Co-reporter:Surin K. Mong, Frank V. Cochran, Hongtao Yu, Zachary Graziano, Yu-Shan Lin, Jennifer R. Cochran, and Bradley L. Pentelute
Biochemistry October 31, 2017 Volume 56(Issue 43) pp:5720-5720
Publication Date(Web):September 27, 2017
DOI:10.1021/acs.biochem.7b00722
Homochirality is a general feature of biological macromolecules, and Nature includes few examples of heterochiral proteins. Herein, we report on the design, chemical synthesis, and structural characterization of heterochiral proteins possessing loops of amino acids of chirality opposite to that of the rest of a protein scaffold. Using the protein Ecballium elaterium trypsin inhibitor II, we discover that selective β-alanine substitution favors the efficient folding of our heterochiral constructs. Solution nuclear magnetic resonance spectroscopy of one such heterochiral protein reveals a homogeneous global fold. Additionally, steered molecular dynamics simulation indicate β-alanine reduces the free energy required to fold the protein. We also find these heterochiral proteins to be more resistant to proteolysis than homochiral l-proteins. This work informs the design of heterochiral protein architectures containing stretches of both d- and l-amino acids.
Co-reporter:Colin M. Fadzen, Justin M. Wolfe, Choi-Fong Cho, E. Antonio Chiocca, Sean E. Lawler, and Bradley L. Pentelute
Journal of the American Chemical Society November 8, 2017 Volume 139(Issue 44) pp:15628-15628
Publication Date(Web):October 9, 2017
DOI:10.1021/jacs.7b09790
Here we describe the utility of peptide macrocyclization through perfluoroaryl-cysteine SNAr chemistry to improve the ability of peptides to cross the blood–brain barrier. Multiple macrocyclic analogues of the peptide transportan-10 were investigated that displayed increased uptake in two different cell lines and improved proteolytic stability. One of these analogues (M13) exhibited substantially increased delivery across a cellular spheroid model of the blood–brain barrier. Through ex vivo imaging of mouse brains, we demonstrated that this perfluoroarene-based macrocycle of TP10 exhibits increased penetration of the brain parenchyma following intravenous administration in mice. Finally, we evaluated macrocyclic analogues of the BH3 domain of the BIM protein to assess if our approach would be applicable to a peptide of therapeutic interest. We identified a BIM BH3 analogue that showed increased penetration of the brain tissue in mice.
Co-reporter:Alexander A. Vinogradov, Zachary P. Gates, Chi Zhang, Anthony J. Quartararo, Kathryn H. Halloran, and Bradley L. Pentelute
ACS Combinatorial Science November 13, 2017 Volume 19(Issue 11) pp:694-694
Publication Date(Web):September 11, 2017
DOI:10.1021/acscombsci.7b00109
A methodology to achieve high-throughput de novo sequencing of synthetic peptide mixtures is reported. The approach leverages shotgun nanoliquid chromatography coupled with tandem mass spectrometry-based de novo sequencing of library mixtures (up to 2000 peptides) as well as automated data analysis protocols to filter away incorrect assignments, noise, and synthetic side-products. For increasing the confidence in the sequencing results, mass spectrometry-friendly library designs were developed that enabled unambiguous decoding of up to 600 peptide sequences per hour while maintaining greater than 85% sequence identification rates in most cases. The reliability of the reported decoding strategy was additionally confirmed by matching fragmentation spectra for select authentic peptides identified from library sequencing samples. The methods reported here are directly applicable to screening techniques that yield mixtures of active compounds, including particle sorting of one-bead one-compound libraries and affinity enrichment of synthetic library mixtures performed in solution.Keywords: de novo sequencing; one-bead one-compound libraries; shotgun nanoliquid chromatography; synthetic peptide mixtures; tandem mass spectrometry;
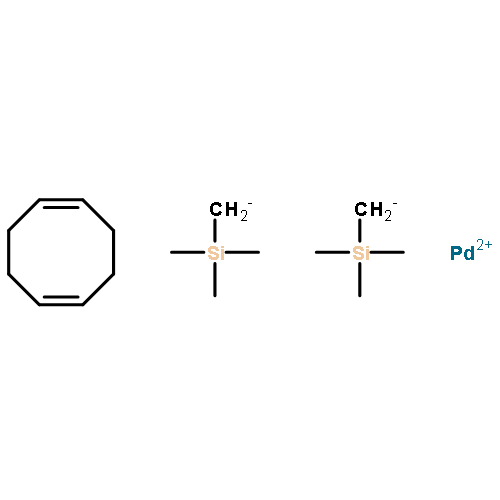
Co-reporter:Anthony J. Rojas, Bradley L. Pentelute, and Stephen L. Buchwald
Organic Letters August 18, 2017 Volume 19(Issue 16) pp:
Publication Date(Web):August 4, 2017
DOI:10.1021/acs.orglett.7b01911
We report the use of a sulfonated biarylphosphine ligand (sSPhos) to promote the chemoselective modification of cysteine containing proteins and peptides with palladium reagents in aqueous medium. The use of sSPhos allowed for the isolation of several air-stable and water-soluble mono- and bis-palladium reagents, which were used in an improved protocol for the rapid S-arylation of cysteines under benign and physiologically relevant conditions. The cosolvent-free aqueous conditions were applied to the conjugation of a variety of biomolecules with affinity tags, heterocycles, fluorophores, and functional handles. Additionally, bis-palladium reagents were used to perform macrocyclization of peptides bearing two cysteine residues.
Co-reporter:Anthony J. Rojas;Chi Zhang;Ekaterina V. Vinogradova;Nathan H. Buchwald;John Reilly;Bradley L. Pentelute;Stephen L. Buchwald
Chemical Science (2010-Present) 2017 vol. 8(Issue 6) pp:4257-4263
Publication Date(Web):2017/05/30
DOI:10.1039/C6SC05454D
Macrocyclic peptides are important therapeutic candidates due to their improved physicochemical properties in comparison to their linear counterparts. Here we detail a method for a divergent macrocyclisation of unprotected peptides by crosslinking two cysteine residues with bis-palladium organometallic reagents. These synthetic intermediates are prepared in a single step from commercially available aryl bis-halides. Two bioactive linear peptides with cysteine residues at i, i + 4 and i, i + 7 positions, respectively, were cyclised to introduce a diverse array of aryl and bi-aryl linkers. These two series of macrocyclic peptides displayed similar linker-dependent lipophilicity, phospholipid affinity, and unique volume of distributions. Additionally, one of the bioactive peptides showed target binding affinity that was predominantly affected by the length of the linker. Collectively, this divergent strategy allowed rapid and convenient access to various aryl linkers, enabling the systematic evaluation of the effect of appending unit on the medicinal properties of macrocyclic peptides.
Co-reporter:Guillaume Lautrette; Fayçal Touti; Hong Geun Lee; Peng Dai;Bradley L. Pentelute
Journal of the American Chemical Society 2016 Volume 138(Issue 27) pp:8340-8343
Publication Date(Web):June 22, 2016
DOI:10.1021/jacs.6b03757
We describe an efficient and mild method for the synthesis of macrocyclic peptides via nitrogen arylation from unprotected precursors. Various electrophiles and lysine-based nucleophiles were investigated and showed high-yielding product formation, even for a macrocyclization scan with 14 variants. We found that nitrogen-linked aryl products were more stable to base and oxidation when compared to thiol arylated species, thereby highlighting the utility of this methodology. Finally, N-aryl macrocyclization was performed on a p53 peptide inhibitor of MDM2 and resulted in identification of a nanomolar binder with improved proteolytic stability and cell permeability.
Co-reporter:Mark D. Simon, Yuta Maki, Alexander A. Vinogradov, Chi Zhang, Hongtao Yu, Yu-Shan Lin, Yasuhiro Kajihara, and Bradley L. Pentelute
Journal of the American Chemical Society 2016 Volume 138(Issue 37) pp:12099-12111
Publication Date(Web):August 5, 2016
DOI:10.1021/jacs.6b03765
A “D-scan” of two small proteins, the disulfide-rich Ecballium elaterium trypsin inhibitor II (EETI-II) and a minimized Z domain of protein A (Z33), is reported. For each protein, the stereochemistry of one amino acid at a time was inverted to generate a series of diastereomers. In much the same way an alanine scan determines necessary residues for protein function, the D-scan elucidated the critical stereocenters of the 30-residue EETI-II and the 33-residue Z33. The folding properties and activity of each variant were investigated. A total of 24 out of 30 EETI-II D-scan analogues folded to give a three-disulfide product. Of the 24 variants that folded, half were high-affinity trypsin inhibitors, and three were as active as the wild type (WT). Of these 12 active variants, most were substantially less stable to reduction than WT EETI-II (WT first reduction potential −270.0 ± 1.5 mV, WT second reduction potential −307.2 ± 1.1 mV). Similarly, ten Z33 analogues retained high binding affinity to IgG (KD < 250 nM, WT: 24 ± 1 nM) and 12 additional analogues had reduced but appreciable IgG binding affinity (KD between 250 nM and 2.5 μM). As with EETI-II, most Z33 analogues were substantially less stable than the WT (ΔG(H2O, 263 K) = 2.4 ± 1.2 kcal/mol). Collectively, our findings show that the D-scan is powerful new strategy for studying how the stereochemistry of amino acids affects the structure and function of proteins.
Co-reporter:Bradley L Pentelute, Lei Wang
Current Opinion in Chemical Biology 2016 Volume 34() pp:v-vi
Publication Date(Web):October 2016
DOI:10.1016/j.cbpa.2016.09.017
Co-reporter:Alexander A. Vinogradov, Mark D. Simon, and Bradley L. Pentelute
Organic Letters 2016 Volume 18(Issue 6) pp:1222-1225
Publication Date(Web):March 7, 2016
DOI:10.1021/acs.orglett.5b03625
A method for chemo- and regioselective conjugation of nucleophiles to fully unprotected peptides and proteins via in situ generation of C-terminal isocyanates is reported. Oxidation of C-terminal peptide hydrazides in aqueous media followed by Curtius rearrangement of acyl azides reliably generates isocyanates, which react with a variety of external nucleophiles, such as hydrazines, hydrazides, aromatic thiols, and hydroxylamines. Multiple peptides and a 53 kDa protein hydrazide were conjugated to different nucleophiles using this reaction.
Co-reporter:Alexander A. Vinogradov, Zi-Ning Choo, Kyle A. Totaro, and Bradley L. Pentelute
Organic Letters 2016 Volume 18(Issue 6) pp:1226-1229
Publication Date(Web):March 7, 2016
DOI:10.1021/acs.orglett.5b03626
A chemistry for the facile two-component macrocyclization of unprotected peptide isocyanates is described. Starting from peptides containing two glutamic acid γ-hydrazide residues, isocyanates can be readily accessed and cyclized with hydrazides of dicarboxylic acids. The choice of a nucleophilic linker allows for the facile modulation of biochemical properties of a macrocyclic peptide. Four cyclic NYAD-1 analogues were synthesized using the described method and displayed a range of biological activities.
Co-reporter:Kyle A. Totaro, Xiaoli Liao, Keshab Bhattacharya, Jari I. Finneman, Justin B. Sperry, Mark A. Massa, Jennifer Thorn, Sa V. Ho, and Bradley L. Pentelute
Bioconjugate Chemistry 2016 Volume 27(Issue 4) pp:994
Publication Date(Web):March 14, 2016
DOI:10.1021/acs.bioconjchem.6b00043
1-Ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) bioconjugations have been utilized in preparing variants for medical research. While there have been advances in optimizing the reaction for aqueous applications, there has been limited focus toward identifying conditions and side reactions that interfere with product formation. We present a systematic investigation of EDC/N-hydroxysulfosuccinimide (sNHS)-mediated bioconjugations on carboxylated peptides and small proteins. We identified yet-to-be-reported side products arising from both the reagents and substrates. Model peptides used in this study illustrate particular substrates are more susceptible to side reactions than others. From our studies, we found that bioconjugations are more efficient with high concentrations of amine nucleophile but not sNHS. Performing bioconjugations on a model affibody protein show that the trends established with model peptides hold for more complex systems.
Co-reporter:Amy E. Rabideau and Bradley Lether Pentelute
ACS Chemical Biology 2016 Volume 11(Issue 6) pp:1490
Publication Date(Web):April 8, 2016
DOI:10.1021/acschembio.6b00169
The intracellular delivery of peptide and protein therapeutics is a major challenge due to the plasma membrane, which acts as a barrier between the extracellular environment and the intracellular milieu. Over the past two decades, a nontoxic PA/LFN delivery platform derived from anthrax lethal toxin has been developed for the transport of non-native cargo into the cytosol of cells in order to understand the translocation process through a protective antigen (PA) pore and to probe intracellular biological functions. Enzyme-mediated ligation using sortase A and native chemical ligation are two facile methods used to synthesize these non-native conjugates, inaccessible by recombinant technology. Cargo molecules that translocate efficiently include enzymes from protein toxins, antibody mimic proteins, and peptides of varying lengths and non-natural amino acid compositions. The PA pore has been found to effectively convey over 30 known cargos other than native lethal factor (LF; i.e., non-native) with diverse sequences and functionalities on the LFN transporter protein. All together these studies demonstrated that non-native cargos must adopt an unfolded or extended conformation and contain a suitable charge composition in order to efficiently pass through the PA pore. This review provides insight into design parameters for the efficient delivery of new cargos using PA and LFN.
Co-reporter:Tessa Lühmann, Surin K. Mong, Mark D. Simon, Lorenz Meinel and Bradley L. Pentelute
Organic & Biomolecular Chemistry 2016 vol. 14(Issue 13) pp:3345-3349
Publication Date(Web):04 Mar 2016
DOI:10.1039/C6OB00208K
H2 relaxin is a pleiotropic peptide hormone with clinical potential. Here we report on the reaction and use of hexafluorobenzene as an intramolecular disulfide replacement between Cys10 and Cys15 in the A-chain of H2 relaxin. Using flow-based Fmoc solid-phase peptide synthesis methodology we were able to obtain high-quality H2 relaxin fragments that were previously reported as challenging to synthesize. Subsequent native chemical ligation and oxidative folding enabled total synthesis of both wild type H2 relaxin and a C6F4 linked analog. Cell-based activity assays revealed modest activity for the C6F4 linked H2 relaxin analog, albeit 100-fold reduced relative to wild type. This work demonstrates how perfluoroarylation-cysteine SNAr chemistry may be a useful tool for the selective replacement of native disulfide bonds in proteins.
Co-reporter:Peng Dai, Chi Zhang, Matthew Welborn, James J. Shepherd, Tianyu Zhu, Troy Van Voorhis, and Bradley L. Pentelute
ACS Central Science 2016 Volume 2(Issue 9) pp:637
Publication Date(Web):August 25, 2016
DOI:10.1021/acscentsci.6b00180
Highly efficient and selective chemical reactions are desired. For small molecule chemistry, the reaction rate can be varied by changing the concentration, temperature, and solvent used. In contrast for large biomolecules, the reaction rate is difficult to modify by adjusting these variables because stringent biocompatible reaction conditions are required. Here we show that adding salts can change the rate constant over 4 orders of magnitude for an arylation bioconjugation reaction between a cysteine residue within a four-residue sequence (π-clamp) and a perfluoroaryl electrophile. Biocompatible ammonium sulfate significantly enhances the reaction rate without influencing the site-specificity of π-clamp mediated arylation, enabling the fast synthesis of two site-specific antibody–drug conjugates that selectively kill HER2-positive breast cancer cells. Computational and structure–reactivity studies indicate that salts may tune the reaction rate through modulating the interactions between the π-clamp hydrophobic side chains and the electrophile. On the basis of this understanding, the salt effect is extended to other bioconjugation chemistry, and a new regioselective alkylation reaction at π-clamp cysteine is developed.
Co-reporter:Daniel T. Cohen; Chi Zhang; Bradley L. Pentelute;Stephen L. Buchwald
Journal of the American Chemical Society 2015 Volume 137(Issue 31) pp:9784-9787
Publication Date(Web):July 30, 2015
DOI:10.1021/jacs.5b05447
Herein we report an umpolung strategy for the bioconjugation of selenocysteine in unprotected peptides. This mild and operationally simple approach takes advantage of the electrophilic character of an oxidized selenocysteine (Se–S bond) to react with a nucleophilic arylboronic acid to provide the arylated selenocysteine within hours. This reaction is amenable to a wide range of boronic acids with different biorelevant functional groups and is unique to selenocysteine. Experimental evidence indicates that under oxidative conditions the arylated derivatives are more stable than the corresponding alkylated selenocysteine.
Co-reporter:Amy E. Rabideau, Xiaoli Liao and Bradley L. Pentelute
Chemical Science 2015 vol. 6(Issue 1) pp:648-653
Publication Date(Web):2014/09/25
DOI:10.1039/C4SC02078B
Mirror image peptides have unique stability and immunogenic properties in mammals, making them attractive agents to investigate. Their properties inside cells have been mostly unexplored because biopolymers are difficult to transport across cellular membranes. Here, we used protective antigen (PA) from anthrax toxin to deliver mirror image polypeptide cargo into the cytosol of mammalian cells when conjugated to the C-terminus of the PA-binding domain of lethal factor, LFN. We found mirror image polypeptides and proteins were translocated as efficiently into cells as their L counterparts. Once in the cytosol, by the use of western blot, we found that D peptides at the C-terminus of LFN were able to achieve higher steady state concentrations when compared to the L-peptide conjugate. With this platform, we delivered a D-peptide MDM2 antagonist to disrupt the p53/MDM2 interaction in cancer cells. For the first time, we show the PA/LFN system is adaptable for the intracellular delivery of mirror image peptides and proteins.
Co-reporter:Alexander A. Vinogradov, Ethan D. Evans and Bradley L. Pentelute
Chemical Science 2015 vol. 6(Issue 5) pp:2997-3002
Publication Date(Web):23 Mar 2015
DOI:10.1039/C4SC03877K
In this study we synthesized and characterized mirror image barnase (B. amyloliquefaciens ribonuclease). D-Barnase was identical to L-barnase, when analyzed by liquid chromatography and mass-spectrometry. Proteolysis of the mirror image enzyme revealed that in contrast to its native counterpart, D-barnase was completely stable to digestive proteases. In enzymatic assays, D-barnase had the reciprocal chiral specificity and was fully active towards mirror image substrates. Interestingly, D-barnase also hydrolyzed the substrate of the native chirality, albeit 4000 times less efficiently. This effect was further confirmed by digesting a native 112-mer RNA with the enzyme. Additional studies revealed that barnase accommodates a range of substrates with various chiralities, but the prime requirement for guanosine remains. These studies point toward using mirror image enzymes as modern agents in biotechnology.
Co-reporter:Amy E. Rabideau and Bradley L. Pentelute
ACS Central Science 2015 Volume 1(Issue 8) pp:423
Publication Date(Web):November 11, 2015
DOI:10.1021/acscentsci.5b00308
Eukaryotes have evolved the ubiquitin (Ub)/proteasome system to degrade polypeptides. The Ub/proteasome system is one way that cells regulate cytosolic protein and amino acids levels through the recognition and ubiquitination of a protein’s N-terminus via E1, E2, and E3 enzymes. The process by which the N-terminus stimulates intracellular protein degradation is referred to as the N-end rule. Characterization of the N-end rule has been limited to only the natural l-amino acids. Using a cytosolic delivery platform derived from anthrax lethal toxin, we probed the stability of mixed chirality proteins, containing one d-amino acid on the N-terminus of otherwise all l-proteins. In all cases, we observed that one N-terminal d-amino acid stabilized the cargo protein to proteasomal degradation with respect to the N-end rule. We found that since the mixed chirality proteins were not polyubiquitinated, they evaded N-end-mediated proteasomal degradation. Evidently, a subtle change on the N-terminus of a natural protein can enhance its intracellular lifetime.
Co-reporter:Chi Zhang, Peng Dai, Alexander M. Spokoyny, and Bradley L. Pentelute
Organic Letters 2014 Volume 16(Issue 14) pp:3652-3655
Publication Date(Web):July 8, 2014
DOI:10.1021/ol501609y
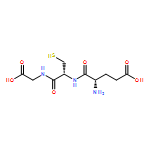
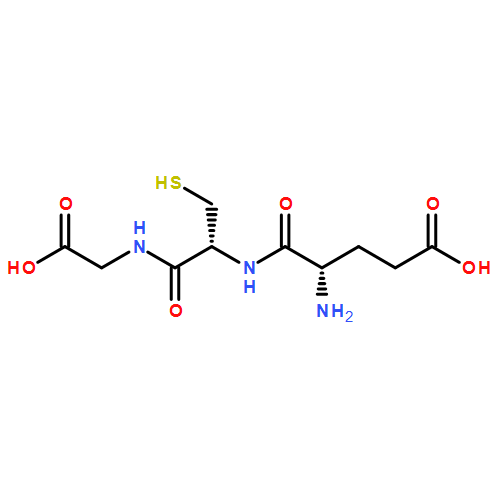
A glutathione S-transferase (GST) catalyzed macrocyclization reaction for peptides up to 40 amino acids in length is reported. GST catalyzes the selective SNAr reaction between an N-terminal glutathione (GSH, γ-Glu-Cys-Gly) tag and a C-terminal perfluoroaryl-modified cysteine on the same polypeptide chain. Cyclic peptides ranging from 9 to 24 residues were quantitatively produced within 2 h in aqueous pH = 8 buffer at room temperature. The reaction was highly selective for cyclization at the GSH tag, enabling the combination of GST-catalyzed ligation with native chemical ligation to generate a large 40-residue peptide macrocycle.
Co-reporter:Yekui Zou, Alexander M. Spokoyny, Chi Zhang, Mark D. Simon, Hongtao Yu, Yu-Shan Lin and Bradley L. Pentelute
Organic & Biomolecular Chemistry 2014 vol. 12(Issue 4) pp:566-573
Publication Date(Web):06 Dec 2013
DOI:10.1039/C3OB42168F
Here we describe a general synthetic platform for side-chain macrocyclization of an unprotected peptide library based on the SNAr reaction between cysteine thiolates and a new generation of highly reactive perfluoroaromatic small molecule linkers. This strategy enabled us to simultaneously “scan” two cysteine residues positioned from i, i + 1 to i, i + 14 sites in a polypeptide, producing 98 macrocyclic products from reactions of 14 peptides with 7 linkers. A complementary reverse strategy was developed; cysteine residues within the polypeptide were first modified with non-bridging perfluoroaryl moieties and then commercially available dithiol linkers were used for macrocyclization. The highly convergent, site-independent, and modular nature of these two strategies coupled with the unique chemoselectivity of a SNAr transformation allows for the rapid diversity-oriented synthesis of hybrid macrocyclic peptide libraries with varied chemical and structural complexities.
Co-reporter:Rocco L. Policarpo;Hansol Kang;Dr. Xiaoli Liao;Amy E. Rabideau;Mark D. Simon ; Bradley L. Pentelute
Angewandte Chemie International Edition 2014 Volume 53( Issue 35) pp:9203-9208
Publication Date(Web):
DOI:10.1002/anie.201403582
Abstract
Sortase-mediated ligation (sortagging) is a versatile, powerful strategy for protein modification. Because the sortase reaction reaches equilibrium, a large excess of polyglycine nucleophile is often employed to drive the reaction forward and suppress sortase-mediated side reactions. A flow-based sortagging platform employing immobilized sortase A within a microreactor was developed that permits efficient sortagging at low nucleophile concentrations. The platform was tested with several reaction partners and used to generate a protein bioconjugate inaccessible by solution-phase batch sortagging.
Co-reporter:Rocco L. Policarpo;Hansol Kang;Dr. Xiaoli Liao;Amy E. Rabideau;Mark D. Simon ; Bradley L. Pentelute
Angewandte Chemie 2014 Volume 126( Issue 35) pp:9357-9362
Publication Date(Web):
DOI:10.1002/ange.201403582
Abstract
Sortase-mediated ligation (sortagging) is a versatile, powerful strategy for protein modification. Because the sortase reaction reaches equilibrium, a large excess of polyglycine nucleophile is often employed to drive the reaction forward and suppress sortase-mediated side reactions. A flow-based sortagging platform employing immobilized sortase A within a microreactor was developed that permits efficient sortagging at low nucleophile concentrations. The platform was tested with several reaction partners and used to generate a protein bioconjugate inaccessible by solution-phase batch sortagging.
Co-reporter:Mark D. Simon;Dr. Patrick L. Heider;Dr. Andrea Adamo;Alexer A. Vinogradov;Surin K. Mong;Xiyuan Li;Tatiana Berger;Rocco L. Policarpo;Chi Zhang;Dr. Yekui Zou;Dr. Xiaoli Liao;Dr. Alexer M. Spokoyny; Klavs F. Jensen; Bradley L. Pentelute
ChemBioChem 2014 Volume 15( Issue 5) pp:713-720
Publication Date(Web):
DOI:10.1002/cbic.201300796
Abstract
A flow-based solid-phase peptide synthesis methodology that enables the incorporation of an amino acid residue every 1.8 min under automatic control or every 3 min under manual control is described. This is accomplished by passing a stream of reagent through a heat exchanger into a low volume, low backpressure reaction vessel, and through a UV detector. These features enable continuous delivery of heated solvents and reagents to the solid support at high flow rate, thereby maintaining maximal concentration of reagents in the reaction vessel, quickly exchanging reagents, and eliminating the need to rapidly heat reagents after they have been added to the vessel. The UV detector enables continuous monitoring of the process. To demonstrate the broad applicability and reliability of this method, it was employed in the total synthesis of a small protein, as well as dozens of peptides. The quality of the material obtained with this method is comparable to that for traditional batch methods, and, in all cases, the desired material was readily purifiable by RP-HPLC. The application of this method to the synthesis of the 113-residue Bacillus amyloliquefaciens RNase and the 130-residue DARPin pE59 is described in the accompanying manuscript.
Co-reporter:Dr. Xiaoli Liao;Amy E. Rabideau;Dr. Bradley L. Pentelute
ChemBioChem 2014 Volume 15( Issue 16) pp:2458-2466
Publication Date(Web):
DOI:10.1002/cbic.201402290
Abstract
Antibody mimics have significant scientific and therapeutic utility for the disruption of protein–protein interactions inside cells; however, their delivery to the cell cytosol remains a major challenge. Here we show that protective antigen (PA), a component of anthrax toxin, efficiently transports commonly used antibody mimics to the cytosol of mammalian cells when conjugated to the N-terminal domain of LF (LFN). In contrast, a cell-penetrating peptide (CPP) was not able to deliver any of these antibody mimics into the cell cytosol. The refolding and binding of a transported tandem monobody to Bcr-Abl (its protein target) in chronic myeloid leukemia cells were confirmed by co-immunoprecipitation. We also observed inhibition of Bcr-Abl kinase activity and induction of apoptosis caused by the monobody. In a separate case, we show disruption of key interactions in the MAPK signaling pathway after PA-mediated delivery of an affibody binder that targets hRaf-1. We show for the first time that PA can deliver bioactive antibody mimics to disrupt intracellular protein–protein interactions. This technology adds a useful tool to expand the applications of these modern agents to the intracellular milieu.
Co-reporter:Surin K. Mong;Alexer A. Vinogradov;Mark D. Simon ; Bradley L. Pentelute
ChemBioChem 2014 Volume 15( Issue 5) pp:721-733
Publication Date(Web):
DOI:10.1002/cbic.201300797
Abstract
We report the convergent total synthesis of two proteins: DARPin pE59 and Bacillus amyloliquefaciens RNase (Barnase). Leveraging our recently developed fast-flow peptide-synthesis platform, we rapidly explored numerous conditions for the assembly of long polypeptides, and were able to mitigate common side reactions, including deletion and aspartimide products. We report general strategies for improving the synthetic quality of difficult peptide sequences with our system. High-quality protein fragments produced under optimal synthetic conditions were subjected to convergent native chemical ligation, which afforded native full-length proteins after a final desulfurization step. Both DARPin and Barnase were folded and found to be as active as their recombinant analogues.
Co-reporter:Alexander M. Spokoyny ; Yekui Zou ; Jingjing J. Ling ; Hongtao Yu ; Yu-Shan Lin ;Bradley L. Pentelute
Journal of the American Chemical Society 2013 Volume 135(Issue 16) pp:5946-5949
Publication Date(Web):April 5, 2013
DOI:10.1021/ja400119t
We report the discovery of a facile transformation between perfluoroaromatic molecules and a cysteine thiolate, which is arylated at room temperature. This new approach enabled us to selectively modify cysteine residues in unprotected peptides, providing access to variants containing rigid perfluoroaromatic staples. This stapling modification performed on a peptide sequence designed to bind the C-terminal domain of an HIV-1 capsid assembly polyprotein (C-CA) showed enhancement in binding, cell permeability, and proteolytic stability properties, as compared to the unstapled analog. Importantly, chemical stability of the formed staples allowed us to use this motif in the native chemical ligation-mediated synthesis of a small protein affibody that is capable of binding the human epidermal growth factor 2 receptor.
Co-reporter:Chi Zhang;Dr. Alexer M. Spokoyny;Dr. Yekui Zou;Mark D. Simon ;Dr. Bradley L. Pentelute
Angewandte Chemie International Edition 2013 Volume 52( Issue 52) pp:14001-14005
Publication Date(Web):
DOI:10.1002/anie.201306430
Co-reporter:Chi Zhang;Dr. Alexer M. Spokoyny;Dr. Yekui Zou;Mark D. Simon ;Dr. Bradley L. Pentelute
Angewandte Chemie 2013 Volume 125( Issue 52) pp:14251-14255
Publication Date(Web):
DOI:10.1002/ange.201306430
Co-reporter:Jingjing J. Ling ; Rocco L. Policarpo ; Amy E. Rabideau ; Xiaoli Liao ;Bradley L. Pentelute
Journal of the American Chemical Society 2012 Volume 134(Issue 26) pp:10749-10752
Publication Date(Web):June 11, 2012
DOI:10.1021/ja302354v
Proteins containing a C-terminal thioester are important intermediates in semisynthesis. Currently there is one main method for the synthesis of protein thioesters that relies upon the use of engineered inteins. Here we report a simple strategy, utilizing sortase A, for routine preparation of recombinant proteins containing a C-terminal αthioester. We used our method to prepare two different anthrax toxin cargo proteins: one containing an αthioester and another containing a D-polypeptide segment situated between two protein domains. We show that both variants can translocate through protective antigen pore. This new method to synthesize a protein thioester allows for interfacing of sortase-mediated ligation and native chemical ligation.
Co-reporter:Alexander A. Vinogradov, Ethan D. Evans and Bradley L. Pentelute
Chemical Science (2010-Present) 2015 - vol. 6(Issue 5) pp:NaN3002-3002
Publication Date(Web):2015/03/23
DOI:10.1039/C4SC03877K
In this study we synthesized and characterized mirror image barnase (B. amyloliquefaciens ribonuclease). D-Barnase was identical to L-barnase, when analyzed by liquid chromatography and mass-spectrometry. Proteolysis of the mirror image enzyme revealed that in contrast to its native counterpart, D-barnase was completely stable to digestive proteases. In enzymatic assays, D-barnase had the reciprocal chiral specificity and was fully active towards mirror image substrates. Interestingly, D-barnase also hydrolyzed the substrate of the native chirality, albeit 4000 times less efficiently. This effect was further confirmed by digesting a native 112-mer RNA with the enzyme. Additional studies revealed that barnase accommodates a range of substrates with various chiralities, but the prime requirement for guanosine remains. These studies point toward using mirror image enzymes as modern agents in biotechnology.
Co-reporter:Tessa Lühmann, Surin K. Mong, Mark D. Simon, Lorenz Meinel and Bradley L. Pentelute
Organic & Biomolecular Chemistry 2016 - vol. 14(Issue 13) pp:NaN3349-3349
Publication Date(Web):2016/03/04
DOI:10.1039/C6OB00208K
H2 relaxin is a pleiotropic peptide hormone with clinical potential. Here we report on the reaction and use of hexafluorobenzene as an intramolecular disulfide replacement between Cys10 and Cys15 in the A-chain of H2 relaxin. Using flow-based Fmoc solid-phase peptide synthesis methodology we were able to obtain high-quality H2 relaxin fragments that were previously reported as challenging to synthesize. Subsequent native chemical ligation and oxidative folding enabled total synthesis of both wild type H2 relaxin and a C6F4 linked analog. Cell-based activity assays revealed modest activity for the C6F4 linked H2 relaxin analog, albeit 100-fold reduced relative to wild type. This work demonstrates how perfluoroarylation-cysteine SNAr chemistry may be a useful tool for the selective replacement of native disulfide bonds in proteins.
Co-reporter:Yekui Zou, Alexander M. Spokoyny, Chi Zhang, Mark D. Simon, Hongtao Yu, Yu-Shan Lin and Bradley L. Pentelute
Organic & Biomolecular Chemistry 2014 - vol. 12(Issue 4) pp:NaN573-573
Publication Date(Web):2013/12/06
DOI:10.1039/C3OB42168F
Here we describe a general synthetic platform for side-chain macrocyclization of an unprotected peptide library based on the SNAr reaction between cysteine thiolates and a new generation of highly reactive perfluoroaromatic small molecule linkers. This strategy enabled us to simultaneously “scan” two cysteine residues positioned from i, i + 1 to i, i + 14 sites in a polypeptide, producing 98 macrocyclic products from reactions of 14 peptides with 7 linkers. A complementary reverse strategy was developed; cysteine residues within the polypeptide were first modified with non-bridging perfluoroaryl moieties and then commercially available dithiol linkers were used for macrocyclization. The highly convergent, site-independent, and modular nature of these two strategies coupled with the unique chemoselectivity of a SNAr transformation allows for the rapid diversity-oriented synthesis of hybrid macrocyclic peptide libraries with varied chemical and structural complexities.
Co-reporter:Anthony J. Rojas, Chi Zhang, Ekaterina V. Vinogradova, Nathan H. Buchwald, John Reilly, Bradley L. Pentelute and Stephen L. Buchwald
Chemical Science (2010-Present) 2017 - vol. 8(Issue 6) pp:NaN4263-4263
Publication Date(Web):2017/03/24
DOI:10.1039/C6SC05454D
Macrocyclic peptides are important therapeutic candidates due to their improved physicochemical properties in comparison to their linear counterparts. Here we detail a method for a divergent macrocyclisation of unprotected peptides by crosslinking two cysteine residues with bis-palladium organometallic reagents. These synthetic intermediates are prepared in a single step from commercially available aryl bis-halides. Two bioactive linear peptides with cysteine residues at i, i + 4 and i, i + 7 positions, respectively, were cyclised to introduce a diverse array of aryl and bi-aryl linkers. These two series of macrocyclic peptides displayed similar linker-dependent lipophilicity, phospholipid affinity, and unique volume of distributions. Additionally, one of the bioactive peptides showed target binding affinity that was predominantly affected by the length of the linker. Collectively, this divergent strategy allowed rapid and convenient access to various aryl linkers, enabling the systematic evaluation of the effect of appending unit on the medicinal properties of macrocyclic peptides.
Co-reporter:Amy E. Rabideau, Xiaoli Liao and Bradley L. Pentelute
Chemical Science (2010-Present) 2015 - vol. 6(Issue 1) pp:NaN653-653
Publication Date(Web):2014/09/25
DOI:10.1039/C4SC02078B
Mirror image peptides have unique stability and immunogenic properties in mammals, making them attractive agents to investigate. Their properties inside cells have been mostly unexplored because biopolymers are difficult to transport across cellular membranes. Here, we used protective antigen (PA) from anthrax toxin to deliver mirror image polypeptide cargo into the cytosol of mammalian cells when conjugated to the C-terminus of the PA-binding domain of lethal factor, LFN. We found mirror image polypeptides and proteins were translocated as efficiently into cells as their L counterparts. Once in the cytosol, by the use of western blot, we found that D peptides at the C-terminus of LFN were able to achieve higher steady state concentrations when compared to the L-peptide conjugate. With this platform, we delivered a D-peptide MDM2 antagonist to disrupt the p53/MDM2 interaction in cancer cells. For the first time, we show the PA/LFN system is adaptable for the intracellular delivery of mirror image peptides and proteins.
.jpg)



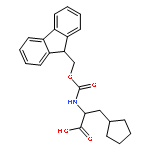
![Cyclobutanepropanoicacid, a-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-,(aS)-](http://img.cochemist.com/ccimg/478200/478183-62-9.png)
![Cyclobutanepropanoicacid, a-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-,(aS)-](http://img.cochemist.com/ccimg/478200/478183-62-9_b.png)


![9-Anthracenepropanoicacid, a-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-,(aS)-](http://img.cochemist.com/ccimg/268800/268734-27-6.png)
![9-Anthracenepropanoicacid, a-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-,(aS)-](http://img.cochemist.com/ccimg/268800/268734-27-6_b.png)


![1-Pyrenepropanoic acid,a-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-,(aS)-](http://img.cochemist.com/ccimg/183100/183071-07-0.png)
![1-Pyrenepropanoic acid,a-[[(9H-fluoren-9-ylmethoxy)carbonyl]amino]-,(aS)-](http://img.cochemist.com/ccimg/183100/183071-07-0_b.png)


![Estra-1,3,5(10)-trien-17-one, 3-[[(trifluoromethyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/92900/92817-04-4.png)
![Estra-1,3,5(10)-trien-17-one, 3-[[(trifluoromethyl)sulfonyl]oxy]-](http://img.cochemist.com/ccimg/92900/92817-04-4_b.png)


![O-[(2,3,4,5,6-PENTAFLUOROPHENYL)METHYL]HYDROXYLAMINE](http://img.cochemist.com/ccimg/73000/72915-12-9.png)
![O-[(2,3,4,5,6-PENTAFLUOROPHENYL)METHYL]HYDROXYLAMINE](http://img.cochemist.com/ccimg/73000/72915-12-9_b.png)
![Benzene,1,1'-[1,2-ethanediylbis(oxy)]bis[4-bromo-](http://img.cochemist.com/ccimg/36600/36506-46-4.png)
![Benzene,1,1'-[1,2-ethanediylbis(oxy)]bis[4-bromo-](http://img.cochemist.com/ccimg/36600/36506-46-4_b.png)







![(2S)-2-[[(2R,3R)-3-methoxy-3-[(2S)-1-[(3R,5S)-3-methoxy-5-methyl-4-[methyl-[(2S)-3-methyl-2-[[(2S)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid](http://img.cochemist.com/ccimg/745100/745017-94-1.png)
![(2S)-2-[[(2R,3R)-3-methoxy-3-[(2S)-1-[(3R,5S)-3-methoxy-5-methyl-4-[methyl-[(2S)-3-methyl-2-[[(2S)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid](http://img.cochemist.com/ccimg/745100/745017-94-1_b.png)





















![Benzene, pentafluoro[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/33300/33288-16-3.png)
![Benzene, pentafluoro[(phenylmethyl)thio]-](http://img.cochemist.com/ccimg/33300/33288-16-3_b.png)


![Benzene, 1,1'-[1,2-ethanediylbis(thio)]bis[2,3,4,5,6-pentafluoro-](http://img.cochemist.com/ccimg/31600/31597-89-4.png)
![Benzene, 1,1'-[1,2-ethanediylbis(thio)]bis[2,3,4,5,6-pentafluoro-](http://img.cochemist.com/ccimg/31600/31597-89-4_b.png)