Co-reporter:Heather R. WilliamsonEsha Sehanobish, Alan M. Shiller, Antonio Sanchez-Amat, Victor L. Davidson
Biochemistry 2017 Volume 56(Issue 7) pp:
Publication Date(Web):January 31, 2017
DOI:10.1021/acs.biochem.6b01137
The first posttranslational modification step in the biosynthesis of the tryptophan-derived quinone cofactors is the autocatalytic hydroxylation of a specific Trp residue at position C-7 on the indole side chain. Subsequent modifications are catalyzed by modifying enzymes, but the mechanism by which this first step occurs is unknown. LodA possesses a cysteine tryptophylquinone (CTQ) cofactor. Metal analysis as well as spectroscopic and kinetic studies of the mature and precursor forms of a D512A LodA variant provides evidence that copper is required for the initial hydroxylation of the precursor protein and that if alternative metals are bound, the modification does not occur and the precursor is unstable. It is shown that the mature native LodA also contains loosely bound copper, which affects the visible absorbance spectrum and quenches the fluorescence spectrum that is attributed to the mature CTQ cofactor. When copper is removed, the fluorescence appears, and when it is added back to the protein, the fluorescence is quenched, indicating that copper reversibly binds in the proximity of CTQ. Removal of copper does not diminish the enzymatic activity of LodA. This distinguishes LodA from enzymes with protein-derived tyrosylquinone cofactors in which copper is present near the cofactor and is absolutely required for activity. Mechanisms are proposed for the role of copper in the hydroxylation of the unactivated Trp side chain. These results demonstrate that the reason that the highly conserved Asp512 is critical for LodA, and possibly all tryptophylquinone enzymes, is not because it is required for catalysis but because it is necessary for CTQ biosynthesis, more specifically to facilitate the initial copper-dependent hydroxylation of a specific Trp residue.
Co-reporter:Zhongxin Ma, Heather R. Williamson, and Victor L. Davidson
Biochemistry 2016 Volume 55(Issue 40) pp:5738
Publication Date(Web):September 13, 2016
DOI:10.1021/acs.biochem.6b00816
In the absence of its substrate, the autoreduction of the high-valent bis-FeIV state of the hemes of MauG to the diferric state proceeds via a Compound I-like and then a Compound II-like intermediate. This process is coupled to oxidative damage to specific methionine residues and inactivation of MauG. The autoreduction of a P107V MauG variant, which is more prone to oxidative damage, proceeds directly from the bis-FeIV to the Compound II-like state with no detectable Compound I intermediate. Comparison of the crystal structures of native and P107V MauG reveals that this mutation alters the positions of amino acid residues in the heme site as well as the water network that delivers protons from the solvent to the hemes during their reduction. Kinetic, spectroscopic, and solvent kinetic isotope effect studies demonstrate that these changes in the heme site affect the protonation state of the ferryl heme and the relative efficiencies of two alternative pathways for the transfer of protons from solvent to the hemes. These changes enhance the rate of autoreduction of P107V MauG such that it competes with the catalytic reaction with substrate and causes the enzyme to inactivate itself during the steady-state reaction with H2O2 and its substrate. Thus, while this mutation has negligible effects on the initial steady-state kinetic parameters of MauG, it is a fatal mutation as it causes inactivation during catalysis.
Co-reporter:Zhongxin Ma;Heather R. Williamson
PNAS 2015 Volume 112 (Issue 35 ) pp:10896-10901
Publication Date(Web):2015-09-01
DOI:10.1073/pnas.1510986112
The high-valent state of the diheme enzyme MauG exhibits charge–resonance (CR) stabilization in which the major species is
a bis-FeIV state with one heme present as FeIV=O and the other as FeIV with axial heme ligands provided by His and Tyr side chains. In the absence of its substrate, the high-valent state is relatively
stable and returns to the diferric state over several minutes. It is shown that this process occurs in two phases. The first
phase is redistribution of the resonance species that support the CR. The second phase is the loss of CR and reduction to
the diferric state. Thermodynamic analysis revealed that the rates of the two phases exhibited different temperature dependencies
and activation energies of 8.9 and 19.6 kcal/mol. The two phases exhibited kinetic solvent isotope effects of 2.5 and 2.3.
Proton inventory plots of each reaction phase exhibited extreme curvature that could not be fit to models for one- or multiple-proton
transfers in the transition state. Each did fit well to a model for two alternative pathways for proton transfer, each involving
multiple protons. In each case the experimentally determined fractionation factors were consistent with one of the pathways
involving tunneling. The percent of the reaction that involved the tunneling pathway differed for the two reaction phases.
Using the crystal structure of MauG it was possible to propose proton–transfer pathways consistent with the experimental data
using water molecules and amino acid side chains in the distal pocket of the high-spin heme.
Co-reporter:María Dolores Chacón-Verdú, Jonatan C. Campillo-Brocal, Patricia Lucas-Elío, Victor L. Davidson, Antonio Sánchez-Amat
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2015 Volume 1854(Issue 9) pp:1123-1131
Publication Date(Web):September 2015
DOI:10.1016/j.bbapap.2014.12.018
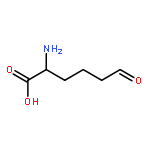
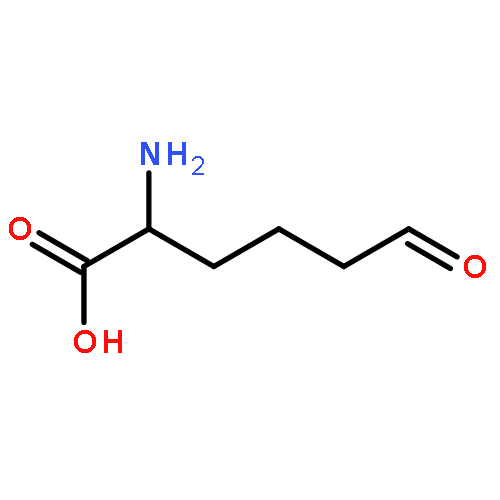
•LodA and GoxA are amino acid oxidases with a cysteine tryptophylquinone cofactor.•The precursor forms of the proteins are monohydroxylated at the Trp in the cofactor.•In the generation of the precursor a conserved Asp residue is critical.•The synthesis of the active protein is catalyzed by a flavoprotein.•The flavoproteins are very specific for the amino acid oxidase that they modify.The lysine-ε-oxidase, LodA, and glycine oxidase, GoxA, from Marinomonas mediteranea each possesses a cysteine tryptophylquinone (CTQ) cofactor. This cofactor is derived from posttranslational modifications which are covalent crosslinking of tryptophan and cysteine residues and incorporation of two oxygen atoms into the indole ring of Trp. In this manuscript, it is shown that the recombinant synthesis of LodA and GoxA containing a fully synthesized CTQ cofactor requires coexpression of a partner flavoprotein, LodB for LodA and GoxB for GoxA, which are not interchangeable. An inactive precursor of LodA or GoxA which contained a monohydroxylated Trp residue and no crosslink to the Cys was isolated from the soluble fraction when they were expressed alone. The structure of LodA revealed an Asp residue close to the cofactor which is conserved in quinohemoprotein amine dehydrogenase (QHNDH), containing CTQ, and methylamine dehydrogenase (MADH) containing tryptophan tryptophylquinone (TTQ) as cofactor. To study the role of this residue in the synthesis of the LodA precursor, Asp-512 was mutated to Ala. When the mutant protein was coexpressed with LodB an inactive protein was isolated which was soluble and contained no modifications at all, suggesting a role for this Asp in the initial LodB-independent hydroxylation of Trp. A similar role had been proposed for this conserved Asp residue in MADH. It is noteworthy that the formation of TTQ in MADH from the precursor also requires an accessory enzyme for its biosynthesis but it is a diheme enzyme MauG and not a flavoprotein. The results presented reveal novel mechanisms of post-translational modification involved in the generation of protein-derived cofactors. This article is part of a Special Issue entitled: Cofactor-dependent proteins: evolution, chemical diversity and bio-applications.
Co-reporter:Sooim Shin, Erik T. Yukl, Esha Sehanobish, Carrie M. Wilmot, and Victor L. Davidson
Biochemistry 2014 Volume 53(Issue 8) pp:
Publication Date(Web):February 11, 2014
DOI:10.1021/bi5000349
The diheme enzyme MauG catalyzes a six-electron oxidation that is required for the posttranslational modification of a precursor of methylamine dehydrogenase (preMADH) to complete the biosynthesis of its protein-derived cofactor, tryptophan tryptophylquinone (TTQ). Crystallographic and computational studies have implicated Gln103 in stabilizing the FeIV═O moiety of the bis-FeIV state by hydrogen bonding. The role of Gln103 was probed by site-directed mutagenesis. Q103L and Q103E mutations resulted in no expression and very little expression of the protein, respectively. Q103A MauG exhibited oxidative damage when isolated. Q103N MauG was isolated at levels comparable to that of wild-type MauG and exhibited normal activity in catalyzing the biosynthesis of TTQ from preMADH. The crystal structure of the Q103N MauG–preMADH complex suggests that a water may mediate hydrogen bonding between the shorter Asn103 side chain and the FeIV═O moiety. The Q103N mutation caused the two redox potentials associated with the diferric/diferrous redox couple to become less negative, although the redox cooperativity of the hemes of MauG was retained. Upon addition of H2O2, Q103N MauG exhibits changes in the absorbance spectrum in the Soret and near-IR regions consistent with formation of the bis-FeIV redox state. However, the rate of spontaneous return of the spectrum in the Soret region was 4.5-fold greater for Q103N MauG than for wild-type MauG. In contrast, the rate of spontaneous decay of the absorbance at 950 nm, which is associated with charge-resonance stabilization of the high-valence state, was similar for wild-type MauG and Q103N MauG. This suggests that as a consequence of the mutation a different distribution of resonance structures stabilizes the bis-FeIV state. These results demonstrate that subtle changes in the structure of the side chain of residue 103 can significantly affect the overall protein stability of MauG and alter the redox properties of the hemes.
Co-reporter:Heather R. Williamson, Brian A. Dow, Victor L. Davidson
Bioorganic Chemistry 2014 Volume 57() pp:213-221
Publication Date(Web):December 2014
DOI:10.1016/j.bioorg.2014.06.006
•The electronic coupling and reorganization energy can be modulated by the protein.•Preceding reaction steps can give rise to gated or coupled electron transfer.•Hole hopping via amino acid residues accelerates the rate of electron transfer.Electron transfer (ET) through and between proteins is a fundamental biological process. The rates and mechanisms of these ET reactions are controlled by the proteins in which the redox centers that donate and accept electrons reside. The protein influences the magnitudes of the ET parameters, the electronic coupling and reorganization energy that are associated with the ET reaction. The protein can regulate the rates of the ET reaction by requiring reaction steps to optimize the system for ET, leading to kinetic mechanisms of gated or coupled ET. Amino acid residues in the segment of the protein through which long range ET occurs can also modulate the ET rate by serving as staging points for hopping mechanisms of ET. Specific examples are presented to illustrate these mechanisms by which proteins control rates of ET reactions.
Co-reporter:Nafez Abu Tarboush, Erik T. Yukl, Sooim Shin, Manliang Feng, Carrie M. Wilmot, and Victor L. Davidson
Biochemistry 2013 Volume 52(Issue 37) pp:
Publication Date(Web):August 18, 2013
DOI:10.1021/bi400905s
The diheme enzyme MauG catalyzes a six-electron oxidation required for post-translational modification of a precursor of methylamine dehydrogenase (preMADH) to complete the biosynthesis of its protein-derived tryptophan tryptophylquinone (TTQ) cofactor. Crystallographic studies have implicated Glu113 in the formation of the bis-FeIV state of MauG, in which one heme is FeIV═O and the other is FeIV with His-Tyr axial ligation. An E113Q mutation had no effect on the structure of MauG but significantly altered its redox properties. E113Q MauG could not be converted to the diferrous state by reduction with dithionite but was only reduced to a mixed valence FeII/FeIII state, which is never observed in wild-type (WT) MauG. Addition of H2O2 to E113Q MauG generated a high valence state that formed more slowly and was less stable than the bis-FeIV state of WT MauG. E113Q MauG exhibited no detectable TTQ biosynthesis activity in a steady-state assay with preMADH as the substrate. It did catalyze the steady-state oxidation of quinol MADH to the quinone, but 1000-fold less efficiently than WT MauG. Addition of H2O2 to a crystal of the E113Q MauG-preMADH complex resulted in partial synthesis of TTQ. Extended exposure of these crystals to H2O2 resulted in hydroxylation of Pro107 in the distal pocket of the high-spin heme. It is concluded that the loss of the carboxylic group of Glu113 disrupts the redox cooperativity between hemes that allows rapid formation of the diferrous state and alters the distribution of high-valence species that participate in charge-resonance stabilization of the bis-FeIV redox state.
Co-reporter:Manliang Feng, Lyndal M. R. Jensen, Erik T. Yukl, Xiaoxi Wei, Aimin Liu, Carrie M. Wilmot, and Victor L. Davidson
Biochemistry 2012 Volume 51(Issue 8) pp:
Publication Date(Web):February 2, 2012
DOI:10.1021/bi201882e
The diheme enzyme MauG catalyzes a six-electron oxidation required for posttranslational modification of a precursor of methylamine dehydrogenase (preMADH) to complete the biosynthesis of its protein-derived tryptophan tryptophylquinone (TTQ) cofactor. Crystallographic studies had shown that Pro107, which resides in the distal pocket of the high-spin heme of MauG, changes conformation upon binding of CO or NO to the heme iron. In this study, Pro107 was converted to Cys, Val, and Ser by site-directed mutagenesis. The structures of each of these MauG mutant proteins in complex with preMADH were determined, as were their physical and catalytic properties. P107C MauG was inactive, and the crystal structure revealed that Cys107 had been oxidatively modified to a sulfinic acid. Mass spectrometry revealed that this modification was present prior to crystallization. P107V MauG exhibited spectroscopic and catalytic properties that were similar to those of wild-type MauG, but P107V MauG was more susceptible to oxidative damage. The P107S mutation caused a structural change that resulted in the five-coordinate high-spin heme being converted to a six-coordinate heme with a distal axial ligand provided by Glu113. EPR and resonance Raman spectroscopy revealed this heme remained high-spin but with greatly increased rhombicity as compared to that of the axial signal of wild-type MauG. P107S MauG was resistant to reduction by dithionite and reaction with H2O2 and unable to catalyze TTQ biosynthesis. These results show that the presence of Pro107 is critical in maintaining the proper structure of the distal heme pocket of the high-spin heme of MauG, allowing exogenous ligands to bind and directing the reactivity of the heme-activated oxygen during catalysis, thus minimizing the oxidation of other residues of MauG.
Co-reporter:Moonsung Choi, Sooim Shin, and Victor L. Davidson
Biochemistry 2012 Volume 51(Issue 35) pp:
Publication Date(Web):August 16, 2012
DOI:10.1021/bi300817d
Respiration, photosynthesis, and metabolism require the transfer of electrons through and between proteins over relatively long distances. It is critical that this electron transfer (ET) occur with specificity to avoid cellular damage, and at a rate that is sufficient to support the biological activity. A multistep hole hopping mechanism could, in principle, enhance the efficiency of long-range ET through proteins as it does in organic semiconductors. To explore this possibility, two different ET reactions that occur over the same distance within the protein complex of the diheme enzyme MauG and different forms of methylamine dehydrogenase (MADH) were subjected to kinetic and thermodynamic analysis. An ET mechanism of single-step direct electron tunneling from diferrous MauG to the quinone form of MADH is consistent with the data. In contrast, the biosynthetic ET from preMADH, which contains incompletely synthesized tryptophan tryptophylquinone, to the bis-Fe(IV) form of MauG is best described by a two-step hole hopping mechanism. Experimentally determined ET distances matched the distances determined from the crystal structure that would be expected for single-step tunneling and multistep hopping. Experimentally determined relative values of electronic coupling (HAB) for the two reactions correlated well with the relative HAB values predicted from computational analysis of the structure. The rate of the hopping-mediated ET reaction is also 10-fold greater than that of the single-step tunneling reaction despite a smaller overall driving force for the hopping-mediated ET reaction. These data provide insight into how the intervening protein matrix and redox potentials of the electron donor and acceptor determine whether the ET reaction proceeds via single-step tunneling or multistep hopping.
Co-reporter:Victor L. Davidson, Aimin Liu
Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2012 Volume 1824(Issue 11) pp:1299-1305
Publication Date(Web):November 2012
DOI:10.1016/j.bbapap.2012.01.008
Protein-derived cofactors are formed by irreversible covalent posttranslational modification of amino acid residues. An example is tryptophan tryptophylquinone (TTQ) found in the enzyme methylamine dehydrogenase (MADH). TTQ biosynthesis requires the cross-linking of the indole rings of two Trp residues and the insertion of two oxygen atoms onto adjacent carbons of one of the indole rings. The diheme enzyme MauG catalyzes the completion of TTQ within a precursor protein of MADH. The preMADH substrate contains a single hydroxyl group on one of the tryptophans and no crosslink. MauG catalyzes a six-electron oxidation that completes TTQ assembly and generates fully active MADH. These oxidation reactions proceed via a high valent bis-Fe(IV) state in which one heme is present as Fe(IV)=O and the other is Fe(IV) with both axial heme ligands provided by amino acid side chains. The crystal structure of MauG in complex with preMADH revealed that catalysis does not involve direct contact between the hemes of MauG and the protein substrate. Rather it is accomplished through long-range electron transfer, which presumably generates radical intermediates. Kinetic, spectrophotometric, and site-directed mutagenesis studies are beginning to elucidate how the MauG protein controls the reactivity of the hemes and mediates the long range electron/radical transfer required for catalysis. This article is part of a Special Issue entitled: Radical SAM enzymes and Radical Enzymology.Highlights►MauG stabilizes a bis-Fe(IV) state with one heme as His–Fe(IV)=O and another as His–Fe(IV)–Tyr. ►MauG post-translationally modifies two tryptophan residues within a protein substrate. ►MauG mediates long range electron transfer from the protein substrate to the high valent hemes.
Co-reporter:Narayanasami Sukumar, Moonsung Choi, Victor L. Davidson
Journal of Inorganic Biochemistry 2011 Volume 105(Issue 12) pp:1638-1644
Publication Date(Web):December 2011
DOI:10.1016/j.jinorgbio.2011.08.002
The mutation of the axial ligand of the type I copper protein amicyanin from Met to Lys results in a protein that is spectroscopically invisible and redox inactive. M98K amicyanin acts as a competitive inhibitor in the reaction of native amicyanin with methylamine dehydrogenase indicating that the M98K mutation has not affected the affinity for its natural electron donor. The crystal structure of M98K amicyanin reveals that its overall structure is very similar to native amicyanin but that the type I binding site is occupied by zinc. Anomalous difference Fourier maps calculated using the data collected around the absorption edges of copper and zinc confirm the presence of Zn2+ at the type I site. The Lys98 NZ donates a hydrogen bond to a well-ordered water molecule at the type I site which enhances the ability of Lys98 to provide a ligand for Zn2+. Attempts to reconstitute M98K apoamicyanin with copper resulted in precipitation of the protein. The fact that the M98K mutation generated such a selective zinc-binding protein was surprising as ligation of zinc by Lys is rare and this ligand set is unique for zinc.The type I site of M98K amicyanin provides a unique ligand set for Zn2+ rather than copper and formation of a hydrogen bond with water enables the lysine to act as a ligand for zinc.Highlights► M98K mutation of the axial copper ligand of the type I site of amicyanin converts it to a Zn-binding protein. ► Anomalous difference Fourier map clearly confirms the presence of zinc and the absence of copper. ► Addition of copper to M98K apoamicyanin results in denaturation of the protein. ► The ligand set for Zn2+ at the type I site of M98K amicyanin is unique. ► A hydrogen bond to water appears to allow Lys98 to ligate Zn2+.
Co-reporter:Nafez Abu Tarboush;Lyndal M. R. Jensen;Erik T. Yukl;Jiafeng Geng;Aimin Liu;Carrie M. Wilmot
PNAS 2011 Volume 108 (Issue 41 ) pp:
Publication Date(Web):2011-10-11
DOI:10.1073/pnas.1109423108
The diheme enzyme MauG catalyzes the posttranslational modification of the precursor protein of methylamine dehydrogenase
(preMADH) to complete biosynthesis of its protein-derived tryptophan tryptophylquinone (TTQ) cofactor. Catalysis proceeds
through a high valent bis-Fe(IV) redox state and requires long-range electron transfer (ET), as the distance between the modified
residues of preMADH and the nearest heme iron of MauG is 19.4 Å. Trp199 of MauG resides at the MauG-preMADH interface, positioned
midway between the residues that are modified and the nearest heme. W199F and W199K mutations did not affect the spectroscopic
and redox properties of MauG, or its ability to stabilize the bis-Fe(IV) state. Crystal structures of complexes of W199F/K
MauG with preMADH showed no significant perturbation of the MauG-preMADH structure or protein interface. However, neither
MauG variant was able to synthesize TTQ from preMADH. In contrast, an ET reaction from diferrous MauG to quinone MADH, which
does not require the bis-Fe(IV) intermediate, was minimally affected by the W199F/K mutations. W199F/K MauGs were able to
oxidize quinol MADH to form TTQ, the putative final two-electron oxidation of the biosynthetic process, but with kcat/Km values approximately 10% that of wild-type MauG. The differential effects of the W199F/K mutations on these three different
reactions are explained by a critical role for Trp199 in mediating multistep hopping from preMADH to bis-Fe(IV) MauG during
the long-range ET that is required for TTQ biosynthesis.
Co-reporter:Sooim Shin, Moonsung Choi, Heather R. Williamson, Victor L. Davidson
Biochimica et Biophysica Acta (BBA) - Bioenergetics (October 2014) Volume 1837(Issue 10) pp:1595-1601
Publication Date(Web):October 2014
DOI:10.1016/j.bbabio.2014.05.354
Co-reporter:Esha Sehanobish, Sooim Shin, Antonio Sanchez-Amat, Victor L. Davidson
FEBS Letters (3 March 2014) Volume 588(Issue 5) pp:752-756
Publication Date(Web):3 March 2014
DOI:10.1016/j.febslet.2014.01.021
•LodA has a tryptophylquinone cofactor but functions as an oxidase not a dehydrogenase.•LodA exhibits a ping-pong kinetic mechanism with lysine and O2 as substrates.•The kinetic mechanism of LodA is consistent with a covalent lysine-CTQ intermediate.LodA is a novel lysine-ε-oxidase which possesses a cysteine tryptophylquinone cofactor. It is the first tryptophylquinone enzyme known to function as an oxidase. A steady-state kinetic analysis shows that LodA obeys a ping-pong kinetic mechanism with values of kcat of 0.22 ± 0.04 s−1, Klysine of 3.2 ± 0.5 μM and KO2 of 37.2 ± 6.1 μM. The kcat exhibited a pH optimum at 7.5 while kcat/Klysine peaked at 7.0 and remained constant to pH 8.5. Alternative electron acceptors could not effectively substitute for O2 in the reaction. A mechanism for the reductive half reaction of LodA is proposed that is consistent with the ping-pong kinetics.
Co-reporter:Nafez Abu Tarboush, Lyndal M.R. Jensen, Carrie M. Wilmot, Victor L. Davidson
FEBS Letters (19 June 2013) Volume 587(Issue 12) pp:1736-1741
Publication Date(Web):19 June 2013
DOI:10.1016/j.febslet.2013.04.047
•A W199E MauG mutation alters hydrogen bonding interactions with preMADH.•A W199E MauG mutation alters the kinetic mechanism of interprotein electron transfer.•Trp199 of MauG plays multiple roles in MauG-catalyzed TTQ biosynthesis.MauG catalyzes posttranslational modifications of a methylamine dehydrogenase precursor (preMADH) to complete the biosynthesis of its protein-derived tryptophan tryptophylquinone (TTQ) cofactor. Trp199 is present at the site of interaction between MauG and preMADH and is critical to this process as it mediates hole hopping during the inter-protein electron transfer that is required for catalysis. Trp199 was converted to Glu and the structure and reactivity of the W199E/preMADH complex were characterized. The results reveal that the nature of residue 199 is also important for productive complex formation between preMADH and MauG.
Co-reporter:Brian A. Dow, Narayanasami Sukumar, Jason O. Matos, Moonsung Choi, Alfons Schulte, Suren A. Tatulian, Victor L. Davidson
Archives of Biochemistry and Biophysics (15 May 2014) Volumes 550–551() pp:
Publication Date(Web):15 May 2014
DOI:10.1016/j.abb.2014.03.010
•Trp45 stabilizes the structure of amicyanin via hydrogen bonding that connects two β-strands.•Trp45 does not influence the electronic properties of copper which quenches its fluorescence.•As temperature is increased disruption of tertiary and secondary structure precedes loss of copper.The cupredoxin amicyanin possesses a single tryptophan residue, Trp45. Its fluorescence is quenched when copper is bound even though it is separated by 10.1 Å. Mutation of Trp45 to Ala, Phe, Leu and Lys resulted in undetectable protein expression. A W45Y amicyanin variant was isolated. The W45Y mutation did not alter the spectroscopic properties or intrinsic redox potential of amicyanin, but increased the pKa value for the pH-dependent redox potential by 0.5 units. This is due to a hydrogen-bond involving the His95 copper ligand which is present in reduced W45Y amicyanin but not in native amicyanin. The W45Y mutation significantly decreased the thermal stability of amicyanin, as determined by changes in the visible absorbance of oxidized amicyanin and in the circular dichroism spectra for oxidized, reduced and apo forms of amicyanin. Comparison of the crystal structures suggests that the decreased stability of W45Y amicyanin may be attributed to the loss of a strong interior hydrogen bond between Trp45 and Tyr90 in native amicyanin which links two of the β-sheets that comprise the overall structure of amicyanin. Thus, Trp45 is critical for stabilizing the structure of amicyanin but it does not influence the electronic properties of the copper which quenches its fluorescence.
Co-reporter:Sooim Shin, Victor L. Davidson
Archives of Biochemistry and Biophysics (15 February 2014) Volume 544() pp:112-118
Publication Date(Web):15 February 2014
DOI:10.1016/j.abb.2013.10.004
Co-reporter:Nafez Abu Tarboush, Sooim Shin, Jiafeng Geng, Aimin Liu, Victor L. Davidson
FEBS Letters (14 December 2012) Volume 586(Issue 24) pp:4339-4343
Publication Date(Web):14 December 2012
DOI:10.1016/j.febslet.2012.10.044
MauG catalyzes posttranslational modifications of methylamine dehydrogenase to complete the biosynthesis of its protein-derived tryptophan tryptophylquinone (TTQ) cofactor. MauG possesses a five-coordinate high-spin and a six-coordinate low-spin ferric heme, the latter with His-Tyr ligation. Replacement of this tyrosine with lysine generates a MauG variant with only high-spin ferric heme and altered spectroscopic and redox properties. Y294K MauG cannot stabilize the bis-Fe(IV) redox state required for TTQ biosynthesis but instead forms a compound I-like species on reaction with peroxide. The results clarify the role of Tyr ligation of the five-coordinate heme in determining the physical and redox properties and reactivity of MauG.Highlights► A Y294K mutation results in loss rather than replacement of the axial heme ligand provided by Tyr294. ► A Y294K mutation alters spectroscopic and redox properties of both hemes of MauG. ► Y294K MauG forms a high valent compound I-like species rather than a bis-Fe(IV) species. ► Y294K MauG is competent in long range electron transfer but has lost TTQ biosynthesis activity.
Co-reporter:Esha Sehanobish, María Dolores Chacón-Verdú, Antonio Sanchez-Amat, Victor L. Davidson
Archives of Biochemistry and Biophysics (1 August 2015) Volume 579() pp:26-32
Publication Date(Web):1 August 2015
DOI:10.1016/j.abb.2015.05.013

![[2,4'-Bi-1H-indole]-3,3'-dipropanoicacid, a3,a3'-diamino-6',7'-dihydro-6',7'-dioxo-, (a3S,a3'S)-](http://img.cochemist.com/ccimg/134700/134645-25-3.png)
![[2,4'-Bi-1H-indole]-3,3'-dipropanoicacid, a3,a3'-diamino-6',7'-dihydro-6',7'-dioxo-, (a3S,a3'S)-](http://img.cochemist.com/ccimg/134700/134645-25-3_b.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8.png)
![Ferrate(2-), [7,12-diethenyl-3,8,13,17-tetramethyl-21H,23H-porphine-2,18-dipropanoato(4-)-κN21,κN22,κN23,κN24]-, hydrogen (1:2), (SP-4-2)-](/data/chemimg/522800/14875-96-8_b.png)