Co-reporter:Alex Baggott;Joanne R. Bass;Stephen A. Hall;Ian Hamerton;Lyndsey Mooring ;David Sparks
Macromolecular Theory and Simulations 2014 Volume 23( Issue 6) pp:369-372
Publication Date(Web):
DOI:10.1002/mats.201300141
Despite their inability to model bond breaking molecular dynamics simulations are shown to predict thermal degradation temperatures of polycyanurate (cyanate ester) homopolymers and nanocomposites in very close agreement with experimental data. Simulated polymer density, used to predict Tg also shows a reduction within the same temperature range as experimental values for the thermal degradation.
Co-reporter:Daniel N. Meijles, Brendan J. Howlin, Jian-Mei Li
Computational Biology and Chemistry 2012 Volume 39() pp:6-13
Publication Date(Web):August 2012
DOI:10.1016/j.compbiolchem.2012.05.001
The p22phox protein is an essential subunit of the cytochrome b558 of the NADPH oxidase (Nox) complex which by generating reactive oxygen species (ROS) plays important role in regulating cellular function. p22phox stabilises the Nox enzyme, assists in catalytic core maturation and in the meantime provides an anchoring site for cytosolic regulatory subunits to bind. However, the protein structure of the p22phox is still uncertain. In this study we use an in silico computational bioinformatic approach to produce a consensus 3-dimensional model of the p22phox. Based on published protein sequence data of human p22phox and by using transmembrane specific protein prediction algorithms, we found that p22phox consists of two domains: an N-terminal transmembrane domain (124 a.a.) and a C-terminal cytoplasmic domain (71 a.a.). In its predicted most stable form, p22phox contains three transmembrane helices leading to an extracellular N-terminus and an extensive (39 a.a.) extracellular loop between helices 2 and 3. Furthermore, we locate the cytosolic domain phosphorylation site at threonine147 which literature shows is capable of priming the p22phox, in order to accept its binding partners. Our results are consistent with the biological characterisation of p22phox derived from experiments using specific antibody or genetic manipulation. Our 3-D protein model provides insights into the biological function of p22phox and cytochrome b558, and can be used as tool to investigate the regulatory mechanism of Nox isoforms.Graphical abstractHighlights► A new model for the P22phox protein is presented. ► A comparison of 2, 3 and 4 helix models is discussed. ► Experimental evidence and modelling studies favour the 3 helix model. ► The new model has potential in drug design investigations.
Co-reporter:Roderic C.E. Green, Alfred E. Thumser, David Povey, José W. Saldanha, Brian S. Potter, Rex A. Palmer, Brendan J. Howlin
Computational Biology and Chemistry 2009 Volume 33(Issue 3) pp:189-195
Publication Date(Web):June 2009
DOI:10.1016/j.compbiolchem.2009.01.001
Recently published X-ray structures of three common forms, A, B and C, of oligomycin, including absolute configurations, are investigated to examine their binding to ATP Synthase. The X-ray studies reveal regions with differences in three-dimensional structure and hydrogen bonding propensity between the oligomycins, which may be associated with their potential to bind to sites on ATP Synthase. Computational docking studies carried out using MOE with the X-ray structures and an homology model of the FO domain of ATP Synthase from Escherichia coli, are used to derive an induced fit pocket. Docking of all oligomycins to this pocket indicate that the B and C forms bind more tightly than the A form. Consideration of the single crystal X-ray data alone indicate the B form may be the best inhibitor and that O(24) is the most important ligating group for binding, this is supported by the docking data. The latter reveals Asn214 and other key proton translocating residues to be the main residues contacted by the inhibitor. These data allow the binding modes of different forms of oligomycin to be deduced from X-ray single crystal data supported by molecular modelling and computational docking studies.
Co-reporter:Anna C. Tanczos, Rex A. Palmer, Brian S. Potter, José W. Saldanha, Brendan J. Howlin
Computational Biology and Chemistry 2004 Volume 28(5–6) pp:375-385
Publication Date(Web):December 2004
DOI:10.1016/j.compbiolchem.2004.09.009
A series of agonists to the rat muscarinic receptor have been docked computationally to the active site of a homology model of rat M1 muscarinic receptor. The agonists were modelled on the X-ray crystal structure of atropine, which is reported here and the docking studies are shown to reproduce correctly the order of experimental binding affinities for the agonists as well as indicate where there appear to be inconsistencies in the experimental data. The crystal and molecular structure of atropine (tropine tropate; α-[hydroxymethyl]benzeneacetic acid 8-methyl[3.2.1]oct-3-yl ester C17H23NO3) has been determined by X-ray crystallography using an automated Patterson search method, and refined by full-matrix least-squares to a final R of 0.0452 for 2701 independent observed reflections and 192 parameters using Mo Kα radiation, λ = 0.71073 Å at 150 K. The compound crystallises in space group Fdd2 with Z = 16 molecules per unit cell.
Co-reporter:R.D Allington, D Attwood, I Hamerton, J.N Hay, B.J Howlin
Composites Part A: Applied Science and Manufacturing 2004 Volume 35(Issue 10) pp:1161-1173
Publication Date(Web):October 2004
DOI:10.1016/j.compositesa.2004.03.009
In this work computer modelling investigations are reported into the chemical functional groups that are believed to contribute to composite fibre/matrix adhesion. The CERIUS [Compos A: Appl Sci Manuf 29 (1998) 1291] and MOPAC computational packages are employed to simulate the sorption interactions of small molecules at a range of functionalised surfaces. The results of these calculations are compared with fibre surface energetic data reported previously, and models of the fibre surface chemistry with varying degrees of surface treatment are developed. There is agreement that surface treatment of carbon fibres improves adhesion and adsorption characteristics, yet the factors controlling this have still to be determined fully. The empirical study using the computational model developed in this work attempts to resolve the changes in fibre adsorption phenomena into contributions arising from specific surface oxygen and nitrogen functional groups. The general trend in surface concentration of these atoms is similar to XPS analyses of the surface treated carbon fibres and is in agreement with previously published work.

![Poly[oxy-1,4-phenylene(1-methylethylidene)-1,4-phenyleneoxy(carboxyp
henylene)carbonylimino-1,3-phenyleneiminocarbonyl(carboxyphenylene
)]](http://img.cochemist.com/ccimg/70600/70563-59-6.png)
![Poly[oxy-1,4-phenylene(1-methylethylidene)-1,4-phenyleneoxy(carboxyp
henylene)carbonylimino-1,3-phenyleneiminocarbonyl(carboxyphenylene
)]](http://img.cochemist.com/ccimg/70600/70563-59-6_b.png)




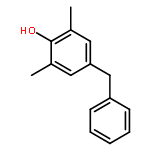

![2,5-Pyrrolidinedione, 3-[3-(2-hydroxyphenyl)-2-propenyl]-1-phenyl-](http://img.cochemist.com/ccimg/197900/197843-91-7.png)
![2,5-Pyrrolidinedione, 3-[3-(2-hydroxyphenyl)-2-propenyl]-1-phenyl-](http://img.cochemist.com/ccimg/197900/197843-91-7_b.png)
![2,5-Pyrrolidinedione, 3-[3-(2-methoxyphenyl)-2-propenyl]-1-phenyl-](http://img.cochemist.com/ccimg/197900/197843-80-4.png)
![2,5-Pyrrolidinedione, 3-[3-(2-methoxyphenyl)-2-propenyl]-1-phenyl-](http://img.cochemist.com/ccimg/197900/197843-80-4_b.png)
![2,5-Pyrrolidinedione, 3-[3-(2-methylphenyl)-2-propenyl]-1-phenyl-](http://img.cochemist.com/ccimg/197900/197843-78-0.png)
![2,5-Pyrrolidinedione, 3-[3-(2-methylphenyl)-2-propenyl]-1-phenyl-](http://img.cochemist.com/ccimg/197900/197843-78-0_b.png)
![Phenol, 4,4'-[sulfonylbis(4,1-phenyleneoxy)]bis-](/data/chemimg/3073100/19896-27-6.png)
![Phenol, 4,4'-[sulfonylbis(4,1-phenyleneoxy)]bis-](/data/chemimg/3073100/19896-27-6_b.png)
![2,5-Furandione, dihydro-3-[3-(4-methoxyphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/1100/1087-24-7.png)
![2,5-Furandione, dihydro-3-[3-(4-methoxyphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/1100/1087-24-7_b.png)
![2,5-Pyrrolidinedione, 3-[3-(4-bromophenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-74-6.png)
![2,5-Pyrrolidinedione, 3-[3-(4-bromophenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-74-6_b.png)
![2,5-Furandione, dihydro-3-[3-(2-phenoxyphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-67-7.png)
![2,5-Furandione, dihydro-3-[3-(2-phenoxyphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-67-7_b.png)
![Butanedioic acid, [3-(2-methoxyphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-65-5.png)
![Butanedioic acid, [3-(2-methoxyphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-65-5_b.png)
![Phenol, 4,4'-[1,2-ethanediylbis(oxy-2,1-ethanediyloxy)]bis-](http://img.cochemist.com/ccimg/29300/29239-84-7.png)
![Phenol, 4,4'-[1,2-ethanediylbis(oxy-2,1-ethanediyloxy)]bis-](http://img.cochemist.com/ccimg/29300/29239-84-7_b.png)
![2,5-Pyrrolidinedione, 3-[3-(4-bromophenyl)-2-propenyl]-1-phenyl-](http://img.cochemist.com/ccimg/197900/197843-82-6.png)
![2,5-Pyrrolidinedione, 3-[3-(4-bromophenyl)-2-propenyl]-1-phenyl-](http://img.cochemist.com/ccimg/197900/197843-82-6_b.png)
![2,5-Furandione, dihydro-3-[3-(4-methylphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-63-3.png)
![2,5-Furandione, dihydro-3-[3-(4-methylphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-63-3_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-[2-[(4,6-DICHLORO-1,3,5-TRIAZIN-2-YL)OXY]PHENYL]-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-90-6.png)
![2,5-PYRROLIDINEDIONE, 3-[3-[2-[(4,6-DICHLORO-1,3,5-TRIAZIN-2-YL)OXY]PHENYL]-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-90-6_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-METHYLPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-70-2.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-METHYLPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-70-2_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-METHYLPHENYL)-2-PROPENYL]-1-(4-PHENOXYPHENYL)-](http://img.cochemist.com/ccimg/197900/197843-86-0.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-METHYLPHENYL)-2-PROPENYL]-1-(4-PHENOXYPHENYL)-](http://img.cochemist.com/ccimg/197900/197843-86-0_b.png)
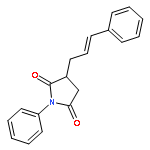
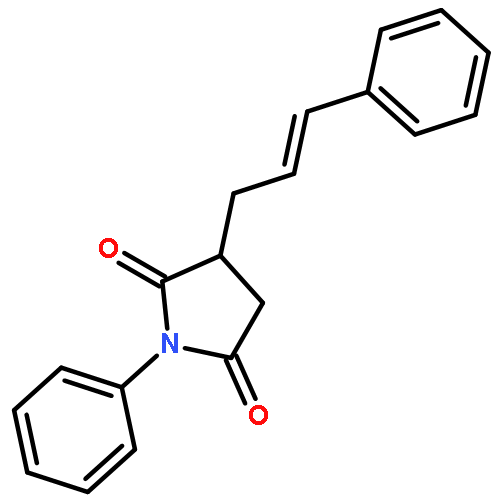
![1,3,5-TRIAZINE, 2,4-DICHLORO-6-[2-(2-PROPENYL)PHENOXY]-](http://img.cochemist.com/ccimg/197900/197843-61-1.png)
![1,3,5-TRIAZINE, 2,4-DICHLORO-6-[2-(2-PROPENYL)PHENOXY]-](http://img.cochemist.com/ccimg/197900/197843-61-1_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-METHYLPHENYL)-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-79-1.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-METHYLPHENYL)-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-79-1_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-METHOXYPHENYL)-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-81-5.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-METHOXYPHENYL)-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-81-5_b.png)
![2,5-FURANDIONE, 3-[3-(4-BROMOPHENYL)-2-PROPENYL]DIHYDRO-](http://img.cochemist.com/ccimg/197900/197843-66-6.png)
![2,5-FURANDIONE, 3-[3-(4-BROMOPHENYL)-2-PROPENYL]DIHYDRO-](http://img.cochemist.com/ccimg/197900/197843-66-6_b.png)
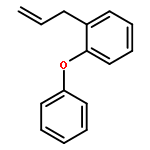
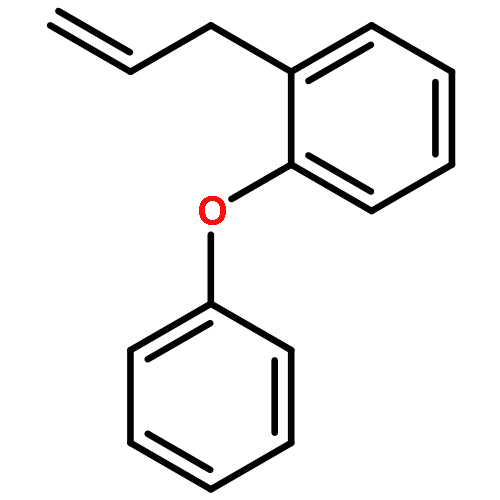

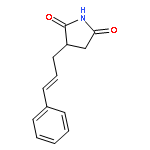
![2,5-Pyrrolidinedione, 3-[3-(4-methylphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-71-3.png)
![2,5-Pyrrolidinedione, 3-[3-(4-methylphenyl)-2-propenyl]-](http://img.cochemist.com/ccimg/197900/197843-71-3_b.png)
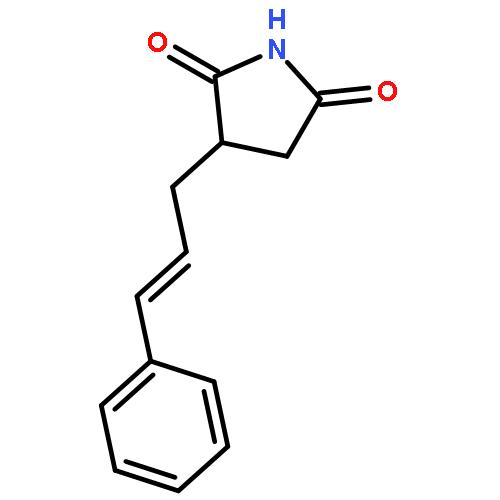
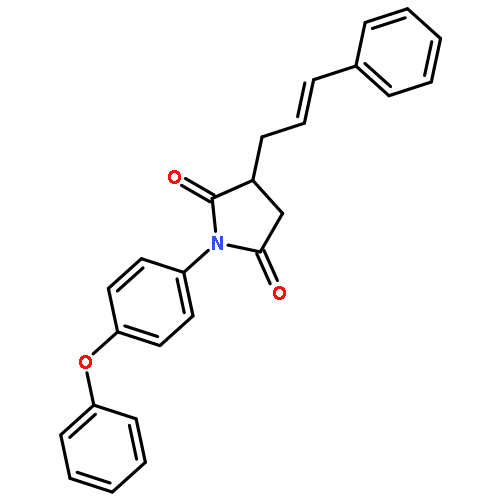
![2,5-PYRROLIDINEDIONE, 3-[3-(4-PHENOXYPHENYL)-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-84-8.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-PHENOXYPHENYL)-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-84-8_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-METHOXYPHENYL)-2-PROPENYL]-1-(4-PHENOXYPHENYL)-](http://img.cochemist.com/ccimg/197900/197843-88-2.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-METHOXYPHENYL)-2-PROPENYL]-1-(4-PHENOXYPHENYL)-](http://img.cochemist.com/ccimg/197900/197843-88-2_b.png)
![3-phenyl-6-[(3-phenyl-2,4-dihydro-1,3-benzoxazin-6-yl)sulfanyl]-2,4-dihydro-1,3-benzoxazine](http://img.cochemist.com/ccimg/228200/228118-04-5.png)
![3-phenyl-6-[(3-phenyl-2,4-dihydro-1,3-benzoxazin-6-yl)sulfanyl]-2,4-dihydro-1,3-benzoxazine](http://img.cochemist.com/ccimg/228200/228118-04-5_b.png)
![Poly[(1,3-dihydro-1,3-dioxo-2H-isoindole-5,2-diyl)-1,4-phenyleneoxy-1,
4-phenylene(1-methylethylidene)-1,4-phenyleneoxy-1,4-phenylene(1,3-d
ihydro-1,3-dioxo-2H-isoindole-2,5-diyl)carbonylimino-1,4-phenyleneoxy
-1,4-phenylene(1-methylethylidene)-1,4-phenyleneoxy-1,4-phenyleneimi
nocarbonyl]](http://img.cochemist.com/ccimg/152200/152108-99-1.png)
![Poly[(1,3-dihydro-1,3-dioxo-2H-isoindole-5,2-diyl)-1,4-phenyleneoxy-1,
4-phenylene(1-methylethylidene)-1,4-phenyleneoxy-1,4-phenylene(1,3-d
ihydro-1,3-dioxo-2H-isoindole-2,5-diyl)carbonylimino-1,4-phenyleneoxy
-1,4-phenylene(1-methylethylidene)-1,4-phenyleneoxy-1,4-phenyleneimi
nocarbonyl]](http://img.cochemist.com/ccimg/152200/152108-99-1_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-[2-[[4,6-BIS[2-(2-PROPENYL)PHENOXY]-1,3,5-TRIAZIN-2-YL]OXY]PHENYL]-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-92-8.png)
![2,5-PYRROLIDINEDIONE, 3-[3-[2-[[4,6-BIS[2-(2-PROPENYL)PHENOXY]-1,3,5-TRIAZIN-2-YL]OXY]PHENYL]-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-92-8_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-PHENOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-75-7.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-PHENOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-75-7_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-METHOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-73-5.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(4-METHOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-73-5_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-PHENOXYPHENYL)-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-83-7.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-PHENOXYPHENYL)-2-PROPENYL]-1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-83-7_b.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-METHOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-72-4.png)
![2,5-PYRROLIDINEDIONE, 3-[3-(2-METHOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-72-4_b.png)
![2,5-PYRROLIDINEDIONE, 3,3'-[[6-[2-(2-PROPENYL)PHENOXY]-1,3,5-TRIAZINE-2,4-DIYL]BIS(OXY-2,1-PHENYLENE-2-PROPENE-3,1-DIYL)]BIS[1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-93-9.png)
![2,5-PYRROLIDINEDIONE, 3,3'-[[6-[2-(2-PROPENYL)PHENOXY]-1,3,5-TRIAZINE-2,4-DIYL]BIS(OXY-2,1-PHENYLENE-2-PROPENE-3,1-DIYL)]BIS[1-PHENYL-](http://img.cochemist.com/ccimg/197900/197843-93-9_b.png)
![2,5-FURANDIONE, DIHYDRO-3-[3-(4-PHENOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-68-8.png)
![2,5-FURANDIONE, DIHYDRO-3-[3-(4-PHENOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-68-8_b.png)
![2,5-FURANDIONE, DIHYDRO-3-[3-(2-METHYLPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-62-2.png)
![2,5-FURANDIONE, DIHYDRO-3-[3-(2-METHYLPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-62-2_b.png)
![2,5-FURANDIONE, DIHYDRO-3-[3-(2-METHOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-64-4.png)
![2,5-FURANDIONE, DIHYDRO-3-[3-(2-METHOXYPHENYL)-2-PROPENYL]-](http://img.cochemist.com/ccimg/197900/197843-64-4_b.png)


![[[2-[p-(oxiranylmethoxy)benzyl]phenoxy]methyl]oxirane](http://img.cochemist.com/ccimg/57500/57469-07-5.png)
![[[2-[p-(oxiranylmethoxy)benzyl]phenoxy]methyl]oxirane](http://img.cochemist.com/ccimg/57500/57469-07-5_b.png)








![2,2'-[methylenebis(o-phenyleneoxymethylene)]bisoxirane](http://img.cochemist.com/ccimg/54300/54208-63-8.png)
![2,2'-[methylenebis(o-phenyleneoxymethylene)]bisoxirane](http://img.cochemist.com/ccimg/54300/54208-63-8_b.png)








![Benzeneacetic acid, a-phenyl-,(3-endo)-8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester](http://img.cochemist.com/ccimg/6900/6878-98-4.png)
![Benzeneacetic acid, a-phenyl-,(3-endo)-8-methyl-8-azabicyclo[3.2.1]oct-3-yl ester](http://img.cochemist.com/ccimg/6900/6878-98-4_b.png)














![8-Azabicyclo[3.2.1]octane,3-(diphenylmethoxy)-8-methyl-, (3-endo)-](http://img.cochemist.com/ccimg/100/86-13-5.png)
![8-Azabicyclo[3.2.1]octane,3-(diphenylmethoxy)-8-methyl-, (3-endo)-](http://img.cochemist.com/ccimg/100/86-13-5_b.png)









![4-[(4-AMINOPHENYL)METHYL]ANILINE;1-[4-[[4-(2,5-DIOXOPYRROL-1-YL)PHENYL]METHYL]PHENYL]PYRROLE-2,5-DIONE](http://img.cochemist.com/data/images/26140-67-0.png)
![4-[(4-AMINOPHENYL)METHYL]ANILINE;1-[4-[[4-(2,5-DIOXOPYRROL-1-YL)PHENYL]METHYL]PHENYL]PYRROLE-2,5-DIONE](http://img.cochemist.com/data/images/26140-67-0_b.png)
![1-METHOXY-4-[4-[4-(4-METHOXYPHENOXY)PHENYL]SULFONYLPHENOXY]BENZENE](http://img.cochemist.com/ccimg/34100/34018-55-8.png)
![1-METHOXY-4-[4-[4-(4-METHOXYPHENOXY)PHENYL]SULFONYLPHENOXY]BENZENE](http://img.cochemist.com/ccimg/34100/34018-55-8_b.png)
![Benzaldehyde,4,4'-[oxybis(2,1-ethanediyloxy)]bis-](http://img.cochemist.com/ccimg/111600/111550-46-0.png)
![Benzaldehyde,4,4'-[oxybis(2,1-ethanediyloxy)]bis-](http://img.cochemist.com/ccimg/111600/111550-46-0_b.png)






![1,2-Propanediol, 3-[3-[3,5,7,9,11,13,15-heptakis(2-methylpropyl)pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxan-1-yl]propoxy]-](/data/chemimg/68400/480439-49-4.png)
![1,2-Propanediol, 3-[3-[3,5,7,9,11,13,15-heptakis(2-methylpropyl)pentacyclo[9.5.1.13,9.15,15.17,13]octasiloxan-1-yl]propoxy]-](/data/chemimg/68400/480439-49-4_b.png)




![N-[5-(4-AMINOPHENOXY)PENTYL]FORMAMIDE](/data/chemimg/56300/484-58-2.png)
![N-[5-(4-AMINOPHENOXY)PENTYL]FORMAMIDE](/data/chemimg/56300/484-58-2_b.png)
![Benzaldehyde,4,4'-[1,2-ethanediylbis(oxy)]bis-](http://img.cochemist.com/ccimg/34100/34074-28-7.png)
![Benzaldehyde,4,4'-[1,2-ethanediylbis(oxy)]bis-](http://img.cochemist.com/ccimg/34100/34074-28-7_b.png)
![1,3,5-Triazine, 2,4,6-tris[4-(1-methyl-1-phenylethyl)phenoxy]-](http://img.cochemist.com/ccimg/109300/109291-48-7.png)
![1,3,5-Triazine, 2,4,6-tris[4-(1-methyl-1-phenylethyl)phenoxy]-](http://img.cochemist.com/ccimg/109300/109291-48-7_b.png)












![Benzaldehyde,4,4'-[1,2-ethanediylbis(oxy-2,1-ethanediyloxy)]bis-](http://img.cochemist.com/ccimg/111600/111550-48-2.png)
![Benzaldehyde,4,4'-[1,2-ethanediylbis(oxy-2,1-ethanediyloxy)]bis-](http://img.cochemist.com/ccimg/111600/111550-48-2_b.png)




![Phenol,4,4'-[1,2-ethanediylbis(oxy)]bis-](http://img.cochemist.com/ccimg/24300/24209-90-3.png)
![Phenol,4,4'-[1,2-ethanediylbis(oxy)]bis-](http://img.cochemist.com/ccimg/24300/24209-90-3_b.png)



![Oxirane,2,2'-[methylenebis(4,1-phenyleneoxymethylene)]bis-](http://img.cochemist.com/ccimg/2100/2095-03-6.png)
![Oxirane,2,2'-[methylenebis(4,1-phenyleneoxymethylene)]bis-](http://img.cochemist.com/ccimg/2100/2095-03-6_b.png)