Co-reporter:Zehua Chen, Du Zhang, Ye Jin, Yang Yang, Neil Qiang Su, and Weitao Yang
The Journal of Physical Chemistry Letters September 21, 2017 Volume 8(Issue 18) pp:4479-4479
Publication Date(Web):August 31, 2017
DOI:10.1021/acs.jpclett.7b01864
To describe static correlation, we develop a new approach to density functional theory (DFT), which uses a generalized auxiliary system that is of a different symmetry, such as particle number or spin, from that of the physical system. The total energy of the physical system consists of two parts: the energy of the auxiliary system, which is determined with a chosen density functional approximation (DFA), and the excitation energy from an approximate linear response theory that restores the symmetry to that of the physical system, thus rigorously leading to a multideterminant description of the physical system. The electron density of the physical system is different from that of the auxiliary system and is uniquely determined from the functional derivative of the total energy with respect to the external potential. Our energy functional is thus an implicit functional of the physical system density, but an explicit functional of the auxiliary system density. We show that the total energy minimum and stationary states, describing the ground and excited states of the physical system, can be obtained by a self-consistent optimization with respect to the explicit variable, the generalized Kohn–Sham noninteracting density matrix. We have developed the generalized optimized effective potential method for the self-consistent optimization. Among options of the auxiliary system and the associated linear response theory, reformulated versions of the particle–particle random phase approximation (pp-RPA) and the spin-flip time-dependent density functional theory (SF-TDDFT) are selected for illustration of principle. Numerical results show that our multireference DFT successfully describes static correlation in bond dissociation and double bond rotation.
Co-reporter:Ye Jin, Du Zhang, Zehua Chen, Neil Qiang Su, and Weitao Yang
The Journal of Physical Chemistry Letters October 5, 2017 Volume 8(Issue 19) pp:4746-4746
Publication Date(Web):September 12, 2017
DOI:10.1021/acs.jpclett.7b02165
A new self-consistent procedure for calculating the total energy with an orbital-dependent density functional approximation (DFA), the generalized optimized effective potential (GOEP), is developed in the present work. The GOEP is a nonlocal Hermitian potential that delivers the sets of occupied and virtual orbitals and minimizes the total energy. The GOEP optimization leads to the same minimum as does the orbital optimization. The GOEP method is promising as an effective optimization approach for orbital-dependent functionals, as demonstrated for the self-consistent calculations of the random phase approximation (RPA) to the correlation functionals in the particle–hole (ph) and particle–particle (pp) channels. The results show that the accuracy in describing the weakly interacting van der Waals systems is significantly improved in the self-consistent calculations. In particular, the important single excitations contribution in non-self-consistent RPA calculations can be captured self-consistently through the GOEP optimization, leading to orbital renormalization, without using the single excitations in the energy functional.
Co-reporter:Guoqing Li, Du Zhang, Yifei Yu, Shengyang Huang, Weitao Yang, and Linyou Cao
Journal of the American Chemical Society November 15, 2017 Volume 139(Issue 45) pp:16194-16194
Publication Date(Web):October 25, 2017
DOI:10.1021/jacs.7b07450
MoS2 presents a promising catalyst for the hydrogen evolution reaction (HER) in water splitting, but its worse catalytic performance in neutral and alkaline media than in acidic environment may be problematic for practical application. This is because the other half reaction of water splitting, i.e., oxygen evolution reaction, often needs to be implemented in alkaline environment. Here we demonstrate a universal strategy that may be used to significantly improve the HER catalysis of MoS2 in all kinds of environments from acidic to alkaline, proton intercalation. Protons may be enabled to intercalate between monolayer MoS2 and underlying substrates or in the interlayer space of thicker MoS2 by two processes: electrochemically polarizing MoS2 at negative potentials (vs RHE) in acidic media or immersing MoS2 into certain acid solutions like TFSI. The improvement in catalytic performance is due to the activity enhancement of the active sites in MoS2 by the intercalated protons, which might be related with the effect of the intercalated protons on electrical conductance and the adsorption energy of hydrogen atoms. The enhancement in catalytic activity by the intercalated proton is very stable even in neutral and alkaline electrolytes.
Co-reporter:Chetan Rupakheti, Rachael Al-Saadon, Yuqi Zhang, Aaron M. Virshup, Peng Zhang, Weitao Yang, and David N. Beratan
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 4) pp:1942-1952
Publication Date(Web):March 7, 2016
DOI:10.1021/acs.jctc.5b00829
Organic light-emitting diodes (OLEDs) have wide-ranging applications, from lighting to device displays. However, the repertoire of organic molecules with efficient blue emission is limited. To address this limitation, we have developed a strategy to design property-optimized, diversity-oriented libraries of structures with favorable fluorescence properties. This approach identifies novel diverse candidate organic molecules for blue emission with strong oscillator strengths and low singlet–triplet energy gaps that favor thermally activated delayed fluorescence (TADF) emission.
Co-reporter:Lin Shen, Jingheng Wu, and Weitao Yang
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 10) pp:4934-4946
Publication Date(Web):August 23, 2016
DOI:10.1021/acs.jctc.6b00663
Molecular dynamics simulation with multiscale quantum mechanics/molecular mechanics (QM/MM) methods is a very powerful tool for understanding the mechanism of chemical and biological processes in solution or enzymes. However, its computational cost can be too high for many biochemical systems because of the large number of ab initio QM calculations. Semiempirical QM/MM simulations have much higher efficiency. Its accuracy can be improved with a correction to reach the ab initio QM/MM level. The computational cost on the ab initio calculation for the correction determines the efficiency. In this paper we developed a neural network method for QM/MM calculation as an extension of the neural-network representation reported by Behler and Parrinello. With this approach, the potential energy of any configuration along the reaction path for a given QM/MM system can be predicted at the ab initio QM/MM level based on the semiempirical QM/MM simulations. We further applied this method to three reactions in water to calculate the free energy changes. The free-energy profile obtained from the semiempirical QM/MM simulation is corrected to the ab initio QM/MM level with the potential energies predicted with the constructed neural network. The results are in excellent accordance with the reference data that are obtained from the ab initio QM/MM molecular dynamics simulation or corrected with direct ab initio QM/MM potential energies. Compared with the correction using direct ab initio QM/MM potential energies, our method shows a speed-up of 1 or 2 orders of magnitude. It demonstrates that the neural network method combined with the semiempirical QM/MM calculation can be an efficient and reliable strategy for chemical reaction simulations.
Co-reporter:Lin Shen and Weitao Yang
Journal of Chemical Theory and Computation 2016 Volume 12(Issue 4) pp:2017-2027
Publication Date(Web):March 1, 2016
DOI:10.1021/acs.jctc.5b01107
We developed a new multiresolution method that spans three levels of resolution with quantum mechanical, atomistic molecular mechanical, and coarse-grained models. The resolution-adapted all-atom and coarse-grained water model, in which an all-atom structural description of the entire system is maintained during the simulations, is combined with the ab initio quantum mechanics and molecular mechanics method. We apply this model to calculate the redox potentials of the aqueous ruthenium and iron complexes by using the fractional number of electrons approach and thermodynamic integration simulations. The redox potentials are recovered in excellent accordance with the experimental data. The speed-up of the hybrid all-atom and coarse-grained water model renders it computationally more attractive. The accuracy depends on the hybrid all-atom and coarse-grained water model used in the combined quantum mechanical and molecular mechanical method. We have used another multiresolution model, in which an atomic-level layer of water molecules around redox center is solvated in supramolecular coarse-grained waters for the redox potential calculations. Compared with the experimental data, this alternative multilayer model leads to less accurate results when used with the coarse-grained polarizable MARTINI water or big multipole water model for the coarse-grained layer.
Co-reporter:Yang Yang; Lin Shen; Du Zhang
The Journal of Physical Chemistry Letters 2016 Volume 7(Issue 13) pp:2407-2411
Publication Date(Web):June 13, 2016
DOI:10.1021/acs.jpclett.6b00936
The particle–particle random phase approximation (pp-RPA) and the particle–particle Tamm–Dancoff approximation (pp-TDA) are applied to the challenging conical intersection problem. Because they describe the ground and excited states on the same footing and naturally take into account the interstate interaction, these particle–particle methods, especially the pp-TDA, can correctly predict the dimensionality of the conical intersection seam as well as describe the potential energy surface in the vicinity of conical intersections. Though the bond length of conical intersections is slightly underestimated compared with the complete-active-space self-consistent field (CASSCF) theory, the efficient particle–particle methods are promising for conical intersections and nonadiabatic dynamics.
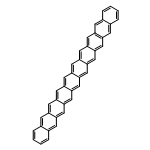
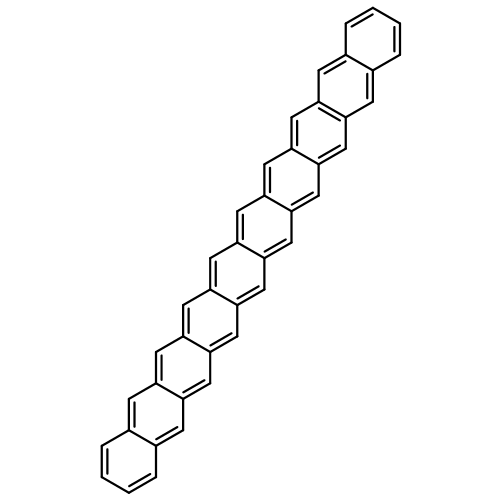
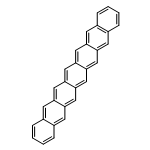
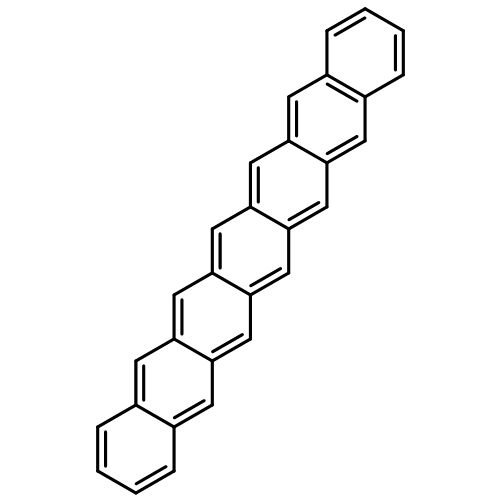
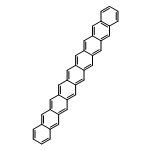
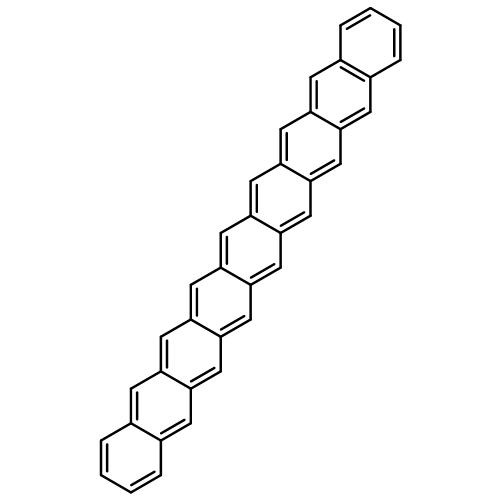
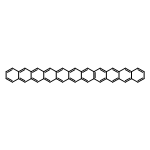
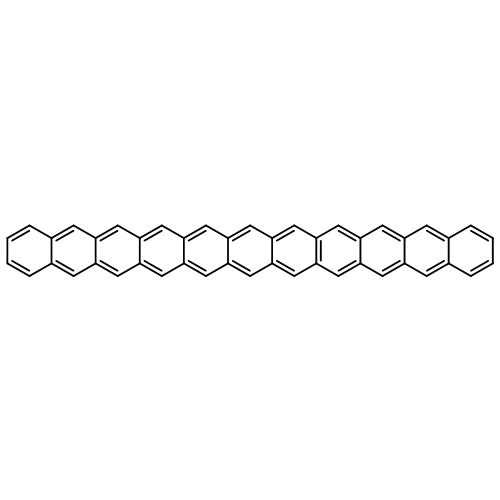
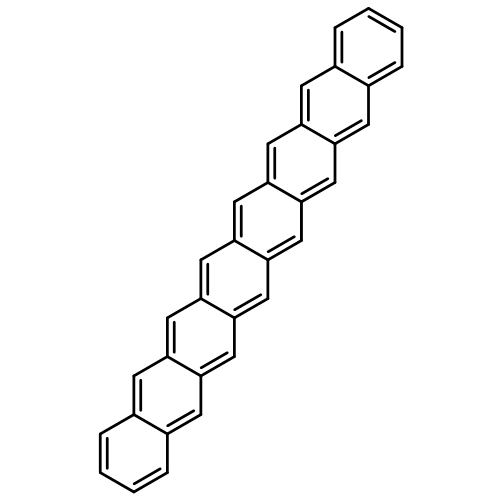
Co-reporter:Yang Yang;Ernest R. Davidson
PNAS 2016 Volume 113 (Issue 35 ) pp:E5098-E5107
Publication Date(Web):2016-08-30
DOI:10.1073/pnas.1606021113
Higher acenes have drawn much attention as promising organic semiconductors with versatile electronic properties. However,
the nature of their ground state and electronic excited states is still not fully clear. Their unusual chemical reactivity
and instability are the main obstacles for experimental studies, and the potentially prominent diradical character, which
might require a multireference description in such large systems, hinders theoretical investigations. Here, we provide a detailed
answer with the particle–particle random-phase approximation calculation. The 1Ag ground states of acenes up to decacene are on the closed-shell side of the diradical continuum, whereas the ground state
of undecacene and dodecacene tilts more to the open-shell side with a growing polyradical character. The ground state of all
acenes has covalent nature with respect to both short and long axes. The lowest triplet state 3B2u is always above the singlet ground state even though the energy gap could be vanishingly small in the polyacene limit. The
bright singlet excited state 1B2u is a zwitterionic state to the short axis. The excited 1Ag state gradually switches from a double-excitation state to another zwitterionic state to the short axis, but always keeps
its covalent nature to the long axis. An energy crossing between the 1B2u and excited 1Ag states happens between hexacene and heptacene. Further energetic consideration suggests that higher acenes are likely to
undergo singlet fission with a low photovoltaic efficiency; however, the efficiency might be improved if a singlet fission
into multiple triplets could be achieved.
Co-reporter:Du Zhang, Degao Peng, Peng Zhang and Weitao Yang
Physical Chemistry Chemical Physics 2015 vol. 17(Issue 2) pp:1025-1038
Publication Date(Web):12 Nov 2014
DOI:10.1039/C4CP04109G
The energy gradient for electronic excited states is of immense interest not only for spectroscopy but also for the theoretical study of photochemical reactions. We present the analytic excited state energy gradient of the particle-particle random phase approximation (pp-RPA). The analytic gradient formula is developed from an approach similar to that of time-dependent density-functional theory (TDDFT). The formula is verified for both the Hartree–Fock and (Generalized) Kohn–Sham reference states via comparison with finite difference results. The excited state potential energy surfaces and optimized geometries of some small molecules are investigated, yielding results of similar or better quality compared to adiabatic TDDFT. The singlet-to-triplet instability in TDDFT resulting in underestimated energies of the lowest triplet states is eliminated by pp-RPA. Charge transfer excitations and double excitations, which are challenging for most adiabatic TDDFT methods, can be reasonably well captured by pp-RPA. Within this framework, ground state potential energy surfaces of stretched single bonds can also be described well.
Co-reporter:Chetan Rupakheti, Aaron Virshup, Weitao Yang, and David N. Beratan
Journal of Chemical Information and Modeling 2015 Volume 55(Issue 3) pp:529-537
Publication Date(Web):January 16, 2015
DOI:10.1021/ci500749q
The small molecule universe (SMU) is defined as a set of over 1060 synthetically feasible organic molecules with molecular weight less than ∼500 Da. Exhaustive enumerations and evaluation of all SMU molecules for the purpose of discovering favorable structures is impossible. We take a stochastic approach and extend the ACSESS framework (Virshup et al. J. Am. Chem. Soc. 2013, 135, 7296–7303) to develop diversity oriented molecular libraries that can generate a set of compounds that is representative of the small molecule universe and that also biases the library toward favorable physical property values. We show that the approach is efficient compared to exhaustive enumeration and to existing evolutionary algorithms for generating such libraries by testing in the NKp fitness landscape model and in the fully enumerated GDB-9 chemical universe containing 3 × 105 molecules.
Co-reporter:Yang Yang, Degao Peng, Ernest R. Davidson, and Weitao Yang
The Journal of Physical Chemistry A 2015 Volume 119(Issue 20) pp:4923-4932
Publication Date(Web):April 20, 2015
DOI:10.1021/jp512727a
The particle–particle random phase approximation (pp-RPA) for calculating excitation energies has been applied to diradical systems. With pp-RPA, the two nonbonding electrons are treated in a subspace configuration interaction fashion while the remaining part is described by density functional theory (DFT). The vertical or adiabatic singlet–triplet energy gaps for a variety of categories of diradicals, including diatomic diradicals, carbene-like diradicals, disjoint diradicals, four-π-electron diradicals, and benzynes are calculated. Except for some excitations in four-π-electron diradicals, where four-electron correlation may play an important role, the singlet–triplet gaps are generally well predicted by pp-RPA. With a relatively low O(r4) scaling, the pp-RPA with DFT references outperforms spin-flip configuration interaction singles. It is similar to or better than the (variational) fractional-spin method. For small diradicals such as diatomic and carbene-like ones, the error of pp-RPA is slightly larger than noncollinear spin-flip time-dependent density functional theory (NC-SF-TDDFT) with LDA or PBE functional. However, for disjoint diradicals and benzynes, the pp-RPA performs much better and is comparable to NC-SF-TDDFT with long-range corrected ωPBEh functional and spin-flip configuration interaction singles with perturbative doubles (SF-CIS(D)). In particular, with a correct asymptotic behavior and being almost free from static correlation error, the pp-RPA with DFT references can well describe the challenging ground state and charge transfer excitations of disjoint diradicals in which almost all other DFT-based methods fail. Therefore, the pp-RPA could be a promising theoretical method for general diradical problems.
Co-reporter:Xiao Zheng;Chen Li;Dadi Zhang
Science China Chemistry 2015 Volume 58( Issue 12) pp:1825-1844
Publication Date(Web):2015 December
DOI:10.1007/s11426-015-5501-z
Delocalization error associated with the presently used density functional approximations is one of the main sources of errors which plague density functional theory calculations. In this paper, we give a comprehensive review on scaling correction (SC) approaches developed recently for reducing the delocalization error. The global and local SC approaches impose the rigorous Perdew-Parr-Levy-Balduz condition that the total electronic energy should scale linearly between integer electron numbers, on systems involving global and local fractional electron distributions, respectively. After presenting the theoretical background and mathematical formulation of scaling corrections, we demonstrate that they lead to universal alleviation of delocalization error. This is exemplified by the substantial improvement for the prediction of a wide range of electronic properties, including Kohn-Sham frontier orbital energies and band gaps, dissociation behavior of molecules, reaction barriers, electric polarizabilities, and charge-transfer species. The encouraging performances of SC approaches highlight their practicality and usefulness, and also affirm that an explicit treatment of fractional electron distributions is essentially important for reducing the intrinsic delocalization error. The existing limitations, the remaining challenges, and the future perspectives of SC are also discussed. Moreover, the SC approaches are compared with some existing methods attempting to remove the self-interaction error, such as the Perdew-Zunger self-interaction correction, the local hybrid hyper-generalized gradient approximations, and the rangeseparated density functional approximations. The unique advantages of SC suggest that it could open a novel and potentially paradigm-changing route for advancing density functional theory methods towards chemical accuracy.
Co-reporter:Aaron M. Virshup ; Julia Contreras-García ; Peter Wipf ; Weitao Yang ;David N. Beratan
Journal of the American Chemical Society 2013 Volume 135(Issue 19) pp:7296-7303
Publication Date(Web):April 2, 2013
DOI:10.1021/ja401184g
The “small molecule universe” (SMU), the set of all synthetically feasible organic molecules of 500 Da molecular weight or less, is estimated to contain over 1060 structures, making exhaustive searches for structures of interest impractical. Here, we describe the construction of a “representative universal library” spanning the SMU that samples the full extent of feasible small molecule chemistries. This library was generated using the newly developed Algorithm for Chemical Space Exploration with Stochastic Search (ACSESS). ACSESS makes two important contributions to chemical space exploration: it allows the systematic search of the unexplored regions of the small molecule universe, and it facilitates the mining of chemical libraries that do not yet exist, providing a near-infinite source of diverse novel compounds.
Co-reporter:Pan Wu, Robin Chaudret, Xiangqian Hu, and Weitao Yang
Journal of Chemical Theory and Computation 2013 Volume 9(Issue 5) pp:2226-2234
Publication Date(Web):March 18, 2013
DOI:10.1021/ct4001087
Noncovalent interactions play a central role in many chemical and biological systems. In a previous study, Johnson et al. developed a noncovalent interaction (NCI) index to characterize and visualize different types of weak interactions. To apply the NCI analysis to fluctuating environments as in the solution phase, we here developed a new averaged noncovalent interaction (i.e., aNCI) index along with a fluctuation index to characterize the magnitude of interactions and fluctuations. We applied aNCI for various systems including solute–solvent and ligand–protein noncovalent interactions. For water and benzene molecules in aqueous solution, solvation structures and the specific hydrogen bond patterns were visualized clearly. For the Cl– + CH3Cl SN2 reaction in aqueous solution, charge reorganization influences over solvation structure along SN2 reaction were revealed. For ligand–protein systems, aNCI can recover several key fluctuating hydrogen bond patterns that have potential applications for drug design. Therefore, aNCI, as a complementary approach to the original NCI method, can extract and visualize noncovalent interactions from thermal noise in fluctuating environments.
Co-reporter:Jun Wang and Weitao Yang
The Journal of Physical Chemistry B 2013 Volume 117(Issue 32) pp:9354-9361
Publication Date(Web):July 22, 2013
DOI:10.1021/jp404948c
Ribose-5-phosphate isomerase (Rpi) catalyzes the interconversion of d-ribose-5-phosphate and d-ribulose-5-phosphate and plays an essential role in the pentose phosphate pathway and the Calvin cycle of photosynthesis. RpiB, one of the two isoforms of Rpi, is also a potential drug target for some pathogenic bacteria. Clostridium thermocellum ribose-5-phosphate isomerase (CtRpi), belonging to the RpiB family, has recently been employed in the industrial production of rare sugars because of its fast reaction kinetics and narrow substrate specificity. It is known that this enzyme adopts a proton transfer mechanism. It was suggested that the deprotonated Cys65 attracts the proton at C2 of the substrate to initiate the isomerization reaction, and this step is the rate-limiting step. However the elaborate catalytic mechanism is still unclear. We have performed quantum mechanical/molecular mechanical simulations of this rate-limiting step of the reaction catalyzed by CtRpi with the substrate d-ribose. Our results demonstrate that the deprotonated Cys65 is not a stable reactant. Instead, our calculations revealed a concerted proton-transfer mechanism: Asp8, a highly conserved residue in the RpiB family, performs as the base to abstract the proton at Cys65 and Cys65 in turn abstracting the proton of the d-ribose simultaneously. Moreover, we found Thr67 cannot catalyze the proton transfer from O2 to O1 of the d-ribose alone. Water molecule(s) may assist this proton transfer with Thr67. Our findings lead to a clear understanding of the catalysis mechanism of the RpiB family and should guide experiments to increase the catalysis efficiency. This study also highlights the importance of initial protonation states of cysteines.
Co-reporter:Aron J. Cohen, Paula Mori-Sánchez, and Weitao Yang
Chemical Reviews 2012 Volume 112(Issue 1) pp:289
Publication Date(Web):December 22, 2011
DOI:10.1021/cr200107z
Co-reporter:Xiancheng Zeng, Xiangqian Hu, and Weitao Yang
Journal of Chemical Theory and Computation 2012 Volume 8(Issue 12) pp:4960-4967
Publication Date(Web):October 12, 2012
DOI:10.1021/ct300758v
A fragment-based fractional number of electrons (FNE) approach is developed to study entire electron transfer (ET) processes from the electron donor region to the acceptor region in the condensed phase. Both regions are described by the density-fragment interaction (DFI) method, while FNE as an efficient ET order parameter is applied to simulate the electron transfer process. In association with the QM/MM energy expression, the DFI-FNE method is demonstrated to describe ET processes robustly with the Ru2+–Ru3+ self-exchange ET as a proof-of-concept example. This method allows for systematic calculations of redox free energies, reorganization energies, electronic couplings and the absolute ET rate constants within the Marcus regime.
Co-reporter:Xiangqian Hu, Yingdi Jin, Xiancheng Zeng, Hao Hu and Weitao Yang
Physical Chemistry Chemical Physics 2012 vol. 14(Issue 21) pp:7700-7709
Publication Date(Web):20 Feb 2012
DOI:10.1039/C2CP23714H
We reformulate the density fragment interaction (DFI) approach [Fujimoto and Yang, J. Chem. Phys., 2008, 129, 054102.] to achieve linear-scaling quantum mechanical calculations for large molecular systems. Two key approximations are developed to improve the efficiency of the DFI approach and thus enable the calculations for large molecules: the electrostatic interactions between fragments are computed efficiently by means of polarizable electrostatic-potential-fitted atomic charges; and frozen fragment pseudopotentials, similar to the effective fragment potentials that can be fitted from interactions between small molecules, are employed to take into account the Pauli repulsion effect among fragments. Our reformulated and parallelized DFI method demonstrates excellent parallel performance based on the benchmarks for the system of 256 water molecules. Molecular dynamics simulations for the structural properties of liquid water also show a qualitatively good agreement with experimental measurements including the heat capacity, binding energy per water molecule, and the radial distribution functions of atomic pairs of O–O, O–H, and H–H. With this approach, large-scale quantum mechanical simulations for water and other liquids become feasible.
Co-reporter:Zuofeng Chen;Xiangqian Hu;Xiangsong Lin;Thomas J. Meyer;Shubin Liu;Javier J. Concepcion
PNAS 2012 Volume 109 (Issue 39 ) pp:
Publication Date(Web):2012-09-25
DOI:10.1073/pnas.1118344109
Water oxidation is a linchpin in solar fuels formation, and catalysis by single-site ruthenium complexes has generated significant
interest in this area. Combining several theoretical tools, we have studied the entire catalytic cycle of water oxidation
for a single-site catalyst starting with [RuII(tpy)(bpm)(OH2)]2+ (i.e., [RuII-OH2]2+; tpy is 2,2′∶6′,2′′-terpyridine and bpm is 2,2′-bypyrimidine) as a representative example of a new class of single-site catalysts. The redox
potentials and pKa calculations for the first two proton-coupled electron transfers (PCETs) from [RuII-OH2]2+ to [RuIV = O]2+ and the following electron-transfer process to [RuV = O]3+ suggest that these processes can proceed readily in acidic or weakly basic conditions. The subsequent water splitting process
involves two water molecules, [RuV = O]3+ to generate [RuIII-OOH]2+, and H3O+ with a low activation barrier (∼10 kcal/mol). After the key O---O bond forming step in the single-site Ru catalysis, another
PECT process oxidizes [RuIII-OOH]2+ to [RuIV-OO]2+ when the pH is lower than 3.7. Two possible forms of [RuIV-OO]2+, open and closed, can exist and interconvert with a low activation barrier (< 7 kcal/mol) due to strong spin-orbital coupling
effects. In Pathway 1 at pH = 1.0, oxygen release is rate-limiting with an activation barrier ∼12 kcal/mol while the electron-transfer
step from [RuIV-OO]2+ to [RuV - OO]3+ becomes rate-determining at pH = 0 (Pathway 2) with Ce(IV) as oxidant. The results of these theoretical studies with atomistic
details have revealed subtle details of reaction mechanisms at several stages during the catalytic cycle. This understanding
is helpful in the design of new catalysts for water oxidation.
Co-reporter:Pan Wu, G. Andrés Cisneros, Hao Hu, Robin Chaudret, Xiangqian Hu, and Weitao Yang
The Journal of Physical Chemistry B 2012 Volume 116(Issue 23) pp:6889-6897
Publication Date(Web):March 14, 2012
DOI:10.1021/jp212643j
4-Oxalocrotonate tautomerase (4-OT), a member of tautomerase superfamily, is an essential enzyme in the degradative metabolism pathway occurring in the Krebs cycle. The proton transfer process catalyzed by 4-OT has been explored previously using both experimental and theoretical methods; however, the elaborate catalytic mechanism of 4-OT still remains unsettled. By combining classical molecular mechanics with quantum mechanics, our results demonstrate that the native hexametric 4-OT enzyme, including six protein monomers, must be employed to simulate the proton transfer process in 4-OT due to protein–protein steric and electrostatic interactions. As a consequence, only three out of the six active sites in the 4-OT hexamer are observed to be occupied by three 2-oxo-4-hexenedioates (2o4hex), i.e., half-of-the-sites occupation. This agrees with experimental observations on negative cooperative effect between two adjacent substrates. Two sequential proton transfers occur: one proton from the C3 position of 2o4hex is initially transferred to the nitrogen atom of the general base, Pro1. Subsequently, the same proton is shuttled back to the position C5 of 2o4hex to complete the proton transfer process in 4-OT. During the catalytic reaction, conformational changes (i.e., 1-carboxyl group rotation) of 2o4hex may occur in the 4-OT dimer model but cannot proceed in the hexametric structure. We further explained that the docking process of 2o4hex can influence the specific reactant conformations and an alternative substrate (2-hydroxymuconate) may serve as reactant under a different reaction mechanism than 2o4hex.
Co-reporter:Julia Contreras-García, Erin R. Johnson, Shahar Keinan, Robin Chaudret, Jean-Philip Piquemal, David N. Beratan, and Weitao Yang
Journal of Chemical Theory and Computation 2011 Volume 7(Issue 3) pp:625-632
Publication Date(Web):January 25, 2011
DOI:10.1021/ct100641a
Noncovalent interactions hold the key to understanding many chemical, biological, and technological problems. Describing these noncovalent interactions accurately, including their positions in real space, constitutes a first step in the process of decoupling the complex balance of forces that define noncovalent interactions. Because of the size of macromolecules, the most common approach has been to assign van der Waals interactions (vdW), steric clashes (SC), and hydrogen bonds (HBs) based on pairwise distances between atoms according to their vdW radii. We recently developed an alternative perspective, derived from the electronic density: the non-covalent interactions (NCI) index [ J. Am. Chem. Soc. 2010, 132, 6498]. This index has the dual advantages of being generally transferable to diverse chemical applications and being very fast to compute, since it can be calculated from promolecular densities. Thus, NCI analysis is applicable to large systems, including proteins and DNA, where analysis of noncovalent interactions is of great potential value. Here, we describe the NCI computational algorithms and their implementation for the analysis and visualization of weak interactions, using both self-consistent fully quantum-mechanical as well as promolecular densities. A wide range of options for tuning the range of interactions to be plotted is also presented. To demonstrate the capabilities of our approach, several examples are given from organic, inorganic, solid state, and macromolecular chemistry, including cases where NCI analysis gives insight into unconventional chemical bonding. The NCI code and its manual are available for download at http://www.chem.duke.edu/∼yang/software.htm.
Co-reporter:Julia Contreras-García and Weitao Yang and Erin R. Johnson
The Journal of Physical Chemistry A 2011 Volume 115(Issue 45) pp:12983-12990
Publication Date(Web):July 25, 2011
DOI:10.1021/jp204278k
Hydrogen bonds are of crucial relevance to many problems in chemistry, biology, and materials science. The recently developed NCI (noncovalent interactions) index enables real-space visualization of both attractive (van der Waals and hydrogen-bonding) and repulsive (steric) interactions based on properties of the electron density. It is thus an optimal index to describe the interplay of stabilizing and destabilizing contributions that determine stable minima on hydrogen-bonding potential-energy surfaces (PESs). In the framework of density-functional theory, energetics are completely determined by the electron density. Consequently, NCI will be shown to allow quantitative treatment of hydrogen-bond energetics. The evolution of NCI regions along a PES follows a well-behaved pattern which, upon integration of the electron density, is capable of mimicking conventional hydrogen-bond interatomic potentials.
Co-reporter:Pan Wu, Xiangqian Hu, and Weitao Yang
The Journal of Physical Chemistry Letters 2011 Volume 2(Issue 17) pp:2099-2103
Publication Date(Web):August 4, 2011
DOI:10.1021/jz200808x
We present a new approach to combine λ dynamics with metadynamics (named λ-metadynamics) to compute free energy surfaces with respect to λ. Particularly, the λ-metadynamics method extends metadynamics to a single virtual variable λ, i.e., the coupling parameter between solute and solvent, to compute absolute solvation free energy as an exemplary application. We demonstrate that λ-metadynamics simulations can recover the accurate potential of mean force surface with respect to λ compared to the benchmark results from traditional λ-dynamics with umbrella sampling. The solvation free energy results for five small organic molecules from λ-metadynamics simulations using the same filling scheme show that the statistical errors are within ±0.5 kcal/mol. The new λ-metadynamics method is general, and other variables such as order parameters to describe conformational changes can be easily combined with λ-metadynamics. This should allow for efficient samplings on high-dimension free energy landscapes.Keywords: metadynamics; QM/MM; solvation free energy; λ dynamics; λ-metadynamics;
Co-reporter:Daniel H. Ess, Erin R. Johnson, Xiangqian Hu, and Weitao Yang
The Journal of Physical Chemistry A 2011 Volume 115(Issue 1) pp:76-83
Publication Date(Web):December 9, 2010
DOI:10.1021/jp109280y
Open-shell singlet diradicals are difficult to model accurately within conventional Kohn−Sham (KS) density-functional theory (DFT). These methods are hampered by spin contamination because the KS determinant wave function is neither a pure spin state nor an eigenfunction of the S2 operator. Here we present a theoretical foray for using single-reference closed-shell ground states to describe diradicals by fractional-spin DFT (FS-DFT). This approach allows direct, self-consistent calculation of electronic properties using the electron density corresponding to the proper spin eigenfunction. The resulting FS-DFT approach is benchmarked against diradical singlet−triplet gaps for atoms and small molecules. We have also applied FS-DFT to the singlet−triplet gaps of hydrocarbon polyacenes.
Co-reporter:Erin R. Johnson ; Shahar Keinan ; Paula Mori-Sánchez ; Julia Contreras-García ; Aron J. Cohen
Journal of the American Chemical Society 2010 Volume 132(Issue 18) pp:6498-6506
Publication Date(Web):April 15, 2010
DOI:10.1021/ja100936w
Molecular structure does not easily identify the intricate noncovalent interactions that govern many areas of biology and chemistry, including design of new materials and drugs. We develop an approach to detect noncovalent interactions in real space, based on the electron density and its derivatives. Our approach reveals the underlying chemistry that compliments the covalent structure. It provides a rich representation of van der Waals interactions, hydrogen bonds, and steric repulsion in small molecules, molecular complexes, and solids. Most importantly, the method, requiring only knowledge of the atomic coordinates, is efficient and applicable to large systems, such as proteins or DNA. Across these applications, a view of nonbonded interactions emerges as continuous surfaces rather than close contacts between atom pairs, offering rich insight into the design of new and improved ligands.
Co-reporter:Xiangqian Hu ; Hao Hu ; Jeffrey A. Melvin ; Kathleen W. Clancy ; Dewey G. McCafferty
Journal of the American Chemical Society 2010 Volume 133(Issue 3) pp:478-485
Publication Date(Web):December 13, 2010
DOI:10.1021/ja107513t
Many Gram-positive pathogens possess external pili or fimbriae with which they adhere to host cells during the infection process. Unusual dual intramolecular isopeptide bonds between Asn and Lys side chains within the N-terminal and C-terminal domains of the pilus subunits have been observed initially in the Streptococcus pyogenes pilin subunit Spy0128 and subsequently in GBS52 from Streptococcus agalactiae, in the BcpA major pilin of Bacillus cereus and in the RrgB pilin of Streptococcus pneumoniae, among others. Within each pilin subunit, intramolecular isopeptide bonds serve to stabilize the protein. These bonds provide a means to withstand large external mechanical forces, as well as possibly assisting in supporting a conformation favored for pilin subunit polymerization via sortase transpeptidases. Genome-wide analyses of pili-containing Gram-positive bacteria are known or suspected to contain isopeptide bonds in pilin subunits. For the autocatalytic formation of isopeptide cross-links, a conservation of three amino acids including Asn, Lys, and a catalytically important acidic Glu (or Asp) residue are responsible. However, the chemical mechanism of how isopeptide bonds form within pilin remains poorly understood. Although it is possible that several mechanistic paths could lead to isopeptide bond formation in pili, the requirement of a conserved glutamate and highly organized positioning of residues within the hydrophobic environment of the active site were found in numerous pilin crystal structures such as Spy0128 and RrgB. This suggests a mechanism involving direct coupling of lysine side chain amine to the asparagine carboxamide mediated by critical acid/base or hydrogen bonding interactions with the catalytic glutamate residue. From this mechanistic perspective, we used the QM/MM minimum free-energy path method to examine the reaction details of forming the isopeptide bonds with Spy0128 as a model pilin, specifically focusing on the role of the glutamate in catalysis. It was determined that the reaction mechanism likely consists of two major steps: the nucleophilic attack on Cγ by nitrogen in the unprotonated Lys ε-amino group and, then two concerted proton transfers occur during the formation of the intramolecular isopeptide bond to subsequently release ammonia. More importantly, within the dual active sites of Spy0128, Glu117, and Glu258 residues function as crucial catalysts for each isopeptide bond formation, respectively, by relaying two proton transfers. This work also suggests that domain−domain interactions within Spy0128 may modulate the reactivity of residues within each active site. Our results may hopefully shed light on the molecular mechanisms of pilin biogenesis in Gram-positive bacteria.
Co-reporter:Ganglong Cui, Weihai Fang and Weitao Yang
Physical Chemistry Chemical Physics 2010 vol. 12(Issue 2) pp:416-421
Publication Date(Web):13 Nov 2009
DOI:10.1039/B916688B
Time-dependent density functional theory (TDDFT) has broad application in the study of electronic response, excitation and transport. To extend such application to large and complex systems, we develop a reformulation of TDDFT equations in terms of non-orthogonal localized molecular orbitals (NOLMOs). NOLMO is the most localized representation of electronic degrees of freedom and has been used in ground state calculations. In atomic orbital (AO) representation, the sparsity of NOLMO is transferred to the coefficient matrix of molecular orbitals (MOs). Its novel use in TDDFT here leads to a very simple form of time propagation equations which can be solved with linear-scaling effort. We have tested the method for several long-chain saturated and conjugated molecular systems within the self-consistent charge density-functional tight-binding method (SCC-DFTB) and demonstrated its accuracy. This opens up pathways for TDDFT applications to large bio- and nano- systems.
Co-reporter:Hao Hu and Weitao Yang
The Journal of Physical Chemistry B 2010 Volume 114(Issue 8) pp:2755-2759
Publication Date(Web):February 2, 2010
DOI:10.1021/jp905886q
Computer simulations of reaction processes in solution in general rely on the definition of a reaction coordinate and the determination of the thermodynamic changes of the system along the reaction coordinate. The reaction coordinate often is constituted of characteristic geometrical properties of the reactive solute species, while the contributions of solvent molecules are implicitly included in the thermodynamics of the solute degrees of freedoms. However, solvent dynamics can provide the driving force for the reaction process, and in such cases explicit description of the solvent contribution in the free energy of the reaction process becomes necessary. We report here a method that can be used to analyze the solvent contributions to the reaction activation free energies from the combined QM/MM minimum free-energy path simulations. The method was applied to the self-exchange SN2 reaction of CH3Cl + Cl−, showing that the importance of solvent−solute interactions to the reaction process. The results were further discussed in the context of coupling between solvent and solute molecules in reaction processes.
Co-reporter:Ganglong Cui, Weihai Fang, and Weitao Yang
The Journal of Physical Chemistry A 2010 Volume 114(Issue 33) pp:8878-8883
Publication Date(Web):June 15, 2010
DOI:10.1021/jp1027838
Localized molecular orbitals (LMOs) are much more compact representations of electronic degrees of freedom than canonical molecular orbitals (CMOs). The most compact representation is provided by nonorthogonal localized molecular orbitals (NOLMOs), which are linearly independent but are not orthogonal. Both LMOs and NOLMOs are thus useful for linear-scaling calculations of electronic structures for large systems. Recently, NOLMOs have been successfully applied to linear-scaling calculations with density functional theory (DFT) and to reformulating time-dependent density functional theory (TDDFT) for calculations of excited states and spectroscopy. However, a challenge remains as NOLMO construction from CMOs is still inefficient for large systems. In this work, we develop an efficient method to accelerate the NOLMO construction by using predefined centroids of the NOLMO and thereby removing the nonlinear equality constraints in the original method ( J. Chem. Phys. 2004, 120, 9458 and J. Chem. Phys. 2000, 112, 4). Thus, NOLMO construction becomes an unconstrained optimization. Its efficiency is demonstrated for the selected saturated and conjugated molecules. Our method for fast NOLMO construction should lead to efficient DFT and NOLMO-TDDFT applications to large systems.
Co-reporter:Xiangqian Hu, Dequan Xiao, Shahar Keinan, Inge Asselberghs, Michael J. Therien, Koen Clays, Weitao Yang and David N. Beratan
The Journal of Physical Chemistry C 2010 Volume 114(Issue 5) pp:2349-2359
Publication Date(Web):January 15, 2010
DOI:10.1021/jp911556x
Successfully predicting the frequency dispersion of electronic hyperpolarizabilities is an unresolved challenge in materials science and electronic structure theory. We show that the generalized Thomas−Kuhn sum rules, combined with linear absorption data and measured hyperpolarizability at one or two frequencies, may be used to predict the entire frequency-dependent electronic hyperpolarizability spectrum. This treatment includes two- and three-level contributions that arise from the lowest two or three excited electronic state manifolds, enabling us to describe the unusual observed frequency dispersion of the dynamic hyperpolarizability in high oscillator strength M-PZn chromophores, where (porphinato)zinc(II) (PZn) and metal(II)polypyridyl (M) units are connected via an ethyne unit that aligns the high oscillator strength transition dipoles of these components in a head-to-tail arrangement. We show that some of these structures can possess very similar linear absorption spectra yet manifest dramatically different frequency-dependent hyperpolarizabilities, because of three-level contributions that result from excited state-to-excited state transition dipoles among charge polarized states. Importantly, this approach provides a quantitative scheme to use linear optical absorption spectra and very limited individual hyperpolarizability measurements to predict the entire frequency-dependent nonlinear optical response.
Co-reporter:Aron J. Cohen, Paula Mori-Sánchez and Weitao Yang
Journal of Chemical Theory and Computation 2009 Volume 5(Issue 4) pp:786-792
Publication Date(Web):March 18, 2009
DOI:10.1021/ct8005419
In this work the behavior of MP2 for fractional occupations is investigated. The consideration of fractional charge behavior gives a simple derivation of an expression for the chemical potential (or the derivative of energy with respect to the number of electrons) of MP2. A generalized optimized effective potential formalism (OEP) has been developed in which the OEP is a nonlocal potential, which can be applied to explicit functionals of the orbitals and eigenvalues and also facilitates the evaluation of the chemical potential. The MP2 derivative improves upon the corresponding Koopmans’ theorem in Hartree−Fock theory for the ionization energy and also gives a good estimate of the electron affinity. In strongly correlated systems with degeneracies and fractional spins, MP2 diverges, and another corrected second-order perturbative method ameliorates this failure for the energy but still does not recapture the correct behavior for the energy derivatives that yield the gap. Overall we present a view of wave function based methods and their behavior for fractional charges and spins that offers insight into the application of these methods to challenging chemical problems.
Co-reporter:Hao Hu, Weitao Yang
Journal of Molecular Structure: THEOCHEM 2009 Volume 898(1–3) pp:17-30
Publication Date(Web):30 March 2009
DOI:10.1016/j.theochem.2008.12.025
Determining the free energies and mechanisms of chemical reactions in solution and enzymes is a major challenge. For such complex reaction processes, combined quantum mechanics/molecular mechanics (QM/MM) method is the most effective simulation method to provide an accurate and efficient theoretical description of the molecular system. The computational costs of ab initio QM methods, however, have limited the application of ab initio QM/MM methods. Recent advances in ab initio QM/MM methods allowed accurate simulation of the free energies for reactions in solution and in enzymes and thus paved the way for broader applications of the ab initio QM/MM methods. We review here the theoretical developments and applications of the ab initio QM/MM methods, focusing on the determination of reaction path and the free energies of the reaction processes in solution and enzymes.
Co-reporter:Jerry M. Parks, Hao Hu, Johannes Rudolph and Weitao Yang
The Journal of Physical Chemistry B 2009 Volume 113(Issue 15) pp:5217-5224
Publication Date(Web):March 20, 2009
DOI:10.1021/jp805137x
Cdc25B is a dual-specificity phosphatase that catalyzes the dephosphorylation of the Cdk2/CycA protein complex. This enzyme is an important regulator of the human cell cycle and has been identified as a potential anticancer target. In general, protein tyrosine phosphatases are thought to bind the dianionic form of the phosphate and employ general acid catalysis via the Asp residue in the highly conserved WPD-loop. However, the Cdc25 phosphatases form a special subfamily based on their distinct differences from other protein tyrosine phosphatases. Although Cdc25B contains the (H/V)CX5R catalytic motif present in all other protein tyrosine phosphatases, it lacks an analogous catalytic acid residue. No crystallographic data currently exist for the complex of Cdc25B with Cdk2/CycA, so in addition to its natural protein substrate, experimental and theoretical studies are often carried out with small molecule substrates. In an effort to gain understanding of the dephosphorylation mechanism of Cdc25B with a commonly used small molecule substrate, we have performed simulations of the rate-limiting step of the reaction catalyzed by Cdc25B with the substrate p-nitrophenyl phosphate using the recently developed QM/MM Minimum Free Energy Path method (Hu et al. J. Chem. Phys. 2008, 034105). We have simulated the first step of the reaction with both the monoanionic and the dianionic forms of the substrate, and our calculations favor a mechanism involving the monoanionic form. Thus, Cdc25 may employ a unique dephosphorylation mechanism among protein tyrosine phosphatases, at least in the case of the small molecule substrate p-nitrophenyl phosphate.
Co-reporter:Shahar Keinan, Michael J. Therien, David N. Beratan and Weitao Yang
The Journal of Physical Chemistry A 2008 Volume 112(Issue 47) pp:12203-12207
Publication Date(Web):October 30, 2008
DOI:10.1021/jp806351d
Nonlinear optical chromophores containing (porphyrinato)Zn(II), proquinoid, and (terpyridyl)metal(II) building blocks were optimized in a library containing ∼106 structures using the linear combination of atomic potentials (LCAP) methodology. We report here the library design and molecular property optimizations. Two basic structural types of large β0 chromophores were examined: linear and T-shaped motifs. These T-shaped geometries suggest a promising NLO chromophoric architecture for experimental investigation and further support the value of performing LCAP searches in large chemical spaces.
Co-reporter:Aron J. Cohen;Paula Mori-Sánchez
Science 2008 Volume 321(Issue 5890) pp:792-794
Publication Date(Web):
DOI:10.1126/science.1158722
Abstract
Density functional theory of electronic structure is widely and successfully applied in simulations throughout engineering and sciences. However, for many predicted properties, there are spectacular failures that can be traced to the delocalization error and static correlation error of commonly used approximations. These errors can be characterized and understood through the perspective of fractional charges and fractional spins introduced recently. Reducing these errors will open new frontiers for applications of density functional theory.
Co-reporter:Whasil Lee, Xiancheng Zeng, Kristina Rotolo, Ming Yang, Christopher J. Schofield, Vann Bennett, Weitao Yang, Piotr E. Marszalek
Biophysical Journal (7 March 2012) Volume 102(Issue 5) pp:
Publication Date(Web):7 March 2012
DOI:10.1016/j.bpj.2012.01.046
Red blood cells are frequently deformed and their cytoskeletal proteins such as spectrin and ankyrin-R are repeatedly subjected to mechanical forces. While the mechanics of spectrin was thoroughly investigated in vitro and in vivo, little is known about the mechanical behavior of ankyrin-R. In this study, we combine coarse-grained steered molecular dynamics simulations and atomic force spectroscopy to examine the mechanical response of ankyrin repeats (ARs) in a model synthetic AR protein NI6C, and in the D34 fragment of native ankyrin-R when these proteins are subjected to various stretching geometry conditions. Our steered molecular dynamics results, supported by AFM measurements, reveal an unusual mechanical anisotropy of ARs: their mechanical stability is greater when their unfolding is forced to propagate from the N-terminus toward the C-terminus (repeats unfold at ∼60 pN), as compared to the unfolding in the opposite direction (unfolding force ∼ 30 pN). This anisotropy is also reflected in the complex refolding behavior of ARs. The origin of this unfolding and refolding anisotropy is in the various numbers of native contacts that are broken and formed at the interfaces between neighboring repeats depending on the unfolding/refolding propagation directions. Finally, we discuss how these complex mechanical properties of ARs in D34 may affect its behavior in vivo.
Co-reporter:Xiancheng Zeng, Hao Hu, Huan-Xiang Zhou, Piotr E. Marszalek, Weitao Yang
Biophysical Journal (17 February 2010) Volume 98(Issue 4) pp:
Publication Date(Web):17 February 2010
DOI:10.1016/j.bpj.2009.11.004
In the studies of force-induced conformational transitions of biomolecules, the large timescale difference from experiments presents the challenge of obtaining convergent sampling for molecular dynamics simulations. To circumvent this fundamental problem, an approach combining the replica-exchange method and umbrella sampling (REM-US) was developed to simulate mechanical stretching of biomolecules under equilibrium conditions. Equilibrium properties of conformational transitions can be obtained directly from simulations without further assumptions. To test the performance, we carried out REM-US simulations of atomic force microscope (AFM) stretching and relaxing measurements on the polysaccharide pustulan, a (1→6)-β-D-glucan, which undergoes well-characterized rotameric transitions in the backbone bonds. With significantly enhanced sampling convergence and efficiency, the REM-US approach closely reproduced the equilibrium force-extension curves measured in AFM experiments. Consistent with the reversibility in the AFM measurements, the new approach generated identical force-extension curves in both stretching and relaxing simulations—an outcome not reported in previous studies, proving that equilibrium conditions were achieved in the simulations. REM-US may provide a robust approach to modeling of mechanical stretching on polysaccharides and even nucleic acids.
Co-reporter:Xiangqian Hu, Yingdi Jin, Xiancheng Zeng, Hao Hu and Weitao Yang
Physical Chemistry Chemical Physics 2012 - vol. 14(Issue 21) pp:NaN7709-7709
Publication Date(Web):2012/02/20
DOI:10.1039/C2CP23714H
We reformulate the density fragment interaction (DFI) approach [Fujimoto and Yang, J. Chem. Phys., 2008, 129, 054102.] to achieve linear-scaling quantum mechanical calculations for large molecular systems. Two key approximations are developed to improve the efficiency of the DFI approach and thus enable the calculations for large molecules: the electrostatic interactions between fragments are computed efficiently by means of polarizable electrostatic-potential-fitted atomic charges; and frozen fragment pseudopotentials, similar to the effective fragment potentials that can be fitted from interactions between small molecules, are employed to take into account the Pauli repulsion effect among fragments. Our reformulated and parallelized DFI method demonstrates excellent parallel performance based on the benchmarks for the system of 256 water molecules. Molecular dynamics simulations for the structural properties of liquid water also show a qualitatively good agreement with experimental measurements including the heat capacity, binding energy per water molecule, and the radial distribution functions of atomic pairs of O–O, O–H, and H–H. With this approach, large-scale quantum mechanical simulations for water and other liquids become feasible.
Co-reporter:Du Zhang, Degao Peng, Peng Zhang and Weitao Yang
Physical Chemistry Chemical Physics 2015 - vol. 17(Issue 2) pp:NaN1038-1038
Publication Date(Web):2014/11/12
DOI:10.1039/C4CP04109G
The energy gradient for electronic excited states is of immense interest not only for spectroscopy but also for the theoretical study of photochemical reactions. We present the analytic excited state energy gradient of the particle-particle random phase approximation (pp-RPA). The analytic gradient formula is developed from an approach similar to that of time-dependent density-functional theory (TDDFT). The formula is verified for both the Hartree–Fock and (Generalized) Kohn–Sham reference states via comparison with finite difference results. The excited state potential energy surfaces and optimized geometries of some small molecules are investigated, yielding results of similar or better quality compared to adiabatic TDDFT. The singlet-to-triplet instability in TDDFT resulting in underestimated energies of the lowest triplet states is eliminated by pp-RPA. Charge transfer excitations and double excitations, which are challenging for most adiabatic TDDFT methods, can be reasonably well captured by pp-RPA. Within this framework, ground state potential energy surfaces of stretched single bonds can also be described well.
Co-reporter:Ganglong Cui, Weihai Fang and Weitao Yang
Physical Chemistry Chemical Physics 2010 - vol. 12(Issue 2) pp:NaN421-421
Publication Date(Web):2009/11/13
DOI:10.1039/B916688B
Time-dependent density functional theory (TDDFT) has broad application in the study of electronic response, excitation and transport. To extend such application to large and complex systems, we develop a reformulation of TDDFT equations in terms of non-orthogonal localized molecular orbitals (NOLMOs). NOLMO is the most localized representation of electronic degrees of freedom and has been used in ground state calculations. In atomic orbital (AO) representation, the sparsity of NOLMO is transferred to the coefficient matrix of molecular orbitals (MOs). Its novel use in TDDFT here leads to a very simple form of time propagation equations which can be solved with linear-scaling effort. We have tested the method for several long-chain saturated and conjugated molecular systems within the self-consistent charge density-functional tight-binding method (SCC-DFTB) and demonstrated its accuracy. This opens up pathways for TDDFT applications to large bio- and nano- systems.
.jpg)