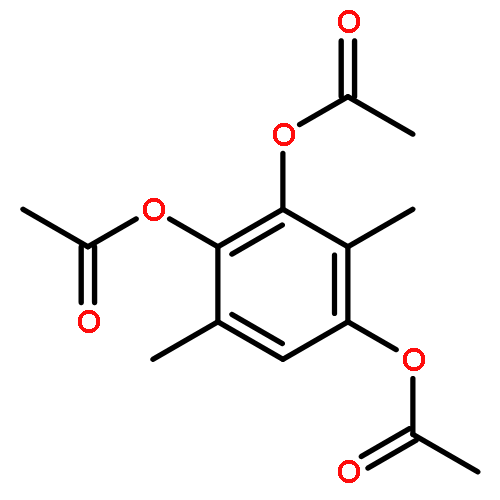
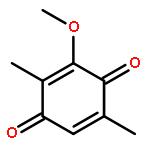
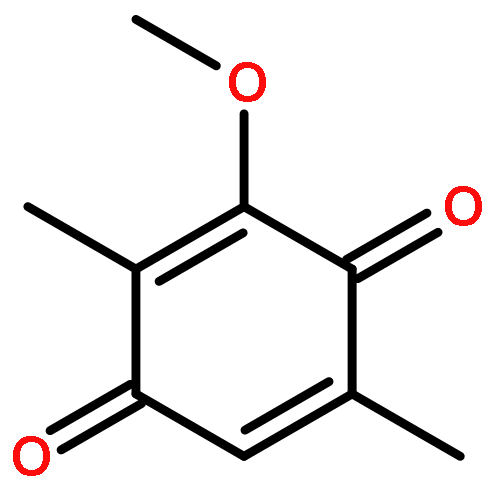
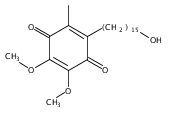
Co-reporter:Jinhong Bai; Chenzhong Liao; Yanghan Liu; Xiaochu Qin; Jiaxuan Chen; Yatao Qiu; Dongguang Qin; Zheng Li; Zheng-Chao Tu
Journal of Medicinal Chemistry 2016 Volume 59(Issue 12) pp:5766-5779
Publication Date(Web):May 25, 2016
DOI:10.1021/acs.jmedchem.6b00324
Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) has the potential to directly limit NAD production in cancer cells and is an effective strategy for cancer treatment. Using a structure-based strategy, we have designed a new class of potent small-molecule inhibitors of NAMPT. Several designed compounds showed promising antiproliferative activities in vitro. (E)-N-(5-((4-(((2-(1H-Indol-3-yl)ethyl)(isopropyl)amino)methyl)phenyl)amino)pentyl)-3-(pyridin-3-yl)acrylamide, 30, bearing an indole moiety, has an IC50 of 25.3 nM for binding to the NAMPT protein and demonstrated promising inhibitory activities in the nanomolar range against several cancer cell lines (MCF-7 GI50 = 0.13 nM; MDA-MB-231 GI50 = 0.15 nM). Triple-negative breast cancer is the most malignant subtype of breast cancer with no effective targeted treatments currently available. Significant antitumor efficacy of compound 30 was achieved (TGI was 73.8%) in an orthotopic MDA-MB-231 triple-negative breast cancer xenograft tumor model. This paper reports promising lead molecules for the inhibition of NAMPT which could serve as a basis for further investigation.
Co-reporter:Yiwu Yao; Zhengchao Tu; Chenzhong Liao; Zhen Wang; Shang Li; Hequan Yao; Zheng Li
Journal of Medicinal Chemistry 2015 Volume 58(Issue 19) pp:7672-7680
Publication Date(Web):September 2, 2015
DOI:10.1021/acs.jmedchem.5b01044
A successful structure-based design of novel cyclic depsipeptides that selectively target class I HDAC isoforms is described. Compound 11 has an IC50 of 2.78 nM for binding to the HDAC1 protein, and the prodrugs 12 and 13 also exhibit promising antiproliferative activities in the nanomolar range against various cancer cell lines. Compounds 12 and 13 show more than 20-fold selectivity toward human cancer cells over human normal cells in comparison with romidepsin (FK228), demonstrating low probability of toxic side effects. In addition, compound 13 exhibits excellent in vivo anticancer activities in a human prostate carcinoma (Du145) xenograft model with no observed toxicity. Thus, prodrug 13 has therapeutic potential as a new class of anticancer agent for further clinical translation.
Co-reporter:Nan Ma, Yiwu Yao, Bing-Xin Zhao, Ying Wang, Wen-Cai Ye and Sheng Jiang
Chemical Communications 2014 vol. 50(Issue 66) pp:9284-9287
Publication Date(Web):27 Jun 2014
DOI:10.1039/C4CC02575J
A consecutive synthetic strategy was developed toward the total synthesis of securinega alkaloids. (−)-Norsecurinine was concisely assembled by addition of a methoxyallene to a ketone for efficient side-chain installation. Ring-closing metathesis was also utilized as a key step. The first total synthesis of (−)-niruroidine was achieved from (−)-norsecurinine in three steps, while the route to (−)-flueggine A featured a 1,3-dipolar cycloaddition to forge the core structure.
Co-reporter:Yiwu Yao, Chenzhong Liao, Zheng Li, Zhen Wang, Qiao Sun, Chunping Liu, Yang Yang, Zhengchao Tu, Sheng Jiang
European Journal of Medicinal Chemistry 2014 Volume 86() pp:639-652
Publication Date(Web):30 October 2014
DOI:10.1016/j.ejmech.2014.09.024
•New HDAC inhibitors demonstrating class I and IIb subtype selectivity have been identified.•Several designed compounds showed much better selectivity and activity than SAHA.•A representative lead compound 22 demonstrated promising class I and IIb HDAC isoforms selectivity.A novel series of HDAC inhibitors demonstrating class I and IIb subtype selectivity have been identified using a scaffold-hopping strategy. Several designed compounds showed better selectivity for class I and IIb over class IIa HDAC isoforms comparing to the FDA approved HDAC targeting drug SAHA. A representative lead compound 22 bearing a biphenyl moiety demonstrated promising class I and IIb HDAC isoforms selectivity and in vitro anticancer activities against several cancer cell lines. This work could serve as a fundamental platform for further exploration of selective HDAC inhibitors using designed molecular scaffold.
Co-reporter:Yanghan Liu, Qicai Xiao, Yongqiang Liu, Zheng Li, Yatao Qiu, Guang-Biao Zhou, Zhu-Jun Yao, Sheng Jiang
European Journal of Medicinal Chemistry 2014 Volume 78() pp:248-258
Publication Date(Web):6 May 2014
DOI:10.1016/j.ejmech.2014.03.062
•A convergent fragment-assembly approach was developed.•A small library of annonaceous acetogenin analogues was established.•Lots of aromatic moieties were introduced using Click chemistry.•Biological evaluation of annonaceous acetogenin analogues was studied.A small library of analogues of annonaceous acetogenins through click linkage with aromatic moieties is established using a convergent modular fragment-assembly approach. These analogues exhibited low micromolar inhibitory activities against the proliferation of several human cancer cell lines. Structure–activity relationship (SAR) of these analogues indicates that replacement of the methoxy groups of ubiquinone ring with methyl groups is proved to be a useful strategy for improving the anticancer activity of quinone–acetogenin hybrids.A variety of aromatic moieties were introduced for the first time into the right part of acetogenins, in which a triazole functionality was employed as the linkage using Click chemistry.
Co-reporter:Yanghan Liu, Yongqiang Liu, Zhen Li, Guang-Biao Zhou, Zhu-Jun Yao, Sheng Jiang
Bioorganic & Medicinal Chemistry Letters 2014 Volume 24(Issue 7) pp:1650-1653
Publication Date(Web):1 April 2014
DOI:10.1016/j.bmcl.2014.02.072
A series of novel bivalent mimetics of annonaceous acetogenins have been designed, synthesized, and evaluated. Among these, compound 7 bearing a homopiperazine ring in the middle region exhibited more potent growth inhibitory activity and higher selectivity against cancer cells over normal cells by comparison with AA005. This work indicates that modification of the middle piperazine ring is a useful optimizing tool for the simplified acetogenin mimetics.
Co-reporter:Jinyue Su;Yatao Qiu;Dayong Zhang
Chinese Journal of Chemistry 2014 Volume 32( Issue 8) pp:685-688
Publication Date(Web):
DOI:10.1002/cjoc.201400147
Abstract
Pyridin-2-ol-N-oxide was designed as an efficient ligand for the coupling reaction of aryl iodides, aryl bromides and aryl chlorides, respectively, with primary amines, cyclic secondary amines or N-containing heterocycles at room or moderate temperature. The catalytic system showed great functional groups tolerance and excellent selective reactivity.
Co-reporter:DingBiao Zou;YaTao Qiu;ZhengChao Tu;ChenZhong Liao
Science China Chemistry 2014 Volume 57( Issue 6) pp:823-832
Publication Date(Web):2014 June
DOI:10.1007/s11426-013-5011-9
We designed a series of 2-methylpyrimidine derivatives as new BCR-ABL inhibitors using scaffold-hopping strategy. These synthetic compounds exhibited significant inhibition against a broad spectrum of Bcr-Abl mutants including the gatekeeper T315I mutant. Compound 7u showed very potent kinase inhibitory activities against Bcr-Abl WT, Bcr-Abl E255K, Bcr-Abl Q252H, Bcr-Abl G250E and Bcr-Abl T315I, with IC50 values of 0.13 nM, 0.17 nM, 0.24 nM, 0.19 nM and 0.65 μM, respectively. This compound also displayed anti-proliferation activity against K562 cell line with an IC50 value of 1.1 nM, thus representing a new lead for further optimization.
Co-reporter:Yatao Qiu, Weijun Jia, Zhiyi Yao, Fanhong Wu and Sheng Jiang
Organic & Biomolecular Chemistry 2013 vol. 11(Issue 9) pp:1502-1510
Publication Date(Web):21 Dec 2012
DOI:10.1039/C2OB26556G
2-Carbomethoxy-3-hydroxyquinoxaline-di-N-oxide was identified as an efficient novel ligand for the copper-catalyzed coupling of aryl halides with various phenols under mild conditions. The catalytic system shows great functional-group tolerance and excellent reactive selectivity.
Co-reporter:Xianlin Li, Zhenchao Tu, Hua Li, Chunping Liu, Zheng Li, Qiao Sun, Yiwu Yao, Jinsong Liu, and Sheng Jiang
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 1) pp:132-136
Publication Date(Web):December 5, 2012
DOI:10.1021/ml300371t
We report the design, synthesis, and biological evaluation of a new series of largazole analogues in which a 4-methylthiazoline moiety was replaced with a triazole and tetrazole ring, respectively. Compound 7 bearing a tetrazole ring was identified to show much better selectivity for HDAC1 over HDAC9 than largazole (10-fold). This work could serve as a foundation for further exploration of selective HDAC inhibitors using a largazole molecular scaffold.Keywords: click chemistry; HDAC inhibitor; largazole; macrocycles; peptides;
Co-reporter:Li-Chuan Wu, Zhe-Sheng Wen, Ya-Tao Qiu, Xiao-Qin Chen, Hao-Bin Chen, Ming-Ming Wei, Zi Liu, Sheng Jiang, and Guang-Biao Zhou
ACS Medicinal Chemistry Letters 2013 Volume 4(Issue 10) pp:921-926
Publication Date(Web):August 12, 2013
DOI:10.1021/ml400093y
Aberration in cell cycle has been shown to be a common occurrence in lung cancer, and cell cycle inhibitor represents an effective therapeutic strategy. In this study, we test the effects of a natural macrocyclic depsipeptide largazole on lung cancer cells and report that this compound potently inhibits the proliferation and clonogenic activity of lung cancer cells but not normal bronchial epithelial cells. Largazole arrests cell cycle at G1 phase with up-regulation of the expression of cyclin-dependent kinase inhibitor p21. Interestingly, largazole enhances the E2F1-HDAC1 binding affinity and induces a proteasomal degradation of E2F1, leading to suppression of E2F1 function in lung cancer but not normal bronchial epithelial cells. Because E2F1 is overexpressed in lung cancer tumor samples, these data indicate that largazole is an E2F1-targeting cell cycle inhibitor, which bears therapeutic potentials for this malignant neoplasm.Keywords: cell cycle; degradation; E2F1; largazole; Lung cancer;
Co-reporter:Qiao Sun, Yiwu Yao, Chunping Liu, Hua Li, Hequan Yao, Xiaowen Xue, Jinsong Liu, Zhengchao Tu, Sheng Jiang
Bioorganic & Medicinal Chemistry Letters 2013 Volume 23(Issue 11) pp:3295-3299
Publication Date(Web):1 June 2013
DOI:10.1016/j.bmcl.2013.03.102
We report the design, synthesis, and biological evaluation of a new series of HDAC1 inhibitors using click chemistry. Compound 17 bearing a phenyl ring at meta-position was identified to show much better selectivity for HDAC1 over HDAC7 than SAHA. The compond 17 also showed better in vitro anticancer activities against several cancer cell lines than that of SAHA. This work could serve as a foundation for further exploration of selective HDAC inhibitors using the compound 17 molecular scaffold.
Co-reporter:Dong Chen, Kai Yang, Hua Xiang, Sheng Jiang
Tetrahedron Letters 2012 Volume 53(Issue 52) pp:7121-7124
Publication Date(Web):26 December 2012
DOI:10.1016/j.tetlet.2012.10.083
2-Carbomethoxy-3-hydroxyquinoxaline-di-N-oxide was identified as an efficient novel ligand for the copper-catalyzed coupling reactions of aryl iodides, bromides, and chlorides with aliphatic amines and N-containing heterocycles under mild conditions. The catalytic system showed great functional-group tolerance and excellent chemoselectivity.
Co-reporter:Yatao Qiu, Yanghan Liu, Kai Yang, Wenkun Hong, Zheng Li, Zhaoyang Wang, Zhiyi Yao, and Sheng Jiang
Organic Letters 2011 Volume 13(Issue 14) pp:3556-3559
Publication Date(Web):June 21, 2011
DOI:10.1021/ol2009208
Several ligands were designed to promote transition-metal-free cross-coupling reactions of aryl halides with benzene derivatives. Among the systems probed, quinoline-1-amino-2-carboxylic acid was found to serve as an excellent catalyst for cross-coupling between aryl halides and unactivated benzene. Reactions using this inexpensive catalytic system displayed a high functional group tolerance as well as excellent chemoselectivities.
Co-reporter:Kai Yang, Zheng Li, Zhaoyang Wang, Zhiyi Yao, and Sheng Jiang
Organic Letters 2011 Volume 13(Issue 16) pp:4340-4343
Publication Date(Web):July 25, 2011
DOI:10.1021/ol2016737
8-Hydroxyquinolin-N-oxide was found to be a very efficient ligand for the copper-catalyzed hydroxylation of aryl iodides, aryl bromides, or aryl chlorides under mild reaction conditions. This methodology provides a direct transformation of aryl halides to phenols and to alkyl aryl ethers. The inexpensive catalytic system showed great functional group tolerance and excellent selectivity.
Co-reporter:Qicai Xiao ; Yongqiang Liu ; Yatao Qiu ; Guangbiao Zhou ; Chan Mao ; Zheng Li ; Zhu-Jun Yao
Journal of Medicinal Chemistry 2011 Volume 54(Issue 2) pp:525-533
Publication Date(Web):December 20, 2010
DOI:10.1021/jm101053k
Annonaceous acetogenins are a large family of naturally occurring polyketides exhibiting remarkable anticancer activities. The first generation of annonaceous acetogenin mimetic (1, AA005) exhibits comparable activity as that of natural products and presents much higher selectivity between cancer and normal cells. In this work, we report the design, synthesis, and evaluation of a new series of compound 1 analogues in which a variety of conformation-constrained fragments were embedded in the left hydrocarbon chain part. Compound 7 bearing a biphenyl moiety was identified to exhibit more potent antiproliferative activity and preferentially target cancer cells over normal cells and thus represents a new lead for further optimization.
Co-reporter:Xin Zeng, Wenming Huang, Yatao Qiu and Sheng Jiang
Organic & Biomolecular Chemistry 2011 vol. 9(Issue 24) pp:8224-8227
Publication Date(Web):20 Sep 2011
DOI:10.1039/C1OB06208E
Under the catalysis of CuI/2-carboxylic acid-quinoline-N-oxide, the cross coupling reactions between aryl iodides or bromides and aqueous ammonia proceed very well to afford N-unprotected aniline derivatives in excellent yields. This inexpensive catalytic system shows great functional group tolerance and excellent reaction selectivity.
Co-reporter:WenKun Hong, Yatao Qiu, Zhiyi Yao, Zhaoyang Wang, Sheng Jiang
Tetrahedron Letters 2011 Volume 52(Issue 38) pp:4916-4919
Publication Date(Web):21 September 2011
DOI:10.1016/j.tetlet.2011.07.046
An efficient palladium-catalyzed direct C–H arylation of unactivated arenes has been discovered. This method employs aryl halides as the direct coupling partners with arenes without using any external ligands. This catalysis opens a new methodology for the preparation of symmetrical as well as unsymmetrical biaryls in a user-friendly approach.
Co-reporter:Qicai Xiao, Yongqiang Liu, Yatao Qiu, Zhiyi Yao, Guangbiao Zhou, Zhu-Jun Yao, Sheng Jiang
Bioorganic & Medicinal Chemistry Letters 2011 Volume 21(Issue 12) pp:3613-3615
Publication Date(Web):15 June 2011
DOI:10.1016/j.bmcl.2011.04.095
A new series of linear dimeric compounds mimicking naturally occurring annonaceous acetogenins have been synthesized by bivalent analogue design, and their cytotoxicities have been evaluated against the growth of cancer cells by MTT method. Most of these compounds show selective action favored to human cancer cell lines over normal cell lines, and compound 9 with bis-terminal benzoquinone functionality exhibits an IC50 = 0.40 μM against MCF7 cell lines. This work mentions that appropriate conformational constraints might be a useful optimizing tool for this unique class of anticancer compounds.
Co-reporter:Wenbin Wu, Zheng Li, Guangbiao Zhou, Sheng Jiang
Tetrahedron Letters 2011 Volume 52(Issue 19) pp:2488-2491
Publication Date(Web):11 May 2011
DOI:10.1016/j.tetlet.2011.03.021
The total synthesis of argyrins A and E were accomplished using a convergent strategy by condensation of one tripeptide and two dipeptide fragments. The synthesis strategy, which was developed for the protection of peptide fragments and identification of the optimum macrocylization site, can be applied to further synthetic studies involving other members of the argyrin family.
Co-reporter:Kai Yang, Yatao Qiu, Zheng Li, Zhaoyang Wang, and Sheng Jiang
The Journal of Organic Chemistry 2011 Volume 76(Issue 9) pp:3151-3159
Publication Date(Web):March 22, 2011
DOI:10.1021/jo1026035
Several new ligands were designed to promote copper-catalyzed Ullman C−N coupling reactions. In this group, 8-hydroxyquinolin-N-oxide was found to serve as a superior ligand for CuBr-catalyzed coupling reactions of aryl iodides, bromides, and chlorides with aliphatic amines and N-heterocycles under a low catalyst loading (1% [Cu] mol). Reactions with the inexpensive catalytic system display a high functional group tolerance as well as excellent chemoselectivity.(1)
Co-reporter:Xin Zeng, Biaolin Yin, Zheng Hu, Chenzhong Liao, Jinglei Liu, Shang Li, Zheng Li, Marc C. Nicklaus, Guangbiao Zhou and Sheng Jiang
Organic Letters 2010 Volume 12(Issue 6) pp:1368-1371
Publication Date(Web):February 25, 2010
DOI:10.1021/ol100308a
The efficient total synthesis of the natural substance largazole is described. Using this strategy, a small library of largazole analogs was developed. Structure−activity relationship studies suggested that the geometry of the alkene in the side chain is critical. While the largazole’s analogues with trans-alkene are potent for the antiproliferative effect, those with cis-alkene are completely inactive. Most importantly, replacement of valine with tyrosine in largazole increased selectivity toward human cancer cells over human normal cells more than 100-fold.
Co-reporter:Sheng Jiang, Chenzhong Liao, Lakshman Bindu, Biaolin Yin, Karen W. Worthy, Robert J. Fisher, Terrence R. Burke Jr., Marc C. Nicklaus, Peter P. Roller
Bioorganic & Medicinal Chemistry Letters 2009 Volume 19(Issue 10) pp:2693-2698
Publication Date(Web):15 May 2009
DOI:10.1016/j.bmcl.2009.03.134
Blocking the interaction between phosphotyrosine (pTyr)-containing activated receptors and the Src homology 2 (SH2) domain of the growth factor receptor-bound protein 2 (Grb 2) is considered to be an effective and non-cytotoxic strategy to develop new anti-proliferate agents due to its potential to shut down the Ras activation pathway. In this study, a series of phosphotyrosine containing cyclic pentapeptides were designed and synthesized based upon the phage library derived cyclopeptide, G1TE. A comprehensive SAR study was also carried out to develop potent Grb2-SH2 domain antagonists based upon this novel template. With both the peptidomimetic optimization of the amino acid side-chains and the constraint of the backbone conformation guided by molecular modeling, we developed several potent antagonists with low micromolar range binding affinity, such as cyclic peptide 15 with an Kd = 0.359 μM, which is providing a novel template for the development of Grb2-SH2 domain antagonists as potential therapeutics for certain cancers.
Co-reporter:Jianlong Zheng, Biaolin Yin, Wenming Huang, Xiaopeng Li, Hequan Yao, Zhaogui Liu, Jiancun Zhang, Sheng Jiang
Tetrahedron Letters 2009 50(36) pp: 5094-5097
Publication Date(Web):
DOI:10.1016/j.tetlet.2009.06.104
Co-reporter:Nan Ma, Yiwu Yao, Bing-Xin Zhao, Ying Wang, Wen-Cai Ye and Sheng Jiang
Chemical Communications 2014 - vol. 50(Issue 66) pp:NaN9287-9287
Publication Date(Web):2014/06/27
DOI:10.1039/C4CC02575J
A consecutive synthetic strategy was developed toward the total synthesis of securinega alkaloids. (−)-Norsecurinine was concisely assembled by addition of a methoxyallene to a ketone for efficient side-chain installation. Ring-closing metathesis was also utilized as a key step. The first total synthesis of (−)-niruroidine was achieved from (−)-norsecurinine in three steps, while the route to (−)-flueggine A featured a 1,3-dipolar cycloaddition to forge the core structure.
Co-reporter:Yatao Qiu, Weijun Jia, Zhiyi Yao, Fanhong Wu and Sheng Jiang
Organic & Biomolecular Chemistry 2013 - vol. 11(Issue 9) pp:NaN1510-1510
Publication Date(Web):2012/12/21
DOI:10.1039/C2OB26556G
2-Carbomethoxy-3-hydroxyquinoxaline-di-N-oxide was identified as an efficient novel ligand for the copper-catalyzed coupling of aryl halides with various phenols under mild conditions. The catalytic system shows great functional-group tolerance and excellent reactive selectivity.
Co-reporter:Xin Zeng, Wenming Huang, Yatao Qiu and Sheng Jiang
Organic & Biomolecular Chemistry 2011 - vol. 9(Issue 24) pp:NaN8227-8227
Publication Date(Web):2011/09/20
DOI:10.1039/C1OB06208E
Under the catalysis of CuI/2-carboxylic acid-quinoline-N-oxide, the cross coupling reactions between aryl iodides or bromides and aqueous ammonia proceed very well to afford N-unprotected aniline derivatives in excellent yields. This inexpensive catalytic system shows great functional group tolerance and excellent reaction selectivity.











![3-fluoro-4-[(4-methyl-1-piperazinyl)methyl]-Benzenamine](http://img.cochemist.com/ccimg/1269700/1269631-99-3.png)
![3-fluoro-4-[(4-methyl-1-piperazinyl)methyl]-Benzenamine](http://img.cochemist.com/ccimg/1269700/1269631-99-3_b.png)
![1H-1,2,3-Triazole-1-octanamide, N-[1,1'-biphenyl]-2-yl-4-(3-pyridinyl)-](http://img.cochemist.com/ccimg/1202600/1202580-59-3.png)
![1H-1,2,3-Triazole-1-octanamide, N-[1,1'-biphenyl]-2-yl-4-(3-pyridinyl)-](http://img.cochemist.com/ccimg/1202600/1202580-59-3_b.png)
![3-chloro-4-[(4-methyl-1-piperazinyl)methyl]-Benzenamine](http://img.cochemist.com/ccimg/943400/943320-67-0.png)
![3-chloro-4-[(4-methyl-1-piperazinyl)methyl]-Benzenamine](http://img.cochemist.com/ccimg/943400/943320-67-0_b.png)
![7H-Furo[3,2-g][1]benzopyran-7-one,4-(4-phenylbutoxy)-](http://img.cochemist.com/ccimg/724800/724709-68-6.png)
![7H-Furo[3,2-g][1]benzopyran-7-one,4-(4-phenylbutoxy)-](http://img.cochemist.com/ccimg/724800/724709-68-6_b.png)
![OXIRANE, [[2-(PHENYLMETHOXY)ETHOXY]METHYL]-, (2S)-](http://img.cochemist.com/ccimg/406300/406218-43-7.png)
![OXIRANE, [[2-(PHENYLMETHOXY)ETHOXY]METHYL]-, (2S)-](http://img.cochemist.com/ccimg/406300/406218-43-7_b.png)
![2-(1H-INDOL-3-YL)-N-[(4-NITROPHENYL)METHYL]ETHANAMINE](http://img.cochemist.com/ccimg/355900/355815-83-7.png)
![2-(1H-INDOL-3-YL)-N-[(4-NITROPHENYL)METHYL]ETHANAMINE](http://img.cochemist.com/ccimg/355900/355815-83-7_b.png)





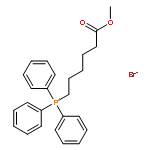
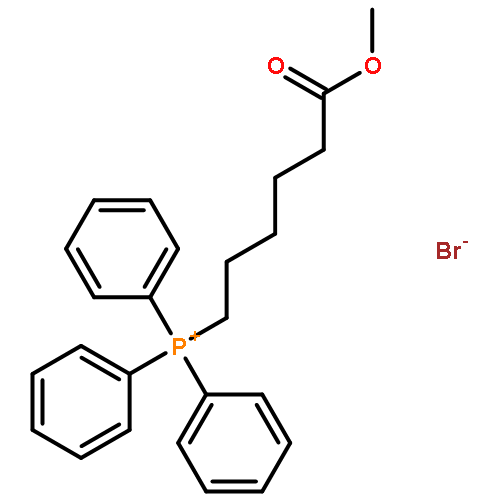


![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7.png)
![(1s,4s,7z,10s,16e,21r)-7-ethylidene-4,21-di(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetrazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone](http://img.cochemist.com/ccimg/128600/128517-07-7_b.png)
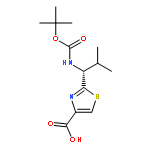
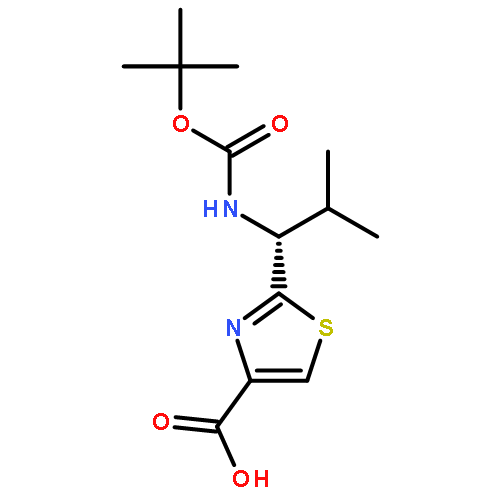
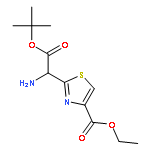
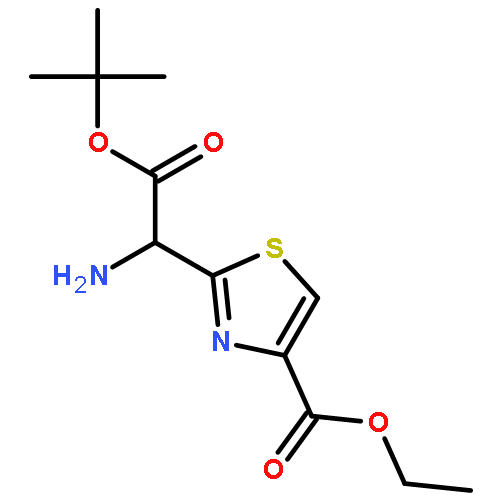




![2-[[(TERT-BUTOXYCARBONYL)AMINO]METHYL]THIAZOLE-4-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/72000/71904-80-8.png)
![2-[[(TERT-BUTOXYCARBONYL)AMINO]METHYL]THIAZOLE-4-CARBOXYLIC ACID](http://img.cochemist.com/ccimg/72000/71904-80-8_b.png)



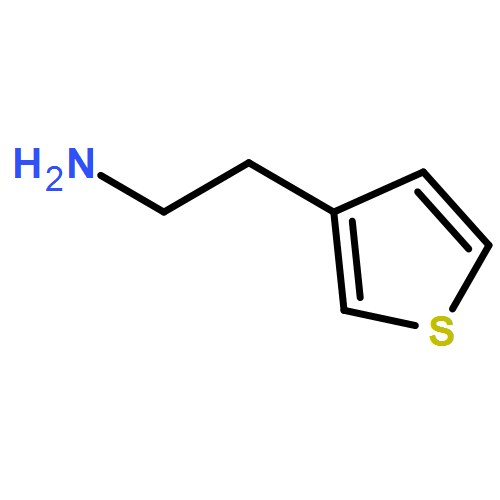
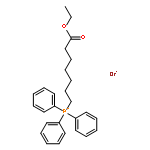
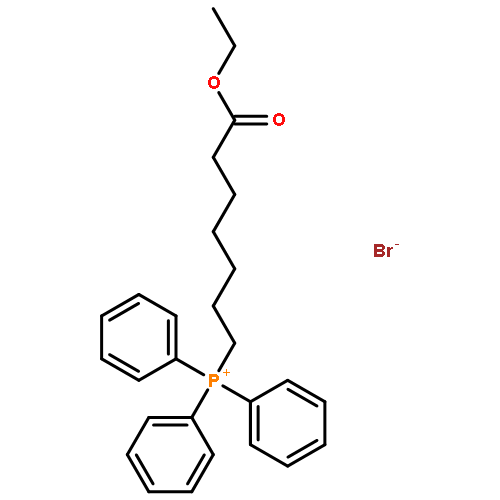

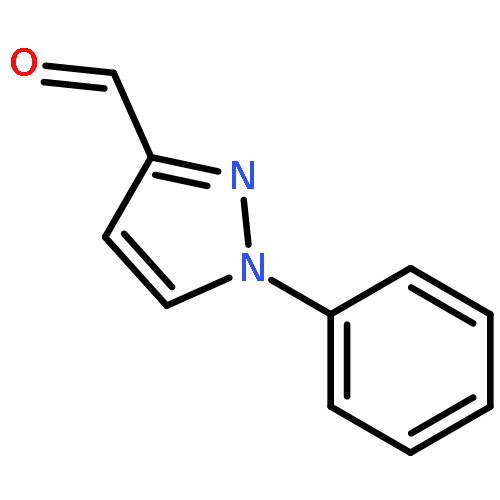
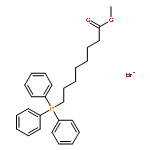
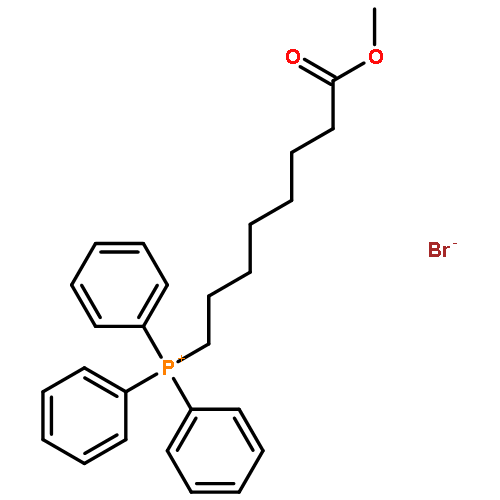









![5-(Bromomethyl)benzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/2700/2606-51-1.png)
![5-(Bromomethyl)benzo[d][1,3]dioxole](http://img.cochemist.com/ccimg/2700/2606-51-1_b.png)