposed that the disruption of a large number of preexisting salt bridges is coupled with the binding process [Holbrook, J. A., et al. (2001) J. Mol. Biol. 310, 379]. To test this proposal, we have conducted explicit solvent MD simulations (multiple ∼25−50 ns trajectories for each salt concentration) to examine the behavior of charged residues in IHF, especially concerning their ability to form salt bridges at different salt concentrations. Of the 17 cationic residues noted by Record and co-workers, most are engaged in salt bridge interactions for a significant portion of the trajectories, especially in the absence of salt. This observation suggests that, from a structural point of view, their proposal is plausible. However, the complex behaviors of charged residues observed in the MD simulations also suggest that the unusual thermodynamic characteristics of IHF−DNA binding likely arise from the interplay between complex dynamics of charged residues both in and beyond the DNA binding site. Moreover, a comparison of MD simulations at different salt concentrations suggests that the strong dependence of the IHF−DNA binding enthalpy on salt concentration may not be due to a significant decrease in the number of stable salt bridges in apo IHF at high salt concentrations. In addition to the Hofmeister effects quantified in more recent studies of IHF−DNA binding, we recommend consideration of the variation of the enthalpy change of salt bridge disruption at different salt concentrations. Finally, the simulation study presented here explicitly highlights the fact that the electrostatic properties of DNA-binding proteins can be rather different in the apo and DNA-bound states, which has important implications for the design of robust methods for predicting DNA binding sites in proteins.
Co-reporter:Puja Goyal, Marcus Elstner, and Qiang Cui
The Journal of Physical Chemistry B 2011 Volume 115(Issue 20) pp:6790-6805
Publication Date(Web):April 28, 2011
DOI:10.1021/jp202259c
The self-consistent charge density functional tight-binding (SCC-DFTB) method has been actively employed to study proton-transfer processes in biological systems. Recent studies in the literature employing SCC-DFTB reported that the method favors the Zundel form of the hydrated proton over the Eigen form, both in gas-phase water clusters and in bulk water, in disagreement with both higher-level calculations and experimental data. In this work, we explore the performance of SCC-DFTB for protonated gas-phase water clusters and bulk water (the latter both with and without an excess proton) with a modified O–H repulsive potential reported in our earlier work and with on-site third-order expansion of the DFT energy. Our results show that, with the proper set of published parameters, SCC-DFTB does correctly favor the Eigen form of the hydrated proton as compared to the Zundel form, both in gas-phase clusters and in the bulk; the amphiphilic character of the hydrated proton discussed in the literature has also been observed. The analyses do, however, bring forth remaining limitations in terms of the solvation structure around the hydrated proton as well as the structure of bulk water, which can guide future improvements of the method.
Co-reporter:Zhe Wu, Qiang Cui, and Arun Yethiraj
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 14) pp:1794-1798
Publication Date(Web):July 5, 2011
DOI:10.1021/jz2006622
The hydrophobic effect plays a central role in many biological processes, including protein folding and aggregation. The hydrophobic interaction between solutes, such as helical peptides, is believed to be of entropic origin and driven by the increase in the entropy of water due to association. In this work, we examine the association between peptides in water using several coarse-grained (CG) models, such as MARTINI (MAR), polarizable MARTINI (POL), and big multipole water (BMW) models, where four atomistic water molecules are grouped into a single CG unit. All models predict that a pair of helical peptides (Ala20 and Leu20) has favorable association free energy. The BMW model is the only model, however, in which this association is entropy-driven, as has been previously established with atomistic simulations and experiments. The MAR and POL models, where the CG water particles do not have a quadrupole moment, predict an enthalpy-driven association, with a negligible entropy change upon association. Similarly, the association of two rigid cylinders in water is found to be enthalpy-driven when the water is described with the CG model of Shinoda et al. that includes a soft-core nonelectrostatic interaction, while BMW predicts an entropy-driven association. These results emphasize the importance of electrostatic interactions in water for the qualitative features of the thermodynamics of biophysical systems.Keywords: coarse-grained models; entropy-driven hydrophobic association; entropy−enthalpy compensation; water quadrupole interactions;
Co-reporter:Li Rao ; Qiang Cui ;Xin Xu
Journal of the American Chemical Society 2010 Volume 132(Issue 51) pp:18092-18102
Publication Date(Web):December 3, 2010
DOI:10.1021/ja103742k
We carry out a theoretical analysis of factors that dictate the binding affinity and selectivity of the copper efflux regulator (CueR) toward different metal ions (Cu+, Ag+, Au+, Zn2+, and Hg2+). In addition to a simplified active-site model, we have established a computational framework based on quantum mechanical/molecular mechanical (QM/MM) and Poisson−Boltzmann approaches that allows us, for the first time, to systematically analyze the protein contribution to transition metal binding affinity and selectivity. We find that the QM/MM model leads to relative binding affinities that are consistent with observations from transcription induction experiments, while an active-site model does not, which highlights the importance of explicitly considering the protein environment for a thorough understanding of metal binding properties of metalloproteins. Regarding the trends in binding affinity, our analysis highlights both intrinsic properties of a metal ion and protein contributions. Specifically, the softness and desolvation penalty of a metal ion make large contributions to the binding affinity; for example, we find that the large desolvation penalty for Zn2+ rather than any stereoelectronic factor (e.g., linear vs tetrahedron coordination) is the key reason that Zn2+ binds much more weakly than Hg2+ to CueR. Moreover, our results explicitly demonstrate that the electrostatic environment of CueR is well-tuned to favor the binding of coinage metal ions over divalent ions. Finally, our analyses highlight the importance of considering the proper solution reference (i.e., the metal ion bound to buffer ligands vs water molecules) when discussing the binding affinity of metal ions to proteins.
Co-reporter:Guanhua Hou, Xiao Zhu and Qiang Cui
Journal of Chemical Theory and Computation 2010 Volume 6(Issue 8) pp:2303-2314
Publication Date(Web):July 7, 2010
DOI:10.1021/ct1001818
Motivated by the need to rapidly explore the potential energy surface of chemical reactions that involve highly charged species, we have developed an implicit solvent model for approximate density functional theory, SCC-DFTB. The solvation free energy is calculated using a popular model that employs Poisson−Boltzmann for electrostatics and a surface-area term for nonpolar contributions. To balance the treatment of species with different charge distributions, we make the atomic radii that define the dielectric boundary and solute cavity depend on the solute charge distribution. Specifically, the atomic radii are assumed to be linearly dependent on the Mulliken charges and solved self-consistently together with the solute electronic structure. Benchmark calculations indicate that the model leads to solvation free energies of comparable accuracy to the SM6 model (especially for ions), which requires much more expensive DFT calculations. With analytical first derivatives and favorable computational speed, the SCC-DFTB-based solvation model can be effectively used, in conjunction with high-level QM calculations, to explore the mechanism of solution reactions. This is illustrated with a brief analysis of the hydrolysis of monomethyl monophosphate ester (MMP) and trimethyl monophosphate ester (TMP). Possible future improvements are also briefly discussed.
Co-reporter:Liang Ma, Laurel Pegram, M. T. Record Jr. and Qiang Cui
Biochemistry 2010 Volume 49(Issue 9) pp:
Publication Date(Web):February 1, 2010
DOI:10.1021/bi9020082
To improve our understanding of the effects of small solutes on protein stability, we conducted atomistic simulations to quantitatively characterize the interactions between two broadly used small solutes, urea and glycine betaine (GB), and a triglycine peptide, which is a good model for a protein backbone. Multiple solute concentrations were analyzed, and each solute−peptide−water ternary system was studied with ∼200−300 ns of molecular dynamics simulations with the CHARMM force field. The comparison between calculated preferential interaction coefficients (Γ23) and experimentally measured values suggests that semiquantitative agreement with experiments can be obtained if care is exercised to balance interactions among the solute, protein, and water. On the other hand, qualitatively incorrect (i.e., wrong sign in Γ23) results can be obtained if a solute model is constructed by directly taking parameters for chemically similar groups from an existing force field. Such sensitivity suggests that small solute thermodynamic data can be valuable in the development of accurate force field models of biomolecules. Further decomposition of Γ23 into group contributions leads to additional insights regarding the effects of small solutes on protein stability. For example, use of the CHARMM force field predicts that urea preferentially interacts with not only amide groups in the peptide backbone but also aliphatic groups, suggesting a role for these interactions in urea-induced protein denaturation; quantitatively, however, it is likely that the CHARMM force field overestimates the interaction between urea and aliphatic groups. The results with GB support a simple thermodynamic model that assumes additivity of preferential interaction between GB and various biomolecular surfaces.
Co-reporter:Zhe Wu, Qiang Cui and Arun Yethiraj
The Journal of Physical Chemistry B 2010 Volume 114(Issue 32) pp:10524-10529
Publication Date(Web):July 27, 2010
DOI:10.1021/jp1019763
A new coarse-grained (CG) model is developed for water. Each CG unit consists of three charged sites, and there is an additional nonelectrostatic soft interaction between central sites on different units. The interactions are chosen to mimic the properties of 4-water clusters in atomistic simulations: the nonelectrostatic component is modeled using a modified Born−Mayer−Huggins potential, and the charges are chosen to reproduce the dipole moment and quadrupole moment tensor of 4-water clusters from atomistic simulations. The parameters are optimized to reproduce experimental data for the compressibility, density, and permittivity of bulk water and the surface tension and interface potential for the air−water interface. This big multipole water (BMW) model represents a qualitative improvement over existing CG water models; for example, it reproduces the dipole potential in membrane−water interface when compared to experiment, with modest additional computational cost as compared to the popular MARTINI CG model.
Co-reporter:Demian Riccardi, Shuo Yang, Qiang Cui
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2010 Volume 1804(Issue 2) pp:342-351
Publication Date(Web):February 2010
DOI:10.1016/j.bbapap.2009.07.026
Recent QM/MM analyses of proton transfer function of human carbonic anhydrase II (CAII) are briefly reviewed. The topics include a preliminary analysis of nuclear quadrupole coupling constant calculations for the zinc ion and more detailed analyses of microscopic pKa of the zinc-bound water and free energy profile for the proton transfer. From a methodological perspective, our results emphasize that performing sufficient sampling is essential to the calculation of all these quantities, which reflects the well solvated nature of CAII active site. From a mechanistic perspective, our analyses highlight the importance of electrostatics in shaping the energetics and kinetics of proton transfer in CAII for its function. We argue that once the pKa for the zinc-bound water is modulated to be in the proper range (~ 7.0), proton transfer through a relatively well solvated cavity towards/from the protein surface (His64) does not require any major acceleration. Therefore, although structural details like the length of the water wire between the donor and acceptor groups still may make a non-negligible contribution, our computational results and the framework of analysis suggest that the significance of such “fine-tuning” is likely secondary to the modulation of pKa of the zinc-bound water. We encourage further experimental analysis with mutation of (charged) residues not in the immediate neighborhood of the zinc ion to quantitatively test this electrostatics based framework; in particular, Φ analysis based on these mutations may shed further light into the relative importance of the classical Grotthus mechanism and the “proton hole” pathway that we have proposed recently for CAII.
Co-reporter:Nilanjan Ghosh, Xavier Prat-Resina, M. R. Gunner and Qiang Cui
Biochemistry 2009 Volume 48(Issue 11) pp:
Publication Date(Web):February 25, 2009
DOI:10.1021/bi8021284
As stringent tests for the molecular model and computational protocol, microscopic pKa calculations are performed for the key residue, Glu286, in cytochrome c oxidase (CcO) using a combined quantum mechanical/molecular mechanical (QM/MM) potential and a thermodynamic integration protocol. The impact of the number of water molecules in the hydrophobic cavity and protonation state of several key residues (e.g., His334, CuB-bound water, and PRDa3) on the computed microscopic pKa values of Glu286 has been systematically examined. To help evaluate the systematic errors in the QM/MM-based protocol, microscopic pKa calculations have also been carried out for sites in a soluble protein (Asp70 in T4 lysozyme) and a better-characterized membrane protein (Asp85 in bacteriorhodopsin). Overall, the results show a significant degree of internal consistency and reproducibility that support the effectiveness of the computational framework. Although the number of water molecules in the hydrophobic cavity does not greatly influence the computed pKa of Glu286, the protonation states of several residues, some of which are rather far away, have more significant impacts. Adopting the standard protonation state for all titratable residues leaves a large net charge on the system and a significantly elevated pKa for Glu286, highlighting that any attempt to address the energetics of proton transfers in CcO at a microscopic level should carefully select the protonation state of residues, even those not in the immediate neighborhood of the active site. The calculations indirectly argue against the deprotonation of His334 for the proton pumping process, although further studies that explicitly compute its pKa are required for a more conclusive statement. Finally, the deprotonated Glu286 is found to be in a stable water-mediated connection with PRDa3 for at least several nanoseconds when this presumed pumping site is protonated. This does not support the proposed role of Glu286 as a robust gating valve that prevents proton leakage, although a conclusive statement awaits a more elaborate characterization of the Glu286−PRDa3 connectivity with free energy simulations and a protonated PRDa3. The large sets of microscopic simulations performed here have provided useful guidance to the establishment of a meaningful molecular model and effective computational protocol for explicitly analyzing the proton transfer kinetics in CcO, which is required for answering key questions regarding the pumping function of this fascinating and complex system.
Co-reporter:Yang Yang and Qiang Cui
The Journal of Physical Chemistry A 2009 Volume 113(Issue 45) pp:12439-12446
Publication Date(Web):June 17, 2009
DOI:10.1021/jp902949f
Combined quantum mechanical/molecular mechanical (QM/MM) calculations with density functional theory are employed to analyze two issues related to the hydrolysis activity of adenosine triphosphate (ATP) in myosin. First, we compare the geometrical properties and electronic structure of ATP in the open (post-rigor) and closed (pre-powerstroke) active sites of the myosin motor domain. Compared to both solution and the open active site cases, the scissile Pγ−O3β bond of ATP in the closed active site is shown to be substantially elongated. Natural bond orbital (NBO) analysis clearly shows that this structural feature is correlated with the stronger anomeric effects in the closed active site, which involve charge transfers from the lone pairs in the nonbridging oxygen in the γ-phosphate to the antibonding orbital of the scissile bond. However, an energetic analysis finds that the ATP molecule is not significantly destabilized by the Pγ−O3β bond elongation. Therefore, despite the notable perturbations in the geometry and electronic structure of ATP as its environment changes from solution to the hydrolysis-competent active site, ground-state destabilization is unlikely to play a major role in enhancing the hydrolysis activity in myosin. Second, two-dimensional potential energy maps are used to better characterize the energetic landscape near the hydrolysis transition state. The results indicate that the transition-state region is energetically flat and a range of structures representative of different mechanisms according to the classical nomenclature (e.g., “associative”, “dissociative”, and “concerted”) are very close in energy. Therefore, at least in the case of ATP hydrolysis in myosin, the energetic distinction between different reaction mechanisms following the conventional nomenclature is likely small. This study highlights the importance of (i) explicitly evaluating the relevant energetic properties for determining whether a factor is essential to catalysis and (ii) broader explorations of the energy landscape beyond saddle points (even on free-energy surface) for characterizing the molecular mechanism of catalysis.
Co-reporter:Yang Yang and Qiang Cui
The Journal of Physical Chemistry B 2009 Volume 113(Issue 14) pp:4930-4939
Publication Date(Web):March 17, 2009
DOI:10.1021/jp810755p
To investigate whether water relay plays an important role in phosphoryl transfer reactions, we have used several theoretical approaches to compare key properties of uridine 3′-m-nitrobenzyl phosphate (UNP) in aqueous and tert-butanol solutions. Previous kinetic experiments found that the isomerization reaction of UNP is abolished in tert-butanol, which was interpreted as the direct evidence that supports the role of water relay in phosphoryl transfer. We have analyzed solute flexibility and solvent structure near the solute using equilibrium molecular dynamics simulations and a combined quantum mechanical/molecular mechanism (QM/MM) potential function for the solute. Snapshots from the simulations are then used in minimum energy path calculations to compare the energetics of direct nucleophilic attack and water-mediated nucleophilic attack pathways. QM/MM simulations are also used to compare the pseudorotation barriers for the pentavalent intermediate formed following the nucleophilic attack, another key step for the isomerization reaction. Combined results from these calculations suggest that water relay does not offer any significant energetic advantage over the direct nucleophilic attack. Unfortunately, the lack of isomerization in tert-butanol solution cannot be straightforwardly explained based on the results we have obtained here and therefore requires additional analysis. This study, nevertheless, has provided new insights into several most commonly discussed possibilities.
Co-reporter:Yang Yang, Haibo Yu, Darrin York, Marcus Elstner and Qiang Cui
Journal of Chemical Theory and Computation 2008 Volume 4(Issue 12) pp:2067-2084
Publication Date(Web):November 6, 2008
DOI:10.1021/ct800330d
Phosphate chemistry is involved in many key biological processes, yet the underlying mechanism often remains unclear. For theoretical analysis to effectively complement experimental mechanistic analysis, it is essential to develop computational methods that can capture the complexity of the underlying potential energy surface and allow for sufficient sampling of the configurational space. To this end, we report the parametrization of an approximate density functional theory, the Self-Consistent-Charge Density-Functional Tight-Binding (SCC-DFTB) method for systems containing phosphorus. Compared to high-level density functional theory and ab initio (MP2 and G3B3) results, the standard second-order parametrization is shown to give reliable structures for a diverse set of phosphate compounds but inaccurate energetics. With the on-site third-order terms included, referred to as SCC-DFTBPA, calculated proton affinities of phosphate compounds are substantially improved, although it remains difficult to obtain reliable proton affinity for both phosphates and compounds that do not contain phosphorus, indicating that further improvement in the formulation of SCC-DFTB is still a challenge to meet. To make SCC-DFTB applicable to phosphate reactions in the current (on-site-third-order-only) formulation, a “reaction-specific” parametrization, referred to as SCC-DFTBPR, is developed based on hydrolysis reactions of model phosphate species. Benchmark calculations in both the gas phase and solution phase indicate that SCC-DFTBPR gives reliable structural properties and semiquantitative energetics for phosphate hydrolysis reactions. Since the number of reaction-specific parameters is small, it is likely that SCC-DFTBPR is applicable to a broad set of phosphate species. Indeed, for 56 reaction exothermicities and 47 energy barriers related to RNA catalysis model reactions collected from the QCRNA database, which involve molecules rather different from those used to parametrize SCC-DFTBPR, the corresponding root-mean-square difference between SCC-DFTBPR and high-level DFT results is only 5.3 kcal/mol. We hope that the parametrized SCC-DFTB models will complement NDDO based reaction-specific models (e.g., AM1-d/PhoT) and high-level ab initio QM/MM methods in better understanding the mechanism of phosphate chemistry in condensed phase, particularly biological systems.
Co-reporter:Prasad Phatak;Nilanjan Ghosh;Haibo Yu;Marcus Elstner
PNAS 2008 Volume 105 (Issue 50 ) pp:19672-19677
Publication Date(Web):2008-12-16
DOI:10.1073/pnas.0810712105
The positions of protons are not available in most high-resolution structural data of biomolecules, thus the identity of proton
storage sites in biomolecules that transport proton is generally difficult to determine unambiguously. Using combined quantum
mechanical/molecular mechanical computations, we demonstrate that a pair of conserved glutamate residues (Glu 194/204) bonded
by a delocalized proton is the proton release group that has been long sought in the proton pump, bacteriorhodopsin. This
model is consistent with all available experimental structural and infrared data for both the wild-type bacteriorhodopsin
and several mutants. In particular, the continuum infrared band in the 1,800- to 2,000-cm−1 region is shown to arise due to the partially delocalized nature of the proton between the glutamates in the wild-type bacteriorhodopsin;
alternations in the flexibility of the glutamates and electrostatic nature of nearby residues in various mutants modulate
the degree of proton delocalization and therefore intensity of the continuum band. The strong hydrogen bond between Glu 194/204
also significantly shifts the carboxylate stretches of these residues well <1,700 cm−1, which explains why carboxylate spectral shift was not observed experimentally in the typical >1,700-cm−1 region upon proton release. By contrast, simulations with the proton restrained on the nearby water cluster, as proposed
by several recent studies [see, for example, Garezarek K, Gerwert K (2006) Functional waters in intraprotein proton transfer
monitored by FTIR difference spectroscopy. Nature 439:109], led to significant structural deviations from available X-ray structures. This study establishes a biological function
for strong, low-barrier hydrogen bonds.
Co-reporter:Nilanjan Ghosh and Qiang Cui
The Journal of Physical Chemistry B 2008 Volume 112(Issue 28) pp:8387-8397
Publication Date(Web):June 10, 2008
DOI:10.1021/jp800168z
A combined quantum mechanical/molecular mechanical (QM/MM) potential function is used in a thermodynamic integration approach to calculate the pKa of residue 66 in two mutants (V66E, V66D) of Staphylococal nuclease relative to solution. Despite the similarity in chemical nature and experimentally measured pKa of the two buried titritable residues, the behaviors of the two mutants and the computed pKa values vary greatly in the simulations. For Glu66, the side chain is consistently observed to spontaneously flip out from the protein interior during titration, and the overall protein structure remains stable throughout the simulations. The computed pKa shifts using conventional sampling techniques with multiple nanoseconds per λ window (Set A and B) are generally close to the experimental value, therefore indicating that large-scale conformational rearrangements are not as important for V66E as suggested by the recent study of Warshel and co-worker. For Asp66, by contrast, flipping of the shorter side chain is not sufficient for getting adequate solvent stabilization of the ionized state. As a result, more complex behaviors such as partial unfolding of a nearby β-sheet region is observed, and the computed pKa shift is substantially higher than the experimental value unless Asp66 is biased to adopt the similar configurations as Glu66 in the V66E simulations. Collectively, these studies suggest that the lack of electronic polarization is not expected to be the dominant source of error in microscopic pKa shift calculations, while the need of enhanced sampling is more compelling for predicting the pKa of buried residues. Furthermore, the comparison between V66E and V66D also highlights that the microscopic interpretation of similar apparent pKa values and effective “dielectric constants” of proteins can vary greatly in terms of the residues that make key contributions and the scale of structural/hydration response to titration, the latter of which is difficult to predict a priori. Perturbative analyses of interactions that contribute to the titration free energy point to mutants that can be used to verify the microscopic mechanisms of titration in V66E/D SNase proteins.
Co-reporter:Yuye Tang;Jejoong Yoo;Arun Yethiraj
Cell Biochemistry and Biophysics 2008 Volume 52( Issue 1) pp:
Publication Date(Web):2008 September
DOI:10.1007/s12013-008-9024-5
Mechanotransduction plays an important role in regulating cell functions and it is an active topic of research in biophysics. Despite recent advances in experimental and numerical techniques, the intrinsic multiscale nature imposes tremendous challenges for revealing the working mechanisms of mechanosensitive channels. Recently, a continuum-mechanics-based hierarchical modeling and simulation framework has been established and applied to study the mechanical responses and gating behaviors of a prototypical mechanosensitive channel, the mechanosensitive channel of large conductance (MscL) in bacteria Escherichia coli (E. coli), from which several putative gating mechanisms have been tested and new insights are deduced. This article reviews these latest findings using the continuum mechanics framework and suggests possible improvements for future simulation studies. This computationally efficient and versatile continuum-mechanics-based protocol is poised to make contributions to the study of a variety of mechanobiology problems.
Co-reporter:Jejoong Yoo, Qiang Cui
Biophysical Journal (8 September 2010) Volume 99(Issue 5) pp:
Publication Date(Web):8 September 2010
DOI:10.1016/j.bpj.2010.06.048
Charged amino acids such as Arginine play important roles in many membrane-mediated biological processes such as voltage gating of ion channels and membrane translocation of cell penetration peptides. It is well established that local membrane deformation and formation of water defects are crucial to the stabilization of charged species in contact with the membrane, which suggests that mechanical properties of the membrane are relevant although a clear connection has not been established. As a quantitative measure, we study how changes in the composition and therefore mechanical properties of a lipid bilayer influence the pKa of Arg in the membrane center using free energy simulations. Compared to previous studies in a single-component lipid bilayer containing saturated lipids or lipids with a modest degree of unsaturation, substantially larger pKa shifts are observed in the presence of highly unsaturated lipid tails and cholesterol. Moreover, the underlying molecular mechanisms for the pKa perturbation are distinct in different systems, with the unsaturated lipid tails mainly destabilizing the charged state of Arg and the cholesterol stabilizing the neutral state of Arg. The observed behaviors in both cases are at odds with predictions based on mechanical considerations at a mesoscopic level—highlighting that, while mechanical considerations are useful for stimulating hypothesis, their applicability to dissecting phenomena at the molecular-length scale is rather limited.
Co-reporter:Jejoong Yoo, Qiang Cui
Biophysical Journal (8 January 2013) Volume 104(Issue 1) pp:
Publication Date(Web):8 January 2013
DOI:10.1016/j.bpj.2012.11.3813
To further foster the connection between particle based and continuum mechanics models for membrane mediated biological processes, we carried out coarse-grained (CG) simulations of gramicidin A (gA) dimer association and analyzed the results based on the combination of potential of mean force (PMF) and stress field calculations. Similar to previous studies, we observe that the association of gA dimers depends critically on the degree of hydrophobic mismatch, with the estimated binding free energy of >10 kcal/mol in a distearoylphosphatidylcholine bilayer. Qualitative trends in the computed PMF can be understood based on the stress field distributions near a single gA dimer and between a pair of gA dimers. For example, the small PMF barrier, which is ∼1 kcal/mol independent of lipid type, can be captured nearly quantitatively by considering membrane deformation energy associated with the region confined by two gA dimers. However, the PMF well depth is reproduced poorly by a simple continuum model that only considers membrane deformation energy beyond the annular lipids. Analysis of lipid orientation, configuration entropy, and stress distribution suggests that the annular lipids make a significant contribution to the association of two gA dimers. These results highlight the importance of explicitly considering contributions from annular lipids when constructing approximate models to study processes that involve a significant reorganization of lipids near proteins, such as protein-protein association and protein insertion into biomembranes. Finally, large-scale CG simulations indicate that multiple gA dimers also form clusters, although the preferred topology depends on the protein concentration. Even at high protein concentrations, every gA dimer requires contact to lipid hydrocarbons to some degree, and at most three to four proteins are in contact with each gA dimer; this observation highlights another aspect of the importance of interactions between proteins and annular lipids.
Co-reporter:Jejoong Yoo, Qiang Cui
Biophysical Journal (8 January 2013) Volume 104(Issue 1) pp:
Publication Date(Web):8 January 2013
DOI:10.1016/j.bpj.2012.11.3812
Using both atomistic and coarse-grained (CG) models, we compute the three-dimensional stress field around a gramicidin A (gA) dimer in lipid bilayers that feature different degrees of negative hydrophobic mismatch. The general trends in the computed stress field are similar at the atomistic and CG levels, supporting the use of the CG model for analyzing the mechanical features of protein/lipid/water interfaces. The calculations reveal that the stress field near the protein-lipid interface exhibits a layered structure with both significant repulsive and attractive regions, with the magnitude of the stress reaching 1000 bar in certain regions. Analysis of density profiles and stress field distributions helps highlight the Trp residues at the protein/membrane/water interface as mechanical anchors, suggesting that similar analysis is useful for identifying tension sensors in other membrane proteins, especially membrane proteins involved in mechanosensation. This work fosters a connection between microscopic and continuum mechanics models for proteins in complex environments and makes it possible to test the validity of assumptions commonly made in continuum mechanics models for membrane mediated processes. For example, using the calculated stress field, we estimate the free energy of membrane deformation induced by the hydrophobic mismatch, and the results for regions beyond the annular lipids are in general consistent with relevant experimental data and previous theoretical estimates using elasticity theory. On the other hand, the assumptions of homogeneous material properties for the membrane and a bilayer thickness at the protein/lipid interface being independent of lipid type (e.g., tail length) appear to be oversimplified, highlighting the importance of annular lipids of membrane proteins. Finally, the stress field analysis makes it clear that the effect of even rather severe hydrophobic mismatch propagates to only about two to three lipid layers, thus putting a limit on the range of cooperativity between membrane proteins in crowded cellular membranes.
Co-reporter:Liang Ma, Arun Yethiraj, Xi Chen, Qiang Cui
Biophysical Journal (6 May 2009) Volume 96(Issue 9) pp:
Publication Date(Web):6 May 2009
DOI:10.1016/j.bpj.2009.01.047
A computational framework is presented for studying the mechanical response of macromolecules. The method combines a continuum mechanics (CM) model for the mechanical properties of the macromolecule with a continuum electrostatic (CE) treatment of solvation. The molecules are represented by their shape and key physicochemical characteristics such as the distribution of materials properties and charge. As a test case, we apply the model to the effect of added salt on the bending of DNA. With a simple representation of DNA, the CM/CE framework using a Debye-Hückel model leads to results that are in good agreement with both analytical theories and recent experiments, including a modified Odijk-Skolnick-Fixman theory that takes the finite length of DNA into consideration. Calculations using a more sophisticated CE model (Poisson-Boltzmann), however, suffer from convergence problems, highlighting the importance of balancing numerical accuracy in the CM and CE models when dealing with very large systems, particularly those with a high degree of symmetry.
Co-reporter:Shuo Yang, Qiang Cui
Biophysical Journal (6 July 2011) Volume 101(Issue 1) pp:
Publication Date(Web):6 July 2011
DOI:10.1016/j.bpj.2011.05.004
One of the key unresolved issues regarding proton pumping in cytochrome c oxidase (CcO) is the identity of the gating element that prevents the backflow of protons. In this study, we analyze two popular proposals for this element: isomerization of the key branching residue (Glu-286) and (re)orientation of water molecules in the hydrophobic cavity. Using a multifaceted set of computational analyses that involve CcO embedded in either an implicit or explicit treatment of lipid membrane, we show that neither Glu-286 nor active-site water likely constitutes the gating element. Detailed energetic and structural analyses of the simulation results indicate that the gating-relevant properties of these structural motifs observed in previous work are likely a result of the simplified computational models employed in those studies.
Co-reporter:Jejoong Yoo, Qiang Cui
Biophysical Journal (21 October 2009) Volume 97(Issue 8) pp:
Publication Date(Web):21 October 2009
DOI:10.1016/j.bpj.2009.07.051
Although many membrane additives are known to modulate the activities of membrane proteins via perturbing the properties of lipid membrane, the underlying mechanism is often not precisely understood. In this study, we investigate the impact of asymmetrically incorporating single-tailed lysophosphatidylcholine (LPC) into a membrane bilayer using coarse-grained molecular dynamics simulations. Using a simple computational protocol designed to approximately mimic a micropipette setting, we show that asymmetric incorporation of LPC can lead to significant curvature in a bilayer. Detailed analysis of geometrical and mechanical properties (pressure profile) of the resulting mound structure indicates that the degree of pressure profile perturbation is determined not by the local curvature per se but by the packing of lipid headgroups (i.e., area-per-lipid). The findings help provide a concrete basis for understanding the activation mechanism of mechanosensitive channels by asymmetric incorporation of LPC into membrane patches in patch-clamp experiments. The calculated local pressure profiles are valuable to the construction of realistic membrane models for the analysis of mechanosensation in a continuum mechanics framework.
Co-reporter:Yang Yang, Haibo Yu, Qiang Cui
Journal of Molecular Biology (19 September 2008) Volume 381(Issue 5) pp:1407-1420
Publication Date(Web):19 September 2008
DOI:10.1016/j.jmb.2008.06.071
Conventional myosin is representative of biomolecular motors in which the hydrolysis of adenosine triphosphate (ATP) is coupled to large-scale structural transitions both in and remote from the active site. The mechanism that underlies such “mechanochemical coupling,” especially the causal relationship between hydrolysis and allosteric structural changes, has remained elusive despite extensive experimental and computational analyses. In this study, using combined quantum mechanical and molecular mechanical simulations and different conformations of the myosin motor domain, we provide evidence to support that regulation of ATP hydrolysis activity is not limited to residues in the immediate environment of the phosphate. Specifically, we illustrate that efficient hydrolysis of ATP depends not only on the proper orientation of the lytic water but also on the structural stability of several nearby residues, especially the Arg238-Glu459 salt bridge (the numbering of residues follows myosin II in Dictyostelium discoideum) and the water molecule that spans this salt bridge and the lytic water. More importantly, by comparing the hydrolysis activities in two motor conformations with very similar active-site (i.e., Switches I and II) configurations, which distinguished this work from our previous study, the results clearly indicate that the ability of these residues to perform crucial electrostatic stabilization relies on the configuration of residues in the nearby N-terminus of the relay helix and the “wedge loop.” Without the structural support from those motifs, residues in a closed active site in the post-rigor motor domain undergo subtle structural variations that lead to consistently higher calculated ATP hydrolysis barriers than in the pre-powerstroke state. In other words, starting from the post-rigor state, turning on the ATPase activity requires not only displacement of Switch II to close the active site but also structural transitions in the N-terminus of the relay helix and the “wedge loop,” which have been proposed previously to be ultimately coupled to the rotation of the converter subdomain 40 Å away.
Co-reporter:Jejoong Yoo, Meyer B. Jackson, Qiang Cui
Biophysical Journal (19 February 2013) Volume 104(Issue 4) pp:
Publication Date(Web):19 February 2013
DOI:10.1016/j.bpj.2012.12.043
To establish the validity of continuum mechanics models quantitatively for the analysis of membrane remodeling processes, we compare the shape and energies of the membrane fusion pore predicted by coarse-grained (MARTINI) and continuum mechanics models. The results at these distinct levels of resolution give surprisingly consistent descriptions for the shape of the fusion pore, and the deviation between the continuum and coarse-grained models becomes notable only when the radius of curvature approaches the thickness of a monolayer. Although slow relaxation beyond microseconds is observed in different perturbative simulations, the key structural features (e.g., dimension and shape of the fusion pore near the pore center) are consistent among independent simulations. These observations provide solid support for the use of coarse-grained and continuum models in the analysis of membrane remodeling. The combined coarse-grained and continuum analysis confirms the recent prediction of continuum models that the fusion pore is a metastable structure and that its optimal shape is neither toroidal nor catenoidal. Moreover, our results help reveal a new, to our knowledge, bowing feature in which the bilayers close to the pore axis separate more from one another than those at greater distances from the pore axis; bowing helps reduce the curvature and therefore stabilizes the fusion pore structure. The spread of the bilayer deformations over distances of hundreds of nanometers and the substantial reduction in energy of fusion pore formation provided by this spread indicate that membrane fusion can be enhanced by allowing a larger area of membrane to participate and be deformed.
Co-reporter:Michael D. Daily, George N. Phillips, Qiang Cui
Journal of Molecular Biology (16 July 2010) Volume 400(Issue 3) pp:618-631
Publication Date(Web):16 July 2010
DOI:10.1016/j.jmb.2010.05.015
Conformational transitions are functionally important in many proteins. In the enzyme adenylate kinase (AK), two small domains (LID and NMP) close over the larger CORE domain; the reverse (opening) motion limits the rate of catalytic turnover. Here, using double-well Gō simulations of Escherichia coli AK, we elaborate on previous investigations of the AK transition mechanism by characterizing the contributions of rigid-body (Cartesian), backbone dihedral, and contact motions to transition-state (TS) properties. In addition, we compare an apo simulation to a pseudo-ligand-bound simulation to reveal insights into allostery. In Cartesian space, LID closure precedes NMP closure in the bound simulation, consistent with prior coarse-grained models of the AK transition. However, NMP-first closure is preferred in the apo simulation. In backbone dihedral space, we find that, as expected, backbone fluctuations are reduced in the O/C transition in parts of all three domains. Among these “quenching” residues, most in the CORE, especially residues 11–13, are rigidified in the TS of the bound simulation, while residues 42–44 in the NMP are flexible in the TS. In contact space, in both apo and bound simulations, one nucleus of closed-state contacts includes parts of the NMP and CORE; CORE–LID contacts are absent in the TS of the apo simulation but formed in the TS of the bound simulation. From these results, we predict mutations that will perturb the opening and/or closing transition rates by changing the entropy of dihedrals and/or the enthalpy of contacts. Furthermore, regarding allostery, the fully closed structure is populated in the apo simulation, but our contact results imply that ligand binding shifts the preferred O/C transition pathway, thus precluding a simple conformational selection mechanism. Finally, the analytical approach and the insights derived from this work may inform the rational design of flexibility and allostery in proteins.
Co-reporter:Jejoong Yoo, Qiang Cui
Biophysical Journal (15 April 2008) Volume 94(Issue 8) pp:
Publication Date(Web):15 April 2008
DOI:10.1529/biophysj.107.122945
Free energy perturbation calculations are carried out to estimate the effective pKa of an arginine (Arg) sidechain as a function of its location in the dipalmitoylphosphatidylcholine bilayer. Similar to previous all-atom simulations of the voltage sensor domain of a potassium channel in the membrane with charged Arg residues, the membrane and water structures deform to stabilize the charge of the Arg analog. As a result, the computed pKa is >7 throughout the membrane although the value is very close to 7 near the center of the bilayer. With additional stabilizations from negatively charged amino acids or lipid molecules, it is reasonable to expect that Arg in membrane proteins (once in the membrane) can adopt the protonated state despite the low dielectric nature of the bulk lipid membrane.
Co-reporter:Adam W. Van Wynsberghe, Qiang Cui
Structure (10 March 2010) Volume 18(Issue 3) pp:281-283
Publication Date(Web):10 March 2010
DOI:10.1016/j.str.2010.02.001
In this issue, Raimondi et al. (2010) obtained interesting insights concerning structural flexibilities in the Ras superfamily that are essential to both function retention and specialization by analyzing the deformation patterns from physical models of protein structure and from crystal structures of homologous proteins.
Co-reporter:Qiang Cui and Marcus Elstner
Physical Chemistry Chemical Physics 2014 - vol. 16(Issue 28) pp:NaN14377-14377
Publication Date(Web):2014/05/13
DOI:10.1039/C4CP00908H
Semi-empirical (SE) methods are derived from Hartree–Fock (HF) or Density Functional Theory (DFT) by neglect and approximation of electronic integrals. Thereby, parameters are introduced which have to be determined from reference calculations and/or by fitting to available experimental data. This leads to computational methods that are about 2–3 orders of magnitude faster than the standard HF/DFT methods using medium sized basis sets while being about 3 orders of magnitude slower than empirical force field methods (Molecular Mechanics: MM). Therefore, SE methods are most appropriate for a specific range of applications. These include the study of systems that contain a large number of atoms and therefore being too large for ab initio or DFT methods and also problems where dynamic or entropic effects are particularly important. In the latter case, the errors made by considering a very limited number of molecular structures or neglecting entropic contributions can be much larger than the accuracy lost due to the use of SE methods. Another area where SE methods are attractive concerns the analysis of systems for which reliable MM models are not readily available. Therefore, even in an era when rapid progress is being made in ab initio methods, there is considerable interest in further developing SE methods. We illustrate this point by focusing on the discussion of recent development and application of the Density Functional Tight Binding method.
Co-reporter:Yuqing Zheng and Qiang Cui
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 20) pp:NaN13698-13698
Publication Date(Web):2015/04/24
DOI:10.1039/C5CP01858G
Histone tails are the short peptide protrusions outside of the nucleosome core particle and they play a critical role in regulating chromatin dynamics and gene activity. A histone H3 N-terminal tail, like other histone tails, can be covalently modified on different residues to activate or repress gene expression. Previous studies have indicated that, despite its intrinsically disordered nature, the histone H3 N-terminal tail has regions of notable secondary structural propensities. To further understand the structure–dynamics–function relationship in this system, we have carried out 75.6 μs long implicit solvent simulations and 29.3 μs long explicit solvent simulations. The extensive samplings allow us to better characterize not only the underlying free energy landscape but also kinetic properties through Markov state models (MSM). Dihedral principal component analysis (dPCA) and locally scaled diffusion map (LSDMap) analysis yield consistent results that indicate an overall flat free energy surface with several shallow basins that correspond to conformations with a high α-helical propensity in two regions of the peptide. Kinetic information extracted from Markov state models reveals rapid transitions between different metastable states with mean first passage times spanning from several hundreds of nanoseconds to hundreds of microseconds. These findings shed light on how the dynamical nature of the histone H3 N-terminal tail is related to its function. The complementary nature of dPCA, LSDMap and MSM for the analysis of biomolecules is also discussed.

![(2s)-2-amino-3-[[(2s)-2,3-di(octadec-9-enoyloxy)propoxy]-hydroxyphosphoryl]oxypropanoic Acid](http://img.cochemist.com/ccimg/6900/6811-55-8.png)
![(2s)-2-amino-3-[[(2s)-2,3-di(octadec-9-enoyloxy)propoxy]-hydroxyphosphoryl]oxypropanoic Acid](http://img.cochemist.com/ccimg/6900/6811-55-8_b.png)
![4-((e)-2-[4-(diethylamino)phenyl]ethenyl)-1-[3-(triethylammonio)propyl]pyridinium Dibromide](http://img.cochemist.com/ccimg/336200/336185-20-7.png)
![4-((e)-2-[4-(diethylamino)phenyl]ethenyl)-1-[3-(triethylammonio)propyl]pyridinium Dibromide](http://img.cochemist.com/ccimg/336200/336185-20-7_b.png)




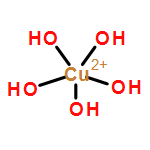
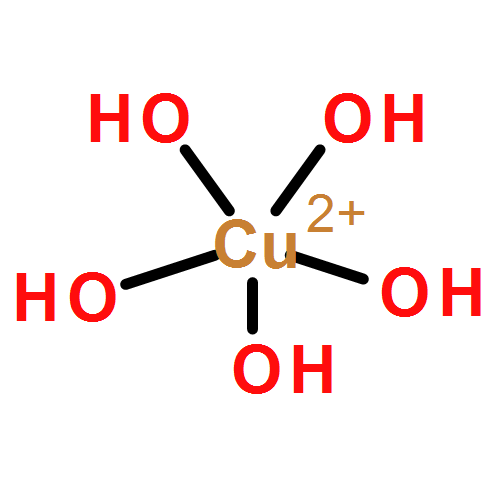


![4-{2-[4-(dipentylamino)phenyl]ethenyl}-1-[3-(triethylammonio)propyl]pyridinium](http://img.cochemist.com/ccimg/303800/303727-66-4.png)
![4-{2-[4-(dipentylamino)phenyl]ethenyl}-1-[3-(triethylammonio)propyl]pyridinium](http://img.cochemist.com/ccimg/303800/303727-66-4_b.png)
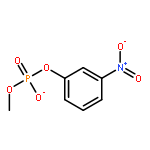
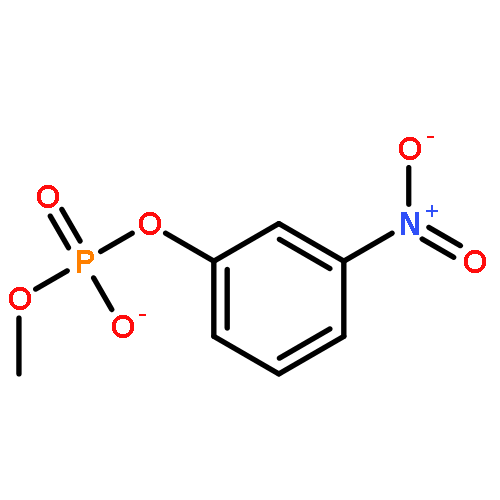


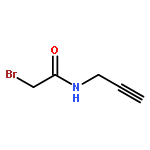
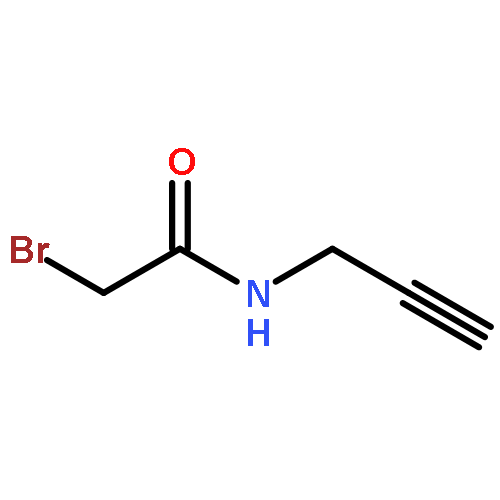


![Pyridinium,4-[2-[4-(dibutylamino)phenyl]ethenyl]-1-[3-(triethylammonio)propyl]-, bromide(1:2)](http://img.cochemist.com/ccimg/149900/149838-22-2.png)
![Pyridinium,4-[2-[4-(dibutylamino)phenyl]ethenyl]-1-[3-(triethylammonio)propyl]-, bromide(1:2)](http://img.cochemist.com/ccimg/149900/149838-22-2_b.png)

















![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1.png)
![9-Octadecenoic acid(9Z)-,1,1'-[(1R)-1-[[[(2-aminoethoxy)hydroxyphosphinyl]oxy]methyl]-1,2-ethanediyl]ester](http://img.cochemist.com/ccimg/4100/4004-05-1_b.png)










![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4.png)
![3,5,9-Trioxa-4-phosphaheptacos-18-en-1-aminium,4-hydroxy-N,N,N-trimethyl-10-oxo-7-[[(9Z)-1-oxo-9-octadecen-1-yl]oxy]-, innersalt, 4-oxide, (7R,18Z)-](http://img.cochemist.com/ccimg/4300/4235-95-4_b.png)
![2,3,7,8-Tetrachlorodibenzo[b,e][1,4]dioxine](http://img.cochemist.com/ccimg/1800/1746-01-6.png)
![2,3,7,8-Tetrachlorodibenzo[b,e][1,4]dioxine](http://img.cochemist.com/ccimg/1800/1746-01-6_b.png)